
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence(Hexafluoroacetylacetonato)copper(I)–cycloalkyne complexes as protected cycloalkynes†
Naoaki
Makio
a,
Yuki
Sakata
a,
Tomoko
Kuribara
a,
Keisuke
Adachi
a,
Yasutomo
Hatakeyama
a,
Tomohiro
Meguro
a,
Kazunobu
Igawa
 b,
Katsuhiko
Tomooka
b,
Takamitsu
Hosoya
a and
Suguru
Yoshida
*a
b,
Katsuhiko
Tomooka
b,
Takamitsu
Hosoya
a and
Suguru
Yoshida
*a
aLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: s-yoshida.cb@tmd.ac.jp
bInstitute for Materials Chemistry and Engineering, Kyushu University, 6-1 Kasuga-koen, Kasuga, Fukuoka 816-8580, Japan
First published on 20th August 2020
Abstract
A protection method for cycloalkynes by the formation of (hexafluoroacetylacetonato)copper(I)–cycloalkyne complexes is disclosed. Various complexes having functional groups were efficiently prepared, which are easily purified by silica-gel column chromatography. Selective click reactions were realized through the complexation of cycloalkynes with copper.
Strain-promoted azide–alkyne cycloaddition (SPAAC) reactions have served as catalyst-free click reactions in broad disciplines including materials chemistry, pharmaceutical sciences, and chemical biology.1–5 To realize efficient molecular conjugations, a variety of cycloalkynes have been developed so far.4 However, controlled SPAAC reactions for preparing multifunctional molecules by assembling a number of functional molecules is still challenging due to the significantly reactive cycloalkynes and immature protecting group chemistry.5,6 In the course of our studies on click chemistry,7 we found transient protection methods of cycloalkynes from triazole formation with azides by complexation with cationic copper, silver, or gold (Fig. 1A).8 Furthermore, we developed an efficient synthetic method of functionalized cycloalkynes from functionalized azides by complexation of cycloalkynes having an ethynyl group with cationic copper followed by copper-catalyzed triazole formation at the ethynyl group and subsequent deprotection with chelators (Fig. 1B).8a,b Herein, we disclose an alternative method for the protection of cycloalkynes by complexation with [bis(trimethylsilyl)acetylene](hexafluoroacetylacetonato)copper(I) ((btmsa)Cu(hfacac)),9 in which the cycloalkyne–copper complexes can be easily purified by silica-gel column chromatography (Fig. 1C).
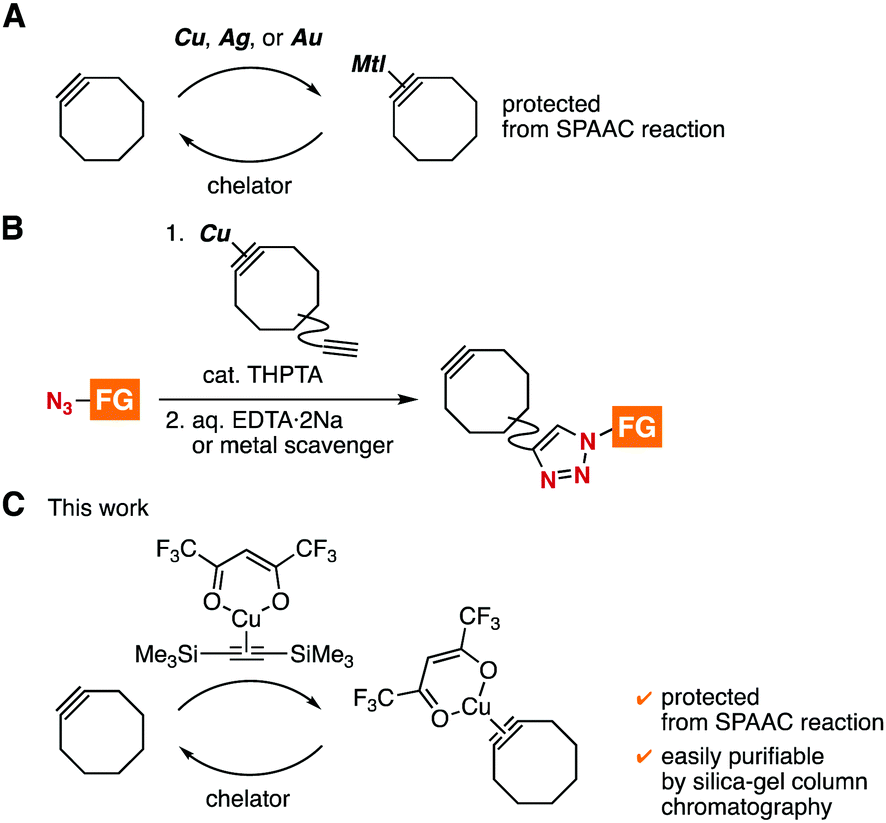 | ||
| Fig. 1 Synthetic methods through cycloalkyne–metal complexes. (A) Transient protection of cycloalkynes by complexation with copper, silver, or gold. (B) Azide-to-cycloalkyne switching approach. (C) This work. FG = functional group. | ||
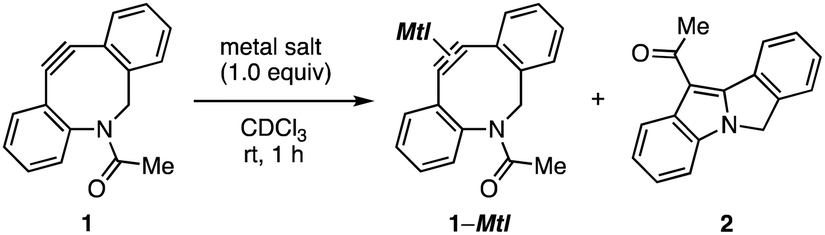
In our recent studies of cycloalkyne–metal complexes,8,10 we faced a limitation in the protection of dibenzo-fused azacyclooctyne (DIBAC)4d by cationic copper, silver, or gold (Table 1). Although we succeeded in the protection of DIBAC 1 with (MeCN)4CuBF4 in methanol,8a,11 cyclization took place to afford tetracyclic compound 2 when treating DIBAC 1 with cationic copper, silver, or gold salt in CDCl3 (entries 1–3). The ring formation providing 2 would proceed through π-activation of cycloalkyne 1 by cationic metal salts followed by intramolecular nucleophilic attack of the nitrogen and acyl migration (Fig. 2).12 This mechanistically intriguing but undesired cyclization motivated us to develop an alternative protection method of cycloalkynes.
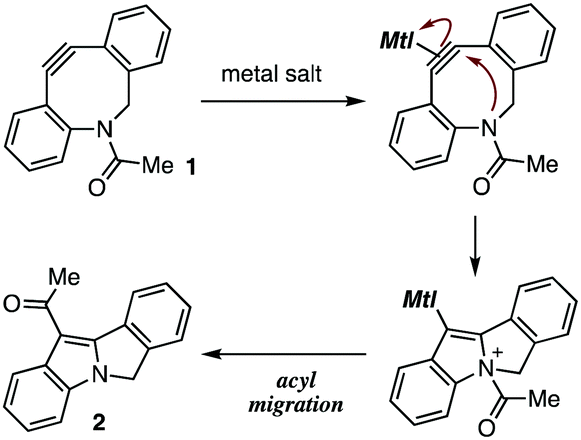 | ||
| Fig. 2 Plausible reaction mechanism of undesired ring-closure. | ||
After screening of alternative methods for the protection using dibenzo-fused cyclooctyne (DIBO) 34c as a model cycloalkyne, we found that ligand exchange of [bis(trimethylsilyl)acetylene](hexafluoroacetylacetonato)copper(I) ((btmsa)Cu(hfacac)) with 3 took place efficiently to provide cycloalkyne–copper complex 4a quantitatively (Fig. 3A). It is worthy to note that complex 4a was easily purified by silica-gel column chromatography owing to the high stability and low polarity.13 Treatment of DIBO–copper complex 4a with azide 5 in dichloromethane resulted in the recovery of azide 5 in excellent yield without triazole formation, clearly showing that cycloalkyne–copper complex 4a worked as a protected cycloalkyne (Fig. 3B, upper).14 Deprotection of 4a successfully proceeded by the treatment with chelators or nitrogen ligands such as aqueous ammonia (Fig. 3B, lower). Increasing the amounts of (btmsa)Cu(hfacac) from 0.1 to 1.0 equiv. in complexation with cyclooctyne 3 resulted in a gradual upfield shift of alkynic sp carbon signals in 13C NMR analyses (Fig. 3C). This result indicates the equilibrium formation of DIBO–copper complex 4a.
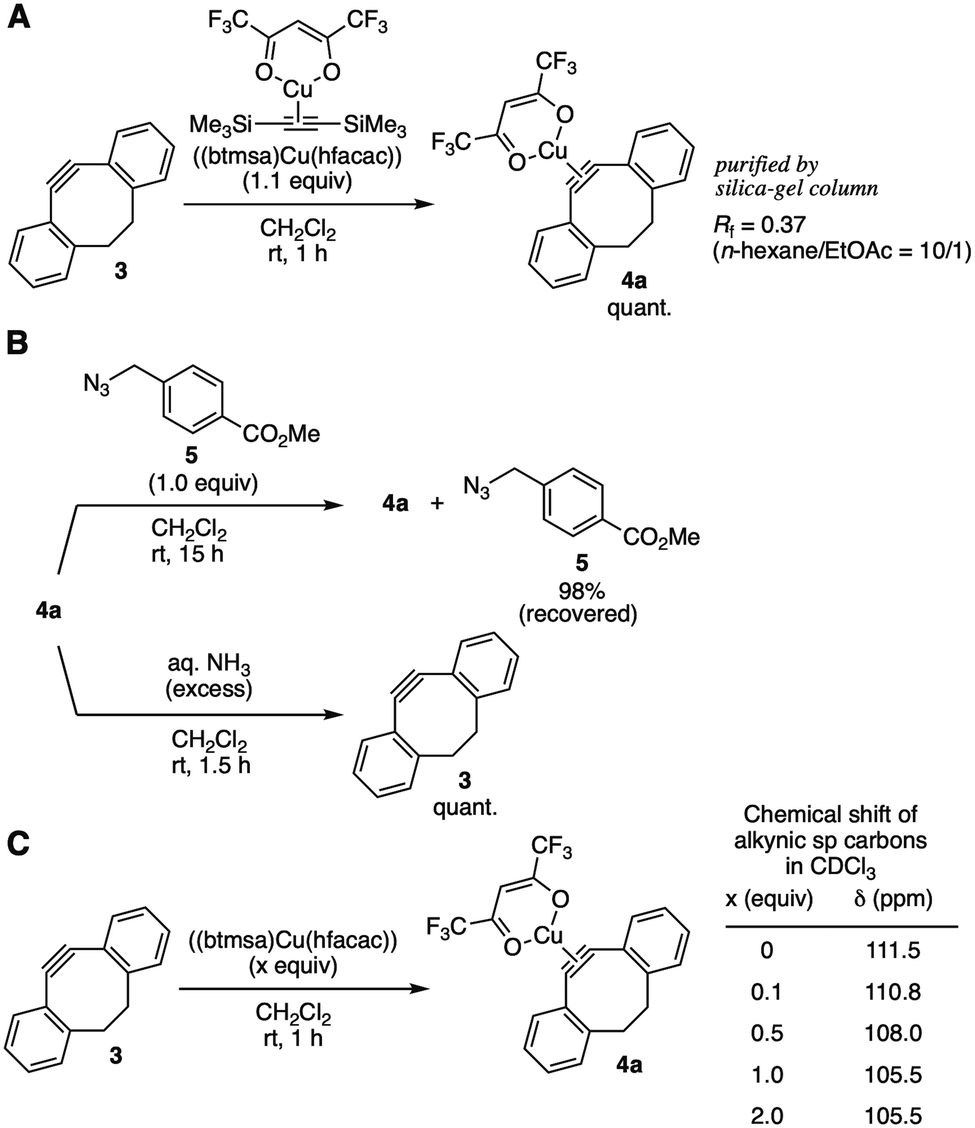 | ||
| Fig. 3 DIBO–(btmsa)Cu(hfacac) complex 4a. (A) Complexation of 3 with (btmsa)Cu(hfacac). (B) Reactivity toward azide 5 and deprotection. (C) NMR study. | ||
A wide range of cycloalkynes involving DIBAC 1 participated in the complexation with (btmsa)Cu(hfacac) (Fig. 4). For instance, the complexation of DIBOs with copper smoothly proceeded to furnish 4b and 4c in high yields without affecting by hydroxy and ethynyl groups. Of note, we succeeded in preparing DIBAC–copper complex 4d without undesired cyclization, clearly demonstrating an advantage of Cu(hfacac) complexes compared to cycloalkyne complexes with cationic metal salts. A variety of bicyclo[6.1.0]nonyne (BCN) complexes 4f–g were prepared without damaging not only hydroxy group but also active carbonate and terminal alkyne moieties.4f We also accomplished the complexation of 4,8-diazacyclononynes (DACNs) with (btmsa)Cu(hfacac) to afford 4h and 4i leaving cycloalkyne, sulfonamide, amide, and terminal alkyne moieties untouched.4k
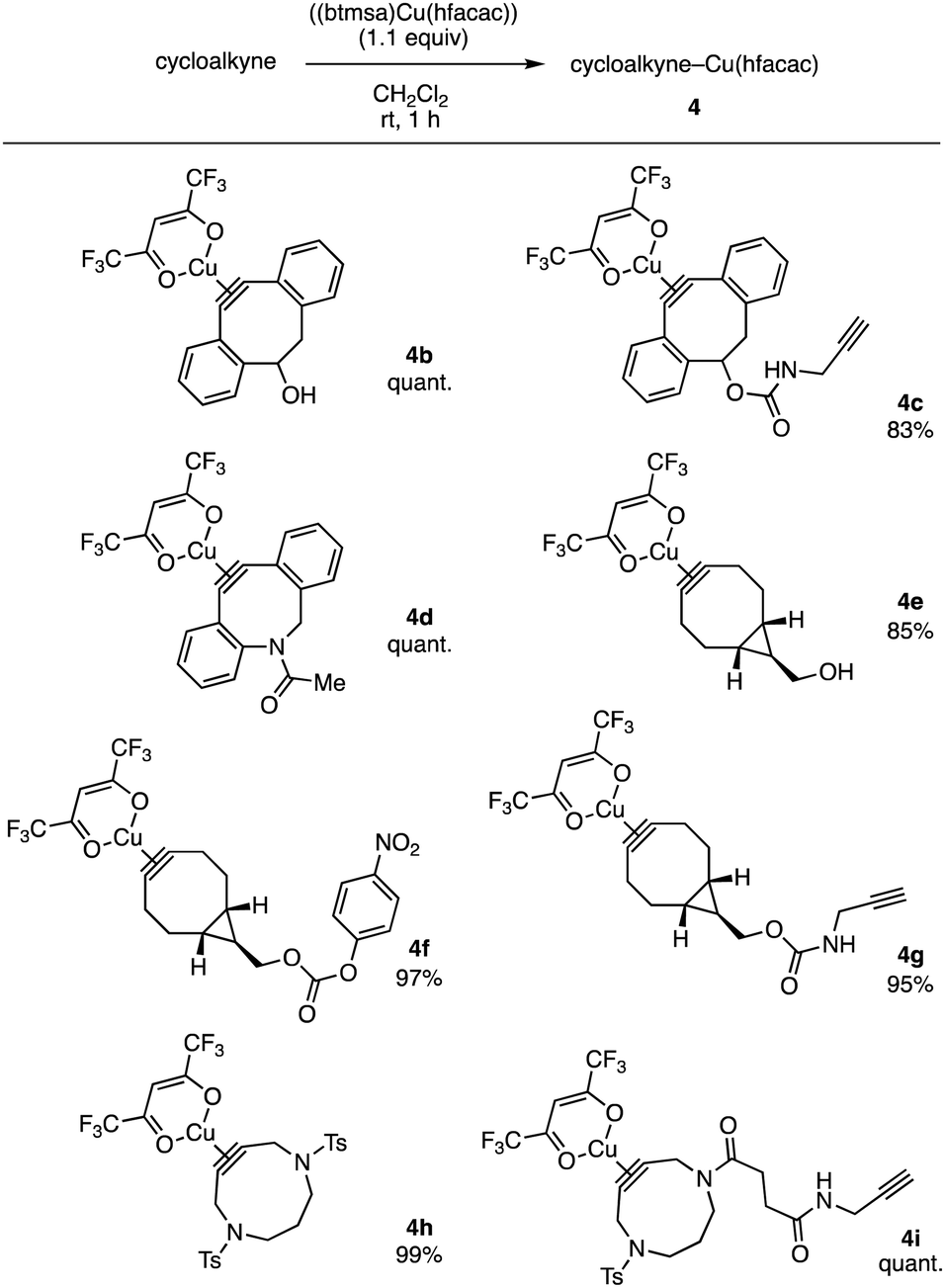 | ||
| Fig. 4 Scope of cycloalkyne(hfacac)copper(I) complexes. See ESI† for structures of the cycloalkynes used. | ||
A synthetic utility of cycloalkyne(hfacac)copper(I) complexes was showcased by preparing BCN–copper complex 4l having an azido group (Fig. 5A).6 Indeed, treatment of BCN 6 with (btmsa)Cu(hfacac) followed by acylation with 4-(azidomethyl)benzoyl chloride (7) in the presence of triethylamine and a catalytic amount of 4-(dimethylamino)pyridine (DMAP) provided azide-containing BCN–Cu(hfacac) complex 4j. We succeeded in click conjugation of complex 4j with alkyne 8 catalyzed by copper at the azido group and subsequent removal of the copper salt with aqueous ammonia to furnish BCN 9 in good yield without reacting the cycloalkyne moiety. These results clearly showed that cycloalkyne(hfacac)copper(I) complexes can be transformed as protected cycloalkynes, which will expand the synthetic utility of cycloalkynes.
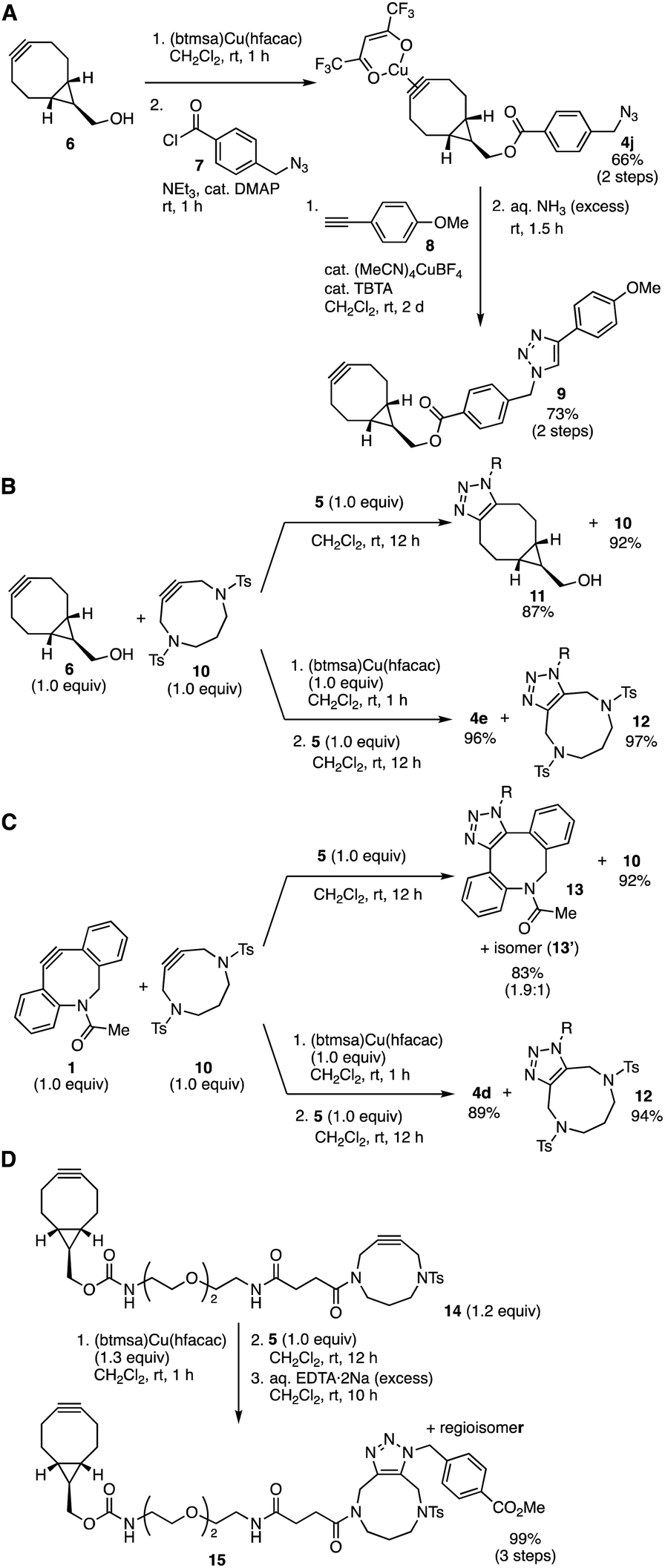 | ||
| Fig. 5 Selective triazole formations. (A) Synthesis of 9. (B) Switchable triazole formations between 6 and 10. (C) Switchable triazole formations between 1 and 10. (D) DACN-selective triazole formation using diyne 14. R = CH2C6H4-p-CO2Me. | ||
The selectivity of the SPAAC reaction between BCN 6 or DIBAC 1 and DACN 10 with azide 5 was switchable through complexation using (btmsa)Cu(hfacac) (Fig. 5B and C). For example, treatment of an equimolar mixture between BCN 6 and DACN 10 with 4-(methoxycarbonyl)benzyl azide (5) in dichloromethane without the addition of (btmsa)Cu(hfacac) afforded triazole 11 in high yield along with recovery of unreacted DACN 10 (Fig. 5B, upper). On the other hand, when azide 5 was added after the complexation of the mixture between 6 and 10 with (btmsa)Cu(hfacac), a selective SPAAC reaction took place efficiently to furnish triazole 12 in excellent yield by virtue of the selective formation of BCN–copper complex 4e similar to the case with AgBF4 (Fig. 5B, lower).8c Since the yield of triazole 12 was remarkably increased compared to the reaction using (MeCN)4CuBF4,8c the use of copper salt with lower Lewis acidity would improve the selective cycloalkyne–copper complexation. Furthermore, switchable click reactivity between DIBAC 1 and DACN 10 was also demonstrated by virtue of the complexation with (btmsa)Cu(hfacac). While a selective SPAAC reaction of DIBAC 1 with azide 5 proceeded without the protection, the pretreatment of the mixture between 1 and 10 with (btmsa)Cu(hfacac) followed by the addition of azide 5 provided triazole 12 along with the formation of DIBAC–Cu(hfacac) complex 4d. This switchable click conjugation using DIBAC 1 and DACN 10 indicates a significant improvement from our previous study with silver and gold.
The DACN-selective triazole formation enabled click conjugation of diyne 14 with an azide keeping BCN moiety intact (Fig. 5D). After pretreatment of diyne 14 possessing BCN and DACN moieties with (btmsa)Cu(hfacac), SPAAC reaction with azide 5 proceeded efficiently at the DACN moiety. Following deprotection with an aqueous solution of EDTA·2Na afforded triazoles 15 leaving the BCN moiety intact. Since the Lewis acidity of Cu(hfacac) complexes was lower than that of cationic transition metal complexes, this method will expand available functional ynophiles in the efficient sequential conjugations of functional molecules from recently reported methods using diynes.8c
In summary, we have developed an alternative protection method of cycloalkynes with (btmsa)Cu(hfacac). A wide range of cycloalkyne–copper complexes were efficiently obtained by treating cycloalkynes with (btmsa)Cu(hfacac) and silica-gel column chromatography. The modest Lewis acidity of the Cu(hfacac) complexes enabled efficient sequential click reactions. Further studies such as expansion of available ynophiles, applications for bimolecular modification, and assembling functional molecules through the selective SPAAC reaction are now ongoing in our laboratory.
The authors thank Assoc. Prof. Dr Keiichi Hirano and Prof. Dr Masanobu Uchiyama at the University of Tokyo for NMR analyses. This work was supported by JSPS KAKENHI Grant Numbers JP19K05451 (C; S. Y.), JP18J11113 (JSPS Research Fellow; T. M.), JP18H02104 (B; T. H.), and JP18H04386 (Middle Molecular Strategy; T. H.); the Naito Foundation (S. Y.); the Japan Agency for Medical Research and Development (AMED) under Grant Number JP20am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS); the Cooperative Research Project of Research Center for Biomedical Engineering; and the Research Program of “Five-Star Alliance” in “NJRC Mater. & Dev.”
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075 CrossRef CAS PubMed; (c) J. Lahann, Click Chemistry for Biotechnology and Materials Science, John Wiley & Sons, West Sussex, 2009 CrossRef.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed.
- (a) M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168 CrossRef CAS PubMed; (b) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC; (c) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666 CrossRef CAS PubMed; (d) S. Arumugam, S. V. Orski, N. E. Mbua, C. McNitt, G.-J. Boons, J. Locklin and V. V. Popik, Pure Appl. Chem., 2013, 85, 1499 CAS; (e) J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- (a) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS PubMed; (b) J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Cgang, I. A. Millar, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793 CrossRef CAS PubMed; (c) X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253 CrossRef CAS PubMed; (d) M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97 RSC; (e) J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688 CrossRef CAS PubMed; (f) J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422 CrossRef CAS PubMed; (g) I. Kii, A. Shiraishi, T. Hiramatsu, T. Matsushita, H. Uekusa, S. Yoshida, M. Yamamoto, A. Kudo, M. Hagiwara and T. Hosoya, Org. Biomol. Chem., 2010, 8, 4051 RSC; (h) G. de Almeida, E. M. Sletten, H. Nakamura, K. Palaniappan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2012, 51, 2443 CrossRef CAS PubMed; (i) B. R. Varga, M. Kμllay, K. Hegyi, S. Beni and P. Kele, Chem. – Eur. J., 2012, 18, 822 CrossRef CAS PubMed; (j) F. Xu, L. Peng, K. Shinohara, T. Morita, S. Yoshida, T. Hosoya, A. Orita and J. Otera, J. Org. Chem., 2014, 79, 11592 CrossRef CAS PubMed; (k) R. Ni, N. Mitsuda, T. Kashiwagi, K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2015, 54, 1190 CrossRef CAS PubMed; (l) C. Gröst and T. Berg, Org. Biomol. Chem., 2015, 13, 3866 RSC; (m) R. R. Ramsubhag and G. B. Dudley, Org. Biomol. Chem., 2016, 14, 5028 RSC; (n) E. G. Burke, B. Gold, T. T. Hoang, R. T. Raines and J. M. Schomaker, J. Am. Chem. Soc., 2017, 139, 8029 CrossRef CAS PubMed; (o) S. Bernard, R. A. Kumar, K. Porte, P. Thuéry, F. Taran and D. Audisio, Eur. J. Org. Chem., 2018, 2000 CrossRef CAS; (p) C. Lis, S. Rubner, C. Gröst, R. Hoffmann, D. Knappe and T. Berg, Chem. – Eur. J., 2018, 24, 13762 CrossRef CAS PubMed; (q) M. Tera, Z. H. Taji and N. W. Luedtke, Angew. Chem., Int. Ed., 2018, 57, 15405 CrossRef CAS PubMed; (r) Y. Kawasaki, Y. Yamanaka, Y. Seto, K. Igawa and K. Tomooka, Chem. Lett., 2019, 49, 495 CrossRef; (s) K. Sharma, A. V. Strizhak, E. Fowler, X. Wang, W. Xu, C. H. Jensen, Y. Wu, H. F. Sore, Y. H. Lau, M. Hyvönen, L. S. Itzhaki and D. R. Spring, Org. Biomol. Chem., 2019, 17, 8014 RSC; (t) K. Sharma, A. V. Strizhak, E. Fowler, W. Xu, B. Chappell, H. F. Sore, W. R. J. D. Galloway, M. N. Grayson, Y. H. Lau, L. S. Itzhaki and D. R. Spring, ACS Omega, 2020, 5, 1157 CrossRef CAS PubMed.
- (a) A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131 RSC; (b) Z.-J. Zheng, D. Wang, Z. Xu and L.-W. Xu, Beilstein J. Org. Chem., 2015, 11, 2557 CrossRef CAS PubMed; (c) S. Yoshida, Bull. Chem. Soc. Jpn., 2018, 91, 1293 CrossRef CAS; (d) S. Yoshida, Org. Biomol. Chem., 2020, 18, 1550 RSC.
- P. Gobbo, T. Romagnoli, S. M. Barbon, J. T. Price, J. Keir, J. B. Gilroy and M. S. Workentin, Chem. Commun., 2015, 51, 6647 RSC.
- (a) S. Yoshida, A. Shiraishi, K. Kanno, T. Matsushita, K. Johmoto, H. Uekusa and T. Hosoya, Sci. Rep., 2011, 1, 82 CrossRef PubMed; (b) S. Yoshida, T. Nonaka, T. Morita and T. Hosoya, Org. Biomol. Chem., 2014, 12, 7489 RSC; (c) T. Meguro, S. Yoshida and T. Hosoya, Chem. Lett., 2017, 46, 1137 CrossRef CAS; (d) S. Yoshida, K. Kanno, I. Kii, Y. Misawa, M. Hagiwara and T. Hosoya, Chem. Commun., 2018, 54, 3705 RSC; (e) T. Meguro, N. Terashima, H. Ito, Y. Koike, I. Kii, S. Yoshida and T. Hosoya, Chem. Commun., 2018, 54, 7904 RSC; (f) S. Yoshida, J. Tanaka, Y. Nishiyama, Y. Hazama, T. Matsushita and T. Hosoya, Chem. Commun., 2018, 54, 13499 RSC; (g) T. Meguro, S. Yoshida, K. Igawa, K. Tomooka and T. Hosoya, Org. Lett., 2018, 20, 4126 CrossRef CAS PubMed; (h) T. Meguro, S. Chen, K. Kanemoto, S. Yoshida and T. Hosoya, Chem. Lett., 2019, 48, 582 CrossRef CAS; (i) S. Yoshida, S. Goto, Y. Nishiyama, Y. Hazama, M. Kondo, T. Matsushita and T. Hosoya, Chem. Lett., 2019, 48, 1038 CrossRef CAS; (j) T. Meguro, Y. Sakata, T. Morita, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 4720 RSC.
- (a) S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, J. Am. Chem. Soc., 2014, 136, 13590 CrossRef CAS PubMed; (b) S. Yoshida, T. Kuribara, H. Ito, T. Meguro, Y. Nishiyama, F. Karaki, Y. Hatakeyama, Y. Koike, I. Kii and T. Hosoya, Chem. Commun., 2019, 55, 3556 RSC; (c) K. Adachi, T. Meguro, Y. Sakata, K. Igawa, K. Tomooka, T. Hosoya and S. Yoshida, Chem. Commun., 2020 10.1039/D0CC04606J.
- (a) K. M. Chi, H. K. Shin, M. J. Hampde-Smith, T. T. Kodas and E. N. Duesler, Inorg. Chem., 1991, 30, 4293 CrossRef CAS; (b) T. H. Baum and C. E. Larson, Chem. Mater., 1992, 4, 365 CrossRef CAS; (c) P. Doppelt and T. H. Baum, J. Organomet. Chem., 1996, 517, 53 CrossRef CAS; (d) V. Pawlowski, A. Strasser and A. Vogler, Z. Naturforsch., B: Chem. Sci., 2003, 58, 950 CAS; (e) A. Ishibashi, S. Kamihigashi, Y. Iwai and S. Sakaguchi, Catalysts, 2019, 9, 780 CrossRef.
- (a) G. Wittig and H.-L. Dorsch, Liebigs Ann. Chem., 1968, 711, 46 CrossRef CAS; (b) G. Wittig and S. Fischer, Chem. Ber., 1972, 105, 3542 CrossRef CAS; (c) G. Gröger, U. Behrens and F. Olbrich, Organometallics, 2000, 19, 3354 CrossRef; (d) M. Shelbourne, X. Chen, T. Brown and A. H. El-Sagheer, Chem. Commun., 2011, 47, 6257 RSC; (e) A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 51, 3940 CrossRef CAS PubMed; (f) A. Das, C. Dash, M. A. Celik, M. Yousufuddin, G. Frenking and H. V. R. Dias, Organometallics, 2013, 32, 3135 CrossRef CAS.
- Treatment of cycloalkyne 1 with (MeCN)4CuBF4 (1.1 equiv.) in methanol followed by the addition of azide 5 (1.0 equiv.) resulted in no triazole formation and the recovery of unreacted azide 5 in 91% yield. When aq. EDTA·2Na (90 equiv.) was added after the pretreatment of cycloalkyne 1 with (MeCN)4CuBF4 (1.1 equiv.) in methanol, cycloalkyne 1 was recovered in 86% yield.
- Ring-closure of DIBAC without acyl migration by hydrogen bromide was reported in ref. 4d.
- Labile nature of a complex from 3 and (MeCN)4CuBF4 rendered the purification by silica-gel column chromatography difficult.
- DIBO–copper complex 4a was stable in methanol, toluene, or N,N-dimethylformamide. In addition, a dichloromethane solution of 4a was stable toward aqueous hydrochloric acid, aqueous ammonium chloride, aqueous sodium bicarbonate, and amino acids such as cysteine, tyrosine, and lysine.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization for new compounds including NMR spectra. See DOI: 10.1039/d0cc05182a |
| This journal is © The Royal Society of Chemistry 2020 |