
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light-mediated Smiles rearrangements and annulations of non-activated aromatics†
Connor A.
Lawson
 *ab,
Andrew P.
Dominey
a,
Glynn D.
Williams‡
a and
John A.
Murphy
*b
*ab,
Andrew P.
Dominey
a,
Glynn D.
Williams‡
a and
John A.
Murphy
*b
aChemical Development, GSK, Gunnels Wood Road, Stevenage, SG1 2NY, UK. E-mail: connor.x.lawson@gsk.com
bDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: john.murphy@strath.ac.uk
First published on 19th August 2020
Abstract
We report the first examples of radical cation Smiles rearrangements. A series of aryloxy alkylamines underwent spontaneous reaction, with the amino group displacing the ipso-alkoxy group through substitution, at ambient temperature and under photoactivation by visible light in the presence of an acridinium catalyst (5 mol%). The study was extended to 3-(2-methoxyphenyl)propan-1-amine derivatives, which lack an appropriate ipso leaving group. Here, efficient cyclisations resulted in displacement of the methoxy group and formation of tetrahydroquinolines.
Development of complementary synthetic methods for the preparation of arylated amines is of high importance, due to their widespread presence in pharmaceuticals, natural products and organic materials. Phenol derivatives have emerged as attractive starting materials due to their abundance, and the ability to derive them from renewable sources.1
Over the last decade, photoredox catalysis has become immensely popular, through its role in engaging in efficient and sustainable transformations.2,3 The exploitation of visible-light has seen a vast number of methodologies developed that are of great synthetic utility due to the mild conditions deployed to access highly reactive species.4 A rising interest in photochemistry has been seen from the pharmaceutical industry.5
Organic photoredox catalysts are growing in popularity due to the avoidance of expensive and toxic metals such as ruthenium and iridium.6 Acridinium salts are among the most popular of these.7 In 2017, Tay and Nicewicz described with great success the use of acridinium salts to catalyse the nucleophilic aromatic substitution (SNAr) of anisoles with N-heteroaromatic amines (Scheme 1a).8 Further expansion of photocatalysed SNAr of anisoles has been reported by the Nicewicz group.9,10
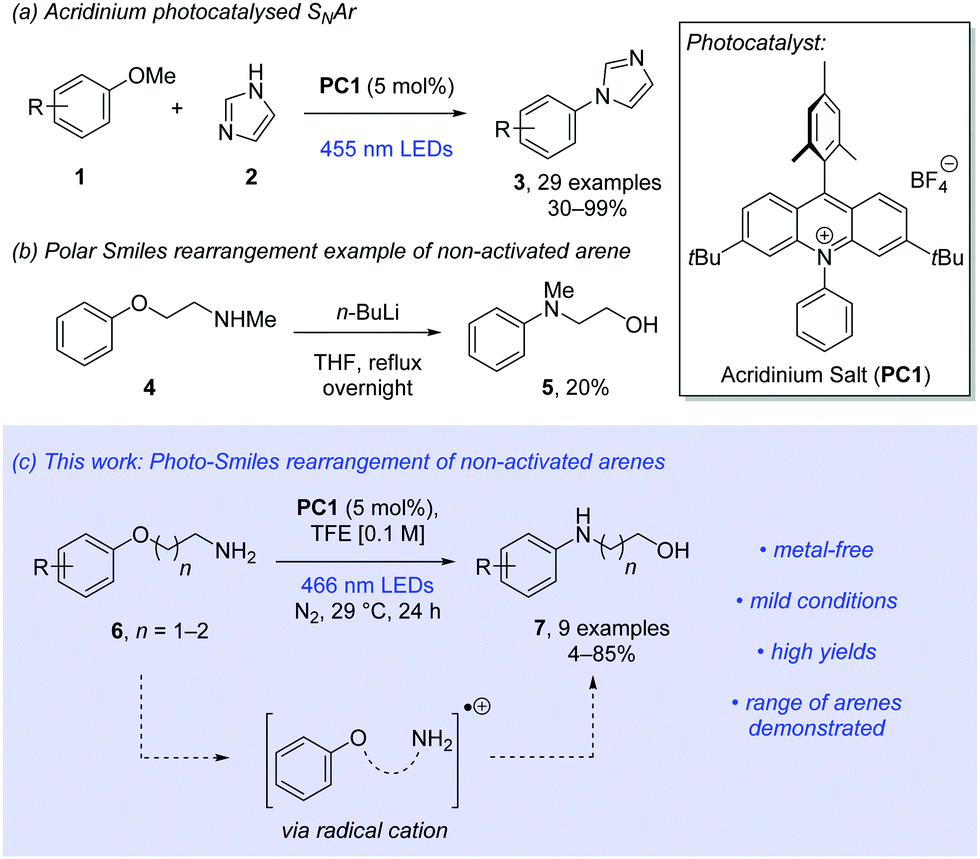 | ||
| Scheme 1 Previous work and our approach to the Smiles rearrangement of non-activated arenes using photoredox catalysis.8–10,13c | ||
We proposed that these conditions could be adapted to allow mild and efficient intramolecular reactions. The Smiles rearrangement is an intramolecular SNAr reaction occurring at the ipso position in an aromatic system.11 The Smiles rearrangement is an important tool in synthetic chemistry, as demonstrated by diverse advances described in the literature.12 Generally, Smiles rearrangements are often limited to electron-poor arenes. Examples of thermally driven Smiles rearrangements of non-activated aromatic systems are sparse and typically require forcing conditions and harsh reagents over prolonged reaction times (Scheme 1b).13
Neutral radical versions of the Smiles rearrangement are also known and have been established by the groups of Speckamp and Motherwell.14 More recently developments by Nevado15 and Stephenson16 are part of an increasing torrent of radical-Smiles rearrangements reported in the literature, including photochemical and electrochemical methods.17
Inspired by recent developments in the Newman–Kwart rearrangement, where chemical, photochemical and electrochemical methods for single-electron oxidation enhanced reactivity, we proposed a radical cation approach to the Smiles rearrangement.18 We now show that a range of aryloxy alkylamines successfully undergo a mild and efficient photo-Smiles rearrangement using an acridinium salt as the organophotoredox catalyst (Scheme 1c).
To our knowledge, this approach yields the first examples of radical cation-promoted Smiles rearrangements. Our publication at this stage is prompted by the very recent report from the Nicewicz lab which details the use of primary alkylamine nucleophiles in intermolecular radical-cation accelerated nucleophilic aromatic substitutions.10
Our studies began with the evaluation of 2-phenoxyethan-1-amine (6a). Pleasingly, the reaction of 6a (0.4 mmol) with 9-mesityl-3,6-di-tert-butyl-10-phenylacridinium tetrafluoroborate (PC1) (5 mol%) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2,2,2-trifluoroethanol (TFE) and 1,2-dichloroethane (DCE) under visible light irradiation with a general purpose 40 W blue LED lamp (see ESI† for light source characterisation) in a nitrogen atmosphere for 24 hours at ambient temperature gave the rearranged product 7a in 87% solution yield (Table 1, entry 1).
:1 mixture of 2,2,2-trifluoroethanol (TFE) and 1,2-dichloroethane (DCE) under visible light irradiation with a general purpose 40 W blue LED lamp (see ESI† for light source characterisation) in a nitrogen atmosphere for 24 hours at ambient temperature gave the rearranged product 7a in 87% solution yield (Table 1, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent [conc.] | Photocatalyst PC (loading) | Variations | 7a yieldb (%) |
| a Unless otherwise stated, all reactions were conducted using 0.4 mmol of 6a in degassed solvent [0.1 M] and irradiated with a Kessil lamp (40 W, A160WE Tuna Blue) for 24 h. b Yields determined by 1H NMR analysis of the crude reaction mixtures using 1,3,5-trimethoxybenzene as an internal standard. c Catalyst showed poor solubility in solvent. d Reaction run at 80 °C. DCE = 1,2-dichloroethane; TFE = 2,2,2-trifluoroethanol; HFIP = 1,1,1,3,3,3-hexafluoro-2-propanol. Catalyst photophysical and electrochemical properties reported in literature.3,6,23 | ||||
| 1 | TFE:DCE (1:1) [0.1 M] |
PC1 (5 mol%) | — | 87 |
| 2 | TFE [0.1 M] | PC1 (5 mol%) | — | 92 |
| 3 | MeCN [0.1 M] | PC1 (5 mol%) | — | 2 |
| 4 | HFIP [0.1 M] | PC1 (5 mol%) | — | 42 |
| 5 | TFE [0.1 M] | PC1 (5 mol%) | — | 29 |
| 6c | TFE [0.1 M] | PC1 (5 mol%) | — | 15 |
| 7 | TFE [0.1 M] | PC1 (1 mol%) | — | 92 |
| 8 | TFE [0.1 M] | PC1 (10 mol%) | — | 90 |
| 9 | TFE [0.05 M] | PC1 (5 mol%) | — | 96 |
| 10 | TFE [0.2 M] | PC1 (5 mol%) | — | 82 |
| 11 | TFE [0.1 M] | — | — | Trace |
| 12d | TFE [0.1 M] | PC1 (5 mol%) | Dark/80 °C | 0 |
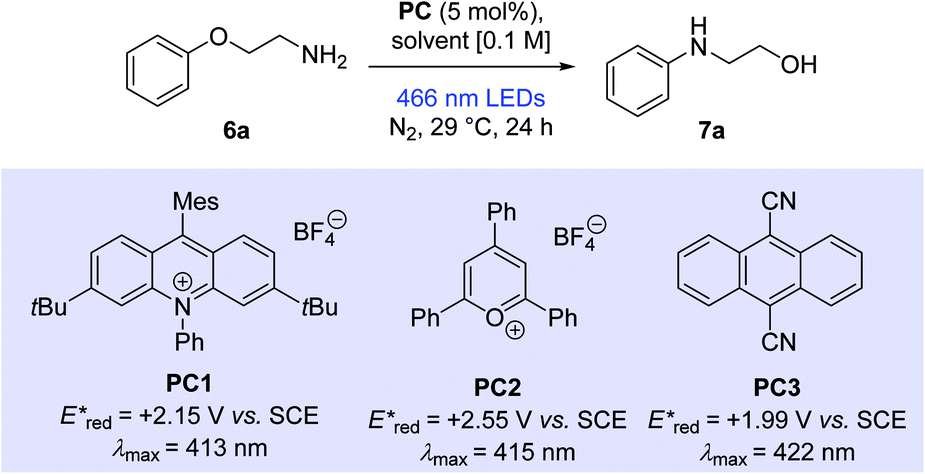
Employing TFE alone as solvent increased the observed solution yield to 92% (entry 2). In related systems, the importance of fluorinated alcohols has been linked to their ability to hydrogen bond key reaction intermediates.19 Changing to MeCN, a solvent with a lower toxicity burden than TFE, proved unsuccessful (entry 3, 2%). Moderate yields were observed when another highly fluorinated alcohol solvent, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), was employed (entry 4, 42%). We also found that these reactions work equally as efficiently if DCE is replaced by α,α,α-trifluorotoluene (TFT) (see ESI,† 86%).
Based on photophysical and electrochemical properties, other commonly used organophotocatalysts were tested. 2,4,6-Triphenylpyrylium tetrafluoroborate (TPT) (PC2) represents an example from an extremely powerful class of photo oxidants.20 However, a moderate solution yield of 29% was observed when PC2 was employed, possibly due to chemical instability (entry 5).21 Dicyanoanthracene (DCA) (PC3) offers an attractive catalyst alternative, owing to its structural simplicity and greater commercial availability.22 Under our conditions, DCA displayed a promising solution yield of 15%, considering that it showed poor solubility in TFE (entry 6). Two other acridinium salts were also tested as photocatalysts (see ESI†). Both showed good reactivity with slightly diminished yields over PC1, however, they have been reported in the literature as suffering from chemical instability3 and were pursued no further.
Once PC1 was established as the optimal catalyst, the loading was adjusted. Organocatalysis has been criticised for its inability to perform at low catalyst loadings.24 In our case, decreasing the catalyst loading to 1 mol% (entry 7, 92%) had no effect on the reaction outcome. Moreover, no noticeable change in yield was observed when catalyst loading was increased to 10 mol% (entry 8, 90%). Dilution of the reaction mixture to 0.05 M showed an improved solution yield (96%) (entry 9 vs. entry 2) and conversely, concentrating the reaction to 0.2 M showed a drop in solution yield (entry 10, 82%). For convenience, 0.1 M was selected as the reaction concentration on this scale for further studies. Control reactions indicated the importance both catalyst and light had on the success of the transformation. In the absence of catalyst, trace amounts of product were observed (entry 11). Heating the reaction mixture to reflux in the dark with catalyst showed no product formation (entry 12).
Following screening, a range of arenes was subjected to the optimum conditions and the reaction products were subsequently isolated (Scheme 2). The product 7a derived from substrate 6a was isolated in 85% yield (1H NMR yield was 92%). para-Alkyl substrates (6b–6d) were successfully rearranged and products isolated respectively in 63%, 73% and 80% yield. The success of isopropyl (6c) and tert-butyl (6d) substrates indicated that sterically bulky groups around the arene impacted little on the excited acridinium's ability for single electron transfer (see ESI† for further discussion). A single rearranged product (7e) was formed in good yield (74%) when ortho-methoxy derivative (6e) was subjected to the reaction conditions. No competing cyclised product was observed as would arise from displacement of the ortho-methoxy group, illustrating the faster kinetics of the ipso-substitution. To answer the question whether ortho-substitution would be possible in the absence of a Smiles rearrangement, we prepared and tested appropriate substrates (see below). Surprisingly, para-methoxy substrate (6f) showed poor conversion to product (4% isolated after 65 h irradiation), with unreacted starting material accounting for the remainder of the mass balance (see ESI†). Biphenyl derivative (6g) gave a moderate yield of the desired Smiles product 7g (32%). Propylene-linked substituent (6h) was smoothly rearranged to the Smiles product (7h) in 83% yield, progressing via a 6-membered cyclic transtion state. The rearrangement also occurred with an electron-withdrawing fluoro present (6i) in excellent yield (81%). However, a nitro-containing substituent (6j, see ESI†) showed no reactivity under our conditions.
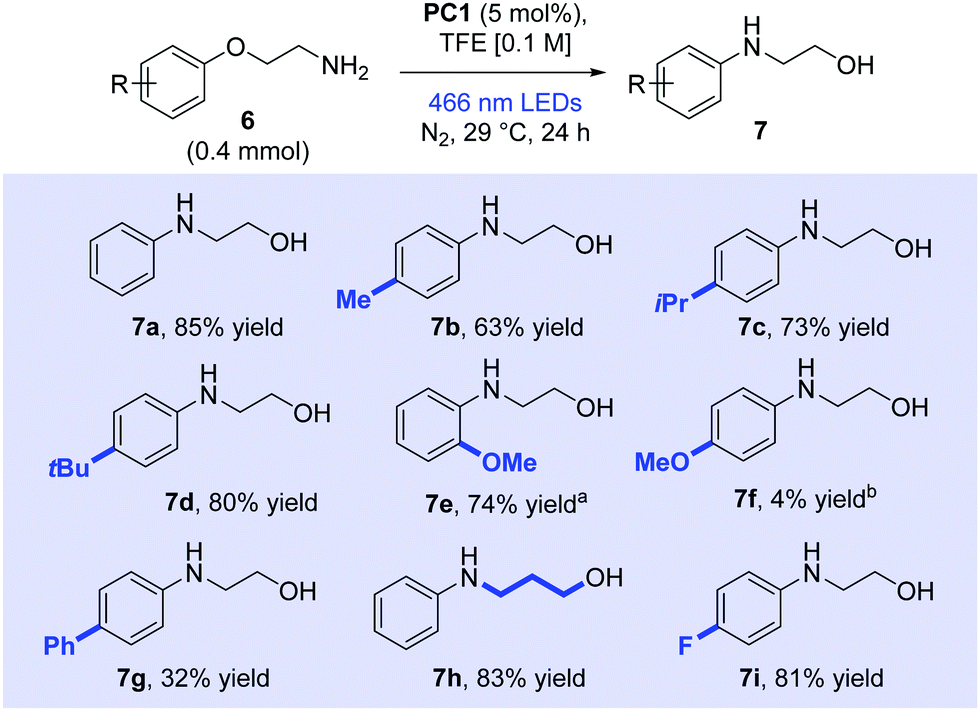 | ||
| Scheme 2 Smiles rearrangement of primary amines. Yields reported as isolated yields following column chromatography. aReaction run in TFE:DCE (1:1) with 0.3 mmol of substrate. bIrradiated for 65 h. DCE = 1,2-dichloroethane; TFE = 2,2,2-trifluoroethanol. | ||
We note in the current results of Nicewicz and Venditto that secondary amines were not successful in intermolecular SNAr reactions.10 Our preliminary investigations into the Smiles rearrangement of secondary amines are noteworthy, showing modest initial success in converting challenging substrate (8) to product (9) and demethylated product (7a) (Scheme 3). It is likely that 7a arose from oxidative demethylation of 9.6
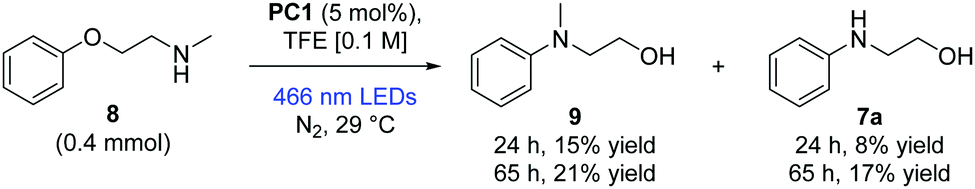 | ||
| Scheme 3 Smiles rearrangement of secondary amines. Yields reported as isolated yields following column chromatography. Unreacted starting material accounted for the remainder of the mass balance. TFE = 2,2,2-trifluoroethanol. | ||
Encouraged by the results of the ipso-substitutions seen above, we now moved to explore ortho-substitutions (Scheme 4). 3-(2-Methoxyphenyl)propan-1-amine (10a) was prepared and was successfully cyclised to form tetrahydroquinoline (11a) in a good yield (66%). Tolyl (10b) and biphenyl (10c) derivatives were also cyclised in very good yields to provide the corresponding tetrahydroquinoline derivatives (75% and 73% respectively). Electron-rich dimethoxy derivative (10d) also cyclised well following prolonged irradiation times (49%). Finally, electron-poor carbomethoxy derivative (10e) afforded desired product 11e in moderate yield (38%). Synthetic applicability was extended to a medicinally relevant core with the efficient preparation of antibacterial imidazo[1,2-a]quinoline 11f (80%).25
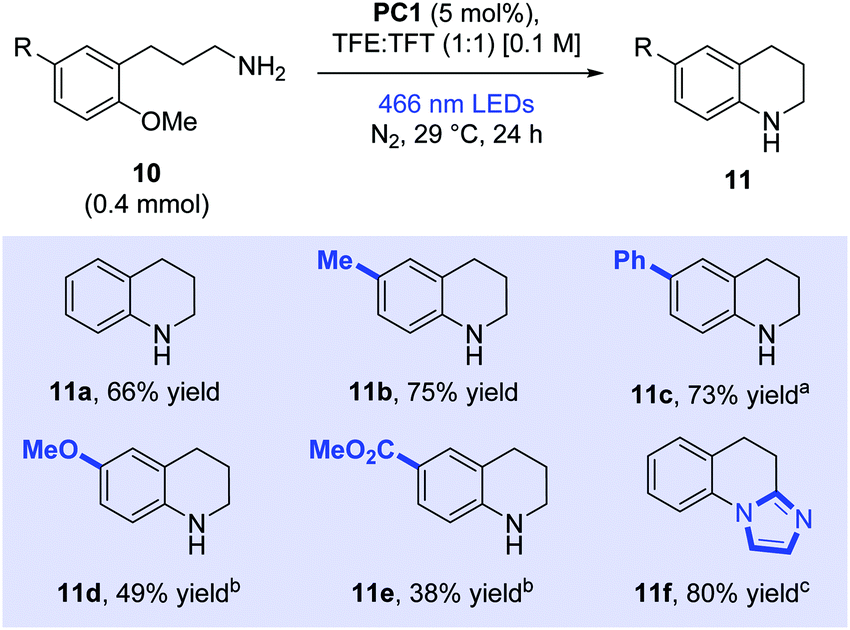 | ||
| Scheme 4 Cyclisation of primary amines. Yields reported as isolated yields following column chromatography. aIrradiated for 72 h. bIrradiated for 40 h. cReaction run in TFE:DCE (1:1). DCE = 1,2-dichloroethane; TFE = 2,2,2-trifluoroethanol; TFT = α,α,α-trifluorotoluene. | ||
Anisole derivatives (ca. +1.8 V vs. SCE) and primary alkylamines (+1.5 to +1.6 V vs. SCE) typically exhibit low oxidation potentials.26 Therefore, it is possible under the conditions employed (PC1, Ered* = +2.15 V vs. SCE) that the substrates undergo single electron oxidation at the amine or the arene.8 In closely related systems, mechanistic probing has suggested arene radical cations are key intermediates.8–10 However, other photoredox transformations involving alkylamines have been proposed to progress via amine radical cation pathways.27 Our proposed mechanism is shown below (Scheme 5). Visible light excitation of the ground-state acridinium salt (PC1) to an excited state (PC1a) enables the oxidation of phenoxyalkylamine (6a) to an arene radical cation (6aa). The tethered primary amine subsequently adds to the ipso-position, giving rise to Meisenheimer complex (6ab). Proton transfer and reduction by the acridinyl radical (PC1b) lead to the Smiles rearrangement product (7a) and regeneration of the ground state catalyst (PC1).
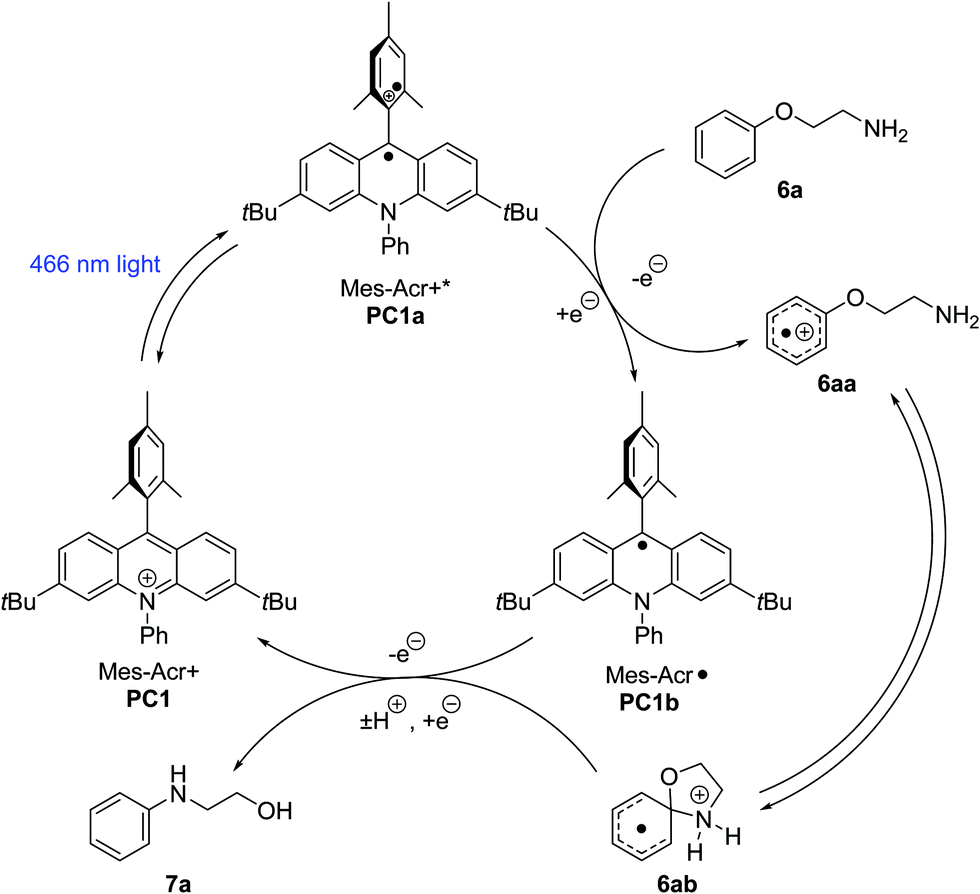 | ||
| Scheme 5 Proposed Smiles rearrangement mechanism (adapted from Tay and Nicewicz).8 | ||
In conclusion, a range of primary aryloxylamines has been successfully reacted in a novel radical cation promoted photo-Smiles reaction using an acridinium salt as an organophotocatalyst. The use of a single electron oxidant has enabled efficient Smiles rearrangements of electron-rich aromatics, that are typically inert and unreactive in this manner in the ground state, leading to substituted anilines via radical cation intermediates. Whereas these reactions show success in ipso rearrangement, ortho cyclisations of appropriate substrates to afford tetrahydroquinolines are also reported. We are currently extending these studies.
C. A. L. is grateful to GlaxoSmithKline for funding and chemical resources. We thank the EPSRC for funding via Prosperity Partnership EP/S035990/1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559 CrossRef CAS PubMed; B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275 CrossRef PubMed.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326 CrossRef CAS PubMed.
- J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Org. Process Res. Dev., 2016, 20, 1134 CrossRef CAS; L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef PubMed; M. K. Bogdos, E. Pinard and J. A. Murphy, Beilstein J. Org. Chem., 2018, 14, 2035 CrossRef PubMed.
- H. E. Bonfield, K. Mercer, A. Diaz-Rodriguez, G. C. Cook, B. S. J. McKay, P. Slade, G. M. Taylor, W. X. Ooi, J. D. Williams, J. P. M. Roberts, J. A. Murphy, L. Schmermund, W. Kroutil, T. Mielke, J. Cartwright, G. Grogan and L. J. Edwards, ChemPhotoChem, 2020, 4, 45 CrossRef CAS; H. E. Bonfield, T. Knauber, F. Lévesque, E. G. Moschetta, F. Susanne and L. J. Edwards, Nat. Commun., 2020, 11, 804 CrossRef PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo, T. Suenobu, K. Kato, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 8459 CrossRef CAS PubMed; A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L.-C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244 CrossRef PubMed.
- N. E. S. Tay and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 16100 CrossRef CAS PubMed.
- N. Holmberg-Douglas and D. A. Nicewicz, Org. Lett., 2019, 21, 7114 CrossRef CAS PubMed.
- N. J. Venditto and D. A. Nicewicz, Org. Lett., 2020, 22, 4817 CrossRef CAS PubMed.
- J. F. Bunnett and R. E. Zahler, Chem. Rev., 1951, 49, 273 CrossRef CAS; W. E. Truce, E. M. Kreider and W. W. Brand, Org. React., 1970, 18, 99 Search PubMed.
- Z.-M. Chen, X.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2015, 44, 5220 RSC; I. Allart-Simon, S. Gérard and J. Sapi, Molecules, 2016, 21, 878 CrossRef CAS PubMed; C. M. Holden and M. F. Greaney, Chem. – Eur. J., 2017, 23, 8992 CrossRef PubMed; A. R. P. Henderson, J. R. Kosowan and T. E. Wood, Can. J. Chem., 2017, 95, 483 CrossRef; H. L. Barlow, P. T. G. Rabet, A. Durie, T. Evans and M. F. Greaney, Org. Lett., 2019, 21, 9033 CrossRef PubMed.
- R. Bayles, M. C. Johnson, R. F. Maisey and R. W. Turner, Synthesis, 1977, 33 CrossRef CAS; I. G. C. Coutts and M. R. Southcott, J. Chem. Soc., Perkin Trans. 1, 1990, 767 RSC; W. ten Hoeve, C. G. Kruse, J. M. Luteyn, J. R. G. Thiecke and H. Wynberg, J. Org. Chem., 1993, 58, 5101 CrossRef; J. J. Weidner, P. M. Weintraub, R. A. Schnettler and N. P. Peet, Tetrahedron, 1997, 53, 6303 CrossRef; C. Bonini, G. Cristiani, M. Funicello and L. Viggiani, Synth. Commun., 2006, 36, 1983 CrossRef.
- R. Loven and W. N. Speckamp, Tetrahedron Lett., 1972, 13, 1567 CrossRef CAS; J. J. Köhler and W. N. Speckamp, Tetrahedron Lett., 1977, 18, 631 CrossRef; W. B. Motherwell and A. M. K. Pennell, J. Chem. Soc., Chem. Commun., 1991, 877 RSC; M. L. E. N. da Mata, W. B. Motherwell and F. Ujjainwalla, Tetrahedron Lett., 1997, 38, 137 CrossRef.
- W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480 CrossRef CAS PubMed; W. Kong, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2014, 53, 5078 Search PubMed; N. Fuentes, W. Kong, L. Fernández-Sánchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964 CrossRef PubMed.
- J. J. Douglas, H. Albright, M. J. Sevrin, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2015, 54, 14898 CrossRef CAS PubMed; T. M. Monos, R. C. McAtee and C. R. J. Stephenson, Science, 2018, 361, 1369 CrossRef.
- T. Zhou, F.-X. Luo, M.-Y. Yang and Z.-J. Shi, J. Am. Chem. Soc., 2015, 137, 14586 CrossRef CAS; E. Brachet, L. Marzo, M. Selkti, B. König and P. Belmont, Chem. Sci., 2016, 7, 5002 RSC; J. C. Gonzalez-Gomez, N. P. Ramirez, T. Lana-Villarreal and P. Bonete, Org. Biomol. Chem., 2017, 15, 9680 RSC; D. M. Whalley, H. A. Duong and M. F. Greaney, Chem. – Eur. J., 2019, 25, 1927 CrossRef PubMed; S. W. Lardy, K. C. Luong and V. A. Schmidt, Chem. – Eur. J., 2019, 25, 15267 CrossRef PubMed; X. Chang, Q. Zhang and C. Guo, Org. Lett., 2019, 21, 10 CrossRef PubMed; Z.-H. Xia, L. Dai, Z.-H. Gao and S. Ye, Chem. Commun., 2020, 56, 1525 RSC; C. Liu, Q. Jiang, Y. Lin, Z. Fang and K. Guo, Org. Lett., 2020, 22, 795 CrossRef PubMed.
- A. J. Perkowski, C. L. Cruz and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 15684 CrossRef CAS PubMed; S. K. Pedersen, A. Ulfkjær, M. N. Newman, S. Yogarasa, A. U. Petersen, T. I. Sølling and M. Pittelkow, J. Org. Chem., 2018, 83, 12000 CrossRef PubMed; T. Broese, A. F. Roesel, A. Prudlik and R. Francke, Org. Lett., 2018, 20, 7483 CrossRef; S. Chiniforoush and C. J. Cramer, J. Org. Chem., 2019, 84, 2148 CrossRef.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366 CrossRef CAS PubMed.
- M. A. Miranda and H. Garcia, Chem. Rev., 1994, 94, 1063 CrossRef CAS.
- T. Zimmermann, G. W. Fischer and M. Reinhardt, Z. Chem., 1986, 26, 400 CrossRef CAS.
- M. Freccero, M. Mella and A. Albini, Tetrahedron, 1994, 50, 2115 CrossRef CAS.
- R. Akaba, H. Sakuragi and K. Tokumaru, J. Chem. Soc., Perkin Trans. 2, 1991, 291 RSC; Y. Wang, O. Haze, J. P. Dinnocenzo, S. Farid, R. S. Farid and I. R. Gould, J. Org. Chem., 2007, 72, 6970 CrossRef CAS PubMed; I. R. Gould, D. Ege, J. E. Moser and S. Farid, J. Am. Chem. Soc., 1990, 112, 4290 CrossRef.
- D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed.
- H. G. Bazin, L. S. Bess and M. T. Livesay, Org. Prep. Proced. Int., 2018, 50, 109 CrossRef CAS.
- J. L. Bourdelande, I. Gallardo and G. Guirado, J. Am. Chem. Soc., 2007, 129, 2817 CrossRef CAS PubMed; H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 714 Search PubMed.
- K. A. Margrey, A. Levens and D. A. Nicewicz, Angew. Chem., Int. Ed., 2017, 56, 15644 CrossRef CAS PubMed; A. J. Musacchio, B. C. Lainhart, X. Zhang, S. G. Naguib, T. C. Sherwood and R. R. Knowles, Science, 2017, 355, 727 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterisation data for new compounds. See DOI: 10.1039/d0cc04666c |
| ‡ Current address: API Development, Aptuit (Oxford) Ltd, an Evotec Company, 111 Innovation Dr, Milton, Abingdon, OX14 4RZ, UK. |
| This journal is © The Royal Society of Chemistry 2020 |