
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A visible light-mediated, decarboxylative, desulfonylative Smiles rearrangement for general arylethylamine syntheses†
David M.
Whalley
ab,
Hung A.
Duong
*a and
Michael F.
Greaney
 *b
*b
aInstitute of Chemical and Engineering Sciences (ICES) Agency for Science, Technology and Research (A*STAR), 8 Biomedical Grove, Neuros, #07-01, 138665, Singapore. E-mail: duong_hung@ices.a-star.edu.sg
bSchool of Chemistry, The University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: michael.greaney@manchester.ac.uk
First published on 21st August 2020
Abstract
A decarboxylative, desulfonylative Smiles rearrangement is presented that employs activated-ester/energy transfer catalysis to decarboxylate β-amino acid derived starting materials at room-temperature under visible light irradiation. The radical Smiles rearrangement gives a range of biologically active arylethylamine products highly relevant to the pharmaceutical industry, chemical biology and materials science. The reaction is then applied to the synthesis of a chiral unnatural amino acid, 2-thienylalanine, used in the treatment of phenylketonuria. We also show how the reaction can proceed under metal-free and catalyst-free conditions.
The desulfonylative Smiles rearrangement is a powerful arylation method that exploits easy to make sulfonamides, to forge more challenging Caryl–Csp3 bonds via ipso-substitution (Scheme 1.1).1 The reaction often features mild and operationally simple conditions, creating versatile pathways to functionalized amino heteroarenes 3 with no requirement for stoichiometric arylmetals. The best exemplified arylations in this regime proceed through a 5-membered transition state with where one or both of the two carbon centres illustrated in red are sp2 hybridised.2–5 Representative examples are shown in Scheme 1.2: seminal work in this area from Motherwell established the reaction for Csp2 radicals in biaryl synthesis,2b the Nevado group have developed an acrylamide system triggered by a wide range of radical conjugate additions,3i,j,k and our own laboratory have used sulfonamide addition to sp-carbons for carbanion aryl transfer.3b,c
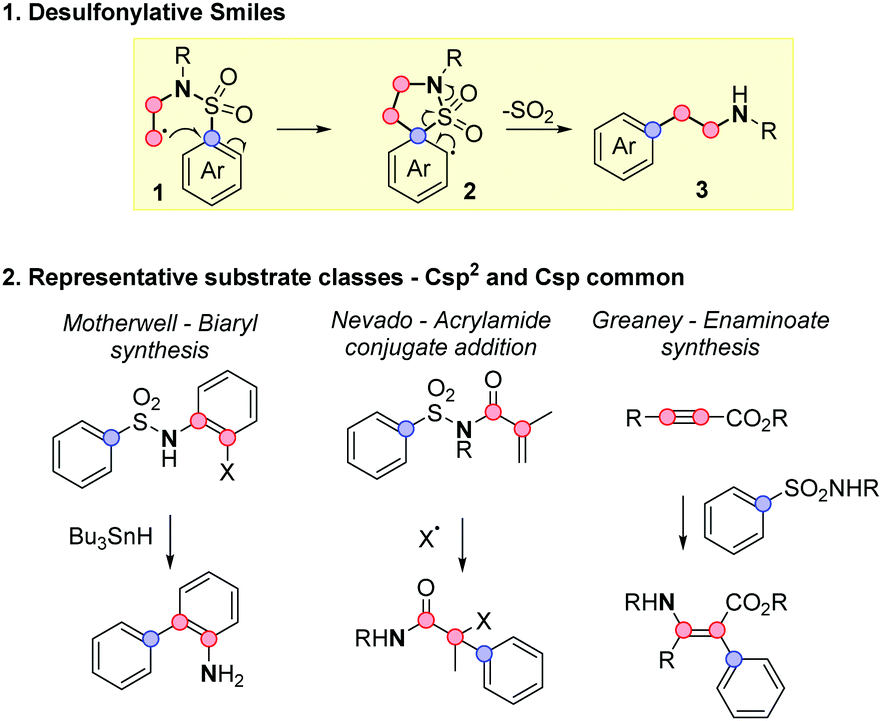 | ||
| Scheme 1 Desulfonylative Smiles rearrangements to access arylethylamine structures. | ||
Smiles reaction on Csp3-tethered substrates, by contrast, is far more limited in the literature, and represents a highly appealing target for method development. The product arylethylamines 3 are amongst the most privileged scaffolds in chemistry and biology, found in endogenous signalling molecules such as melatonin, adrenaline, serotonin and dopamine, and have been widely exploited in medicines for central nervous system (CNS) disorders (e.g. antidepressants agomelatine and venlafaxine, Parkinson's treatment baclofen, and stimulants such as amphetamine and Ritalin).6
A small number of aryl transfer routes to arylethylamines are known, including some of the earliest reports from Speckamp on the α-halomethylpiperidinyl system.2,7 Zard established the reaction for xanthates 4, using thermal cleavage with a peroxide initiator to give the substituted arylethylamines 5 (Scheme 2.1).8 Recent work by Stephenson used the visible light SET oxidation of p-methoxystyrenes to trigger sulfonamide addition, followed by Smiles rearrangement to give 1,1-diarylethylamines 8.3a Our laboratory has effected radical Smiles rearrangement using visible light-mediated addition of bromodifluoroacetate to alkenes 9, giving the gem-difluoro substituted arylethylamines 11.9 In most cases, the reaction design generates secondary radicals for desulfonylative Smiles rearrangement, producing branched products. A notable exception from Reiser and co-workers used decarbonylative Smiles rearrangement from the tert-butylated benzoylamides 12 to access the parent phenylethylamines 13.5c The more demanding amide decarbonylation step necessitated an intramolecular single electron transfer mechanism, requiring electron rich migrating aryl groups for efficient reaction.
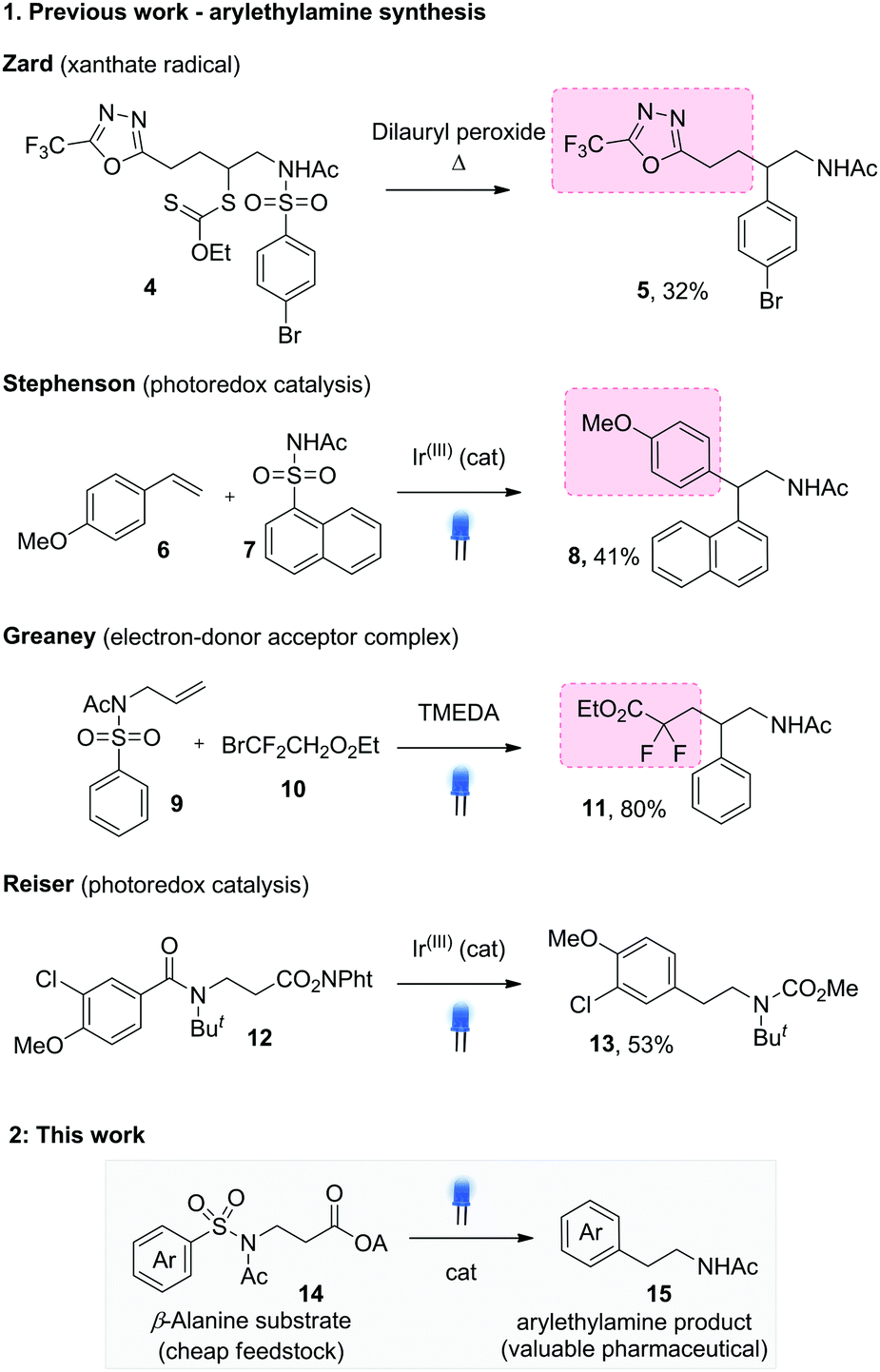 | ||
| Scheme 2 Extrusion Smiles rearrangements to access arylethylamine structures. | ||
We were interested in establishing a general approach to unsubstituted and substituted arylethylamines under mild, catalytic conditions. Our reaction design is shown in Scheme 2.2 and represents the first example of a desulfonylative, decarboxylative Truce–Smiles rearrangement. The extrusion of CO2 provides expedient access to the key Csp3 radical species (1 in Scheme 1.1) and uses readily available amino acid starting materials 14, exploiting their low cost, high availability and in certain cases, enantiopurity. On the other hand, the extrusion of SO2 to irreversibly drive the Truce–Smiles rearrangement has proven to be an efficient means to incorporate a plethora of aryl species including electron rich and poor arene rings as well heterocycles. We hypothesised that the synergy of these two strategies would provide arylethylamine examples largely inaccessible by other reports in the field of Smiles chemistry.
We began by preparing a series of β-alanine oxime esters 16 (three steps, no purification of intermediates required, see ESI†). Recent work from Glorius has used these moieties in the room-temperature decarboxylation of aliphatic carboxylic acids via visible light-mediated energy transfer catalysis.10 This approach is attractive as it removes the need for stoichiometric reductants, commonly necessary for redox-active phthalimide based strategies.11 We used thiophenes as the migrating arenes in our initial optimization studies, owing to their prevalence in pharmaceuticals12 and their success in literature aryl transfer reactions (Table 1).3a,g,5d Using Ir(dF(CF3)ppy)2(dtbbpy)PF6 as catalyst, we were delighted to observe successful reaction for various oxime esters, with the p-fluoro derivative giving the best conversion.
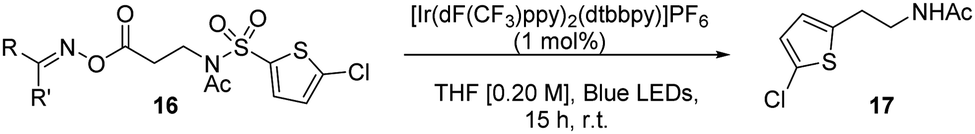
A screen of catalysts, solvents, and concentrations established the iridium photosensitizer (1 mol%) in a tetrahydrofuran (THF) solvent 0.08 M under blue LED irradiation, successfully delivered the arylethylamine product in 56% yield (17a). Notably, we also found that we could lower the catalytic loading to 0.25 mol% with only a small reduction in reaction efficiency (see ESI†).
We explored the scope of the reaction for thiophenylethylamines, initially. Compound 16b, containing the 3-chlorothiophene substituent, gave a substantially higher yield, possibly due to prevention of side-reactions through ortho addition to the thiophene. It was possible to carry out this reaction under metal free conditions using an organic thioxanthone photosensitizer (10 mol%), albeit at reduced efficiency. Both the brominated and un-substituted thiophene rings reacted in moderate yield (17c and 17d), along with the Boc-protected substrate 17e, establishing an orthogonal nitrogen-protecting group pathway.14
Substitution on the ethyl backbone was easily tolerated, with the α and β-methyl substrates rearranging in excellent yield (17f and 17g). Pleasingly, we were able to show for 17g that reaction occurred without racemization, with both R and S starting materials delivering the enantiopure heterocyclic amphetamine-type structures, biosteres of other biologically active amines.13
Moving away from thiophene, the transformation could facilitate the migration of phenyl rings, creating a collection of biologically active phenylethylamines 17h–17o. Fluorinated aryl rings have gained much attention within the medicinal chemistry community and we were keen to demonstrate the applicability to our system. Hydrolysis products of 17h–17j have all been used in the literature as components for pharmaceutically relevant compounds as well as active components in organic semiconductors.14 Whilst the unsubstituted phenyl ring proved to be an ineffective migrating group, 2-fluorophenylethylamine 17h could be incorporated with moderate yield by increasing the electrophilicity of the ipso position. This is further demonstrated in example 17i where both the 2 and 6 positions are blocked, preventing deleterious alkyl radical addition products. 2-Trifluoromethoxyphenyl 17k also acted as a good substrate for desulfonylative Smiles chemistry.15 Additional examples with 2-substitution included 17l, showing that we could incorporate anisyl rings in our chemistry, as well as meta substitution for further functionalisation of the arene system (17m). 1-Naphthylethylamine, a known tryptamine analogue, could be synthesized (17n), potentially opening up new synthetic routes to the antidepressant agomelatine.16 In accordance with other reports on radical ipso substitution, the reaction was tolerant of steric hindrance demonstrated with compound 17o, containing a mesityl ring.17
Finally, we were keen to demonstrate the methodology in the synthesis of a phenylalanine pharmaceutical. β-2-Thienylalanine has been used to treat infants with phenylketonuria, a genetic disorder resulting in the accumulation of phenylalanine in the blood, leading to brain damage if left untreated.18 Using the oxime ester derived from cheap and readily available L-methyl aspartate, photocatalytic Smiles rearrangement gave the protected β-2-thienylalanine 17p in 68% yield and 98% ee. Our approach compliments existing routes to this class of molecule, which use asymmetric Rh hydrogenation19 or enzymatic catalysis with phenylalanine ammonia lyase.20
The mechanism for the Smiles arylation likely begins with an energy transfer (EnT) event between the excited iridium catalyst and the activated imine I giving II, a diradical species (Scheme 4). Homolytic N–O bond cleavage can then take place, releasing iminyl radical III which can be quenched via hydrogen atom abstraction (HAT),21 from the solvent's relatively weak C–H bond (α-CH: BDE = 385 kJ mol−1).22IV can spontaneously decarboxylate to give a primary alkyl radical V.23 This can then undergo a radical desulfonylative-Smiles rearrangement via ipso substitution, forming a spirocyclic intermediate VI, releasing SO2 to irreversibly drive the process to completion. One further HAT event between amidyl radical VII and the solvent gives the product, VIII. Owing to the inability of similarly reducing photocatalyst (see ESI†) to yield any product, the absence of a suitable stoichiometric reductant, and the irreversible reduction potential of −2.0 V (vs. SCE)10 we can rule out a photoredox mechanism as a viable pathway for N–O bond cleavage. Furthermore the reaction can proceed with reasonable efficiency using the organic photosensitizer thioxanthone (17b, Scheme 3). In the dark no conversion is observed but under blue LED irradiation (catalyst free), a 20% NMR yield of the product is observed.
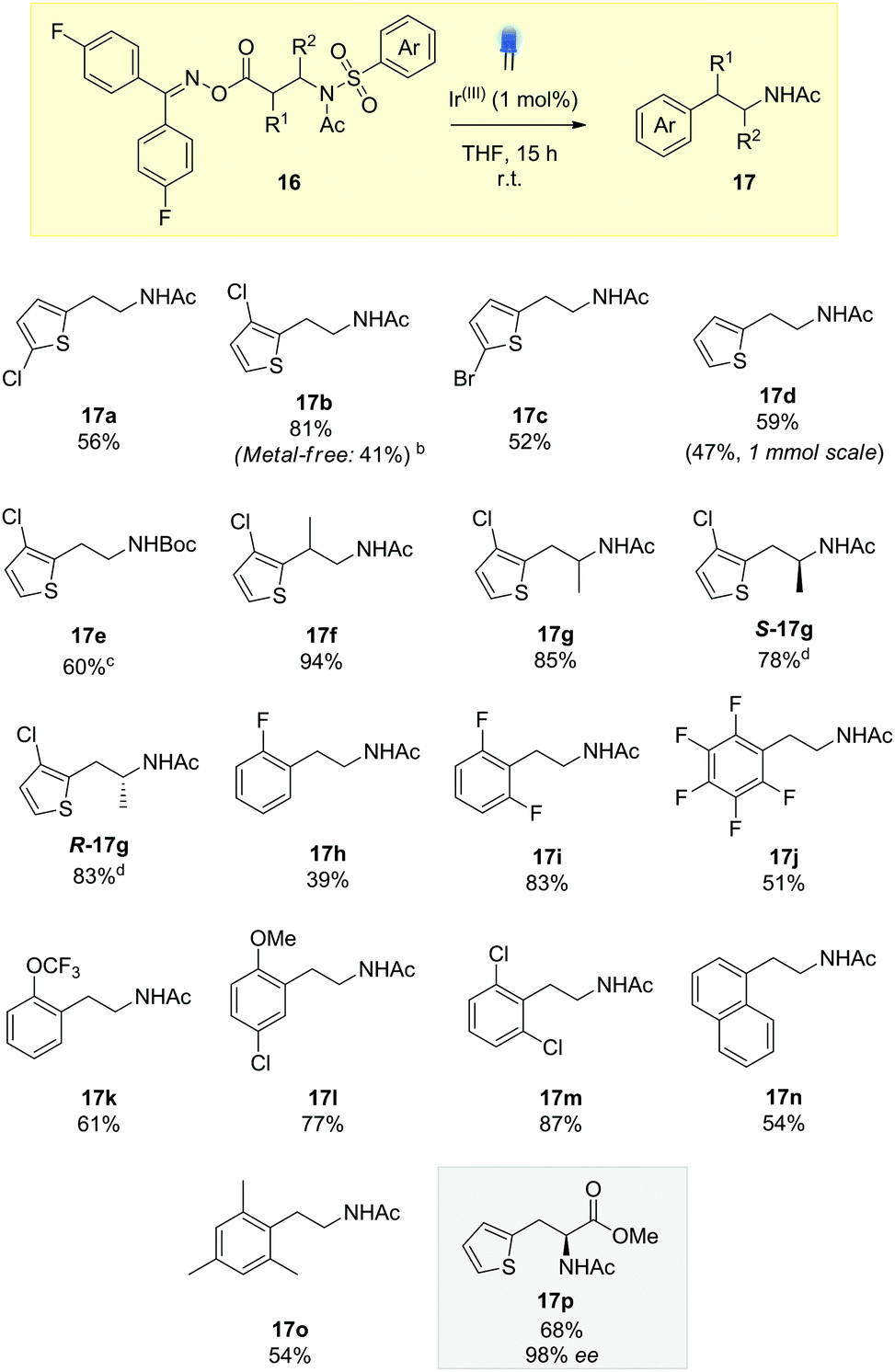 | ||
Scheme 3 Scope of the decarboxylative, desulfonylative Truce–Smiles rearrangement.a,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a0.20 mmol scale, Ir(III) = Ir(dF(CF3)ppy)2(dtbbpy)PF6, 2.5 mL THF. b10 mol% thioxanthone catalyst, blue LED irradiation. cNMR yield using 1,3,5-trimethoxybenzene as an internal standard. dNo evidence of racemization, isolated as single enantiomers. a0.20 mmol scale, Ir(III) = Ir(dF(CF3)ppy)2(dtbbpy)PF6, 2.5 mL THF. b10 mol% thioxanthone catalyst, blue LED irradiation. cNMR yield using 1,3,5-trimethoxybenzene as an internal standard. dNo evidence of racemization, isolated as single enantiomers. | ||
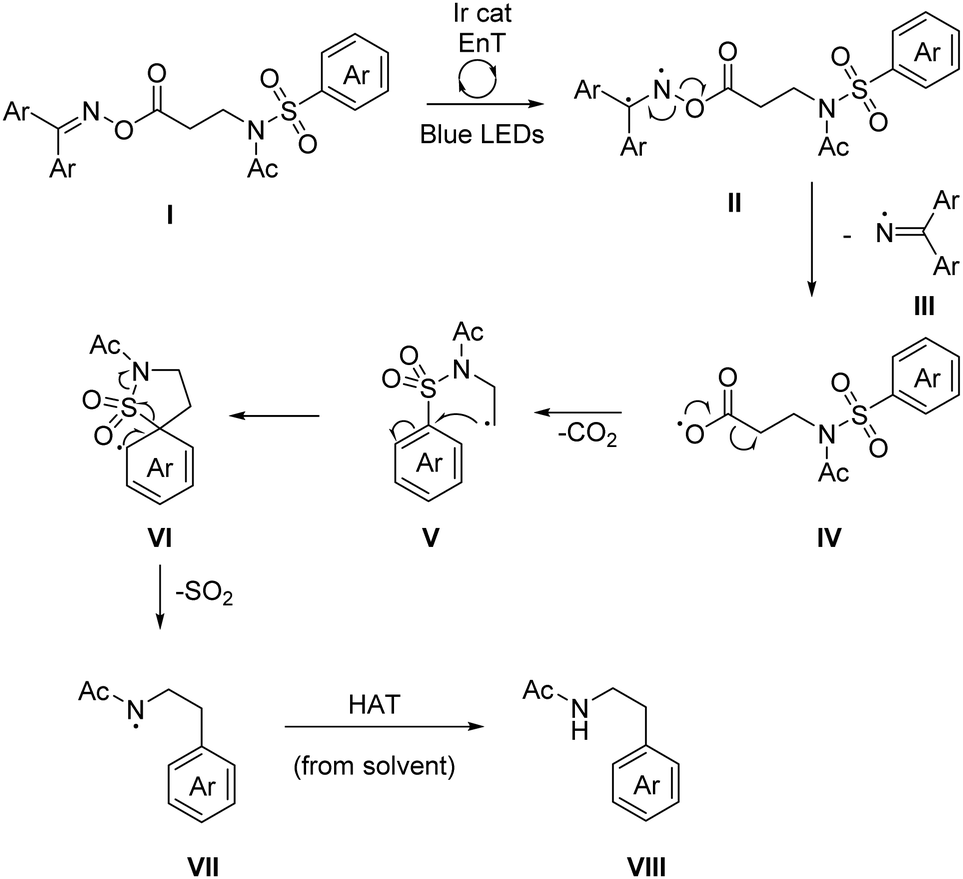 | ||
| Scheme 4 Proposed mechanism of the decarboxylative, desulfonylative radical Truce–Smiles rearrangement. Ar = 4-F-phenyl. | ||
In view of this last observation, we were intrigued to see if the reaction could be carried out catalyst-free by direct excitation of the oxime moiety using UVA radiation at room temperature (Scheme 5). This would allow for a reagent-free approach to sp3–sp2 C–C bond formation, at room temperature. We were pleased to find that we obtained a 41% NMR yield of the final product (cf. example 17a, Scheme 3), indicating the viability of this approach.24
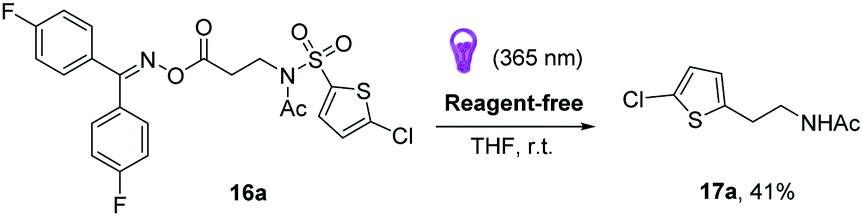 | ||
| Scheme 5 Reagent-free desulfonylative, decarboxylative Truce–Smiles rearrangement. | ||
In conclusion, we have developed a new decarboxylative, desulfonylative Truce–Smiles rearrangement enabled by visible light energy transfer catalysis. The reaction uses starting materials derived from commercially available β-amino acids, giving extensive control over the aryl species, ethyl chain substitution and amino protection for the synthesis of new arylethylamines. The reaction can be carried out metal-free and catalyst-free and applied to the synthesis of chiral unnatural amino acids.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Holden and M. F. Greaney, Chem. – Eur. J., 2017, 23, 8992–9008 CrossRef CAS PubMed.
- Early work: (a) R. Loven and W. N. Speckamp, Tetrahedron Lett., 1972, 13, 1567 CrossRef; (b) W. B. Motherwell and A. M. K. Pennell, Chem. Commun., 1991, 877 RSC.
- Recent desulfonylative Smiles reports: (a) T. Monos, R. McAtee and C. Stephenson, Science, 2018, 361, 1369 CrossRef CAS PubMed; (b) P. G. T. Rabet, S. Boyd and M. F. Greaney, Angew. Chem., Int. Ed., 2017, 56, 4183 CrossRef CAS PubMed; (c) C. A. Holden, S. M. A. Sohel and M. F. Greaney, Angew. Chem., Int. Ed., 2016, 55, 2450 CrossRef CAS PubMed; (d) E. Brachet, L. Marzo, M. Selkti, B. König and P. Belmont, Chem. Sci., 2016, 7, 5002 RSC; (e) S. Coulibali, T. Godou and S. Canesi, Org. Lett., 2016, 18, 4348 CrossRef CAS PubMed; (f) S. W. Crossley, R. M. Martinez, S. Guevara-Zuluaga and R. A. Shenvi, Org. Lett., 2016, 18, 2620 CrossRef CAS PubMed; (g) J. J. Douglas, H. Albright, M. J. Sevrin, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2015, 54, 14898 CrossRef CAS PubMed; (h) O. K. Rasheed, I. R. Hardcastle, J. Raftery and P. Quayle, Org. Biomol. Chem., 2015, 13, 8048 RSC; (i) N. Fuentes, W. Kong, L. Fernandez-Sanchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964 CrossRef CAS PubMed; (j) W. Kong, N. Fuentes, A. Garcia-Dominguez, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 2487 CrossRef CAS PubMed; (k) W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480 CrossRef CAS PubMed.
- For desulfonylative Smiles that proceed through 4-membered transition states, see: (a) M. W. Wilson, S. E. Ault-Justus, J. C. Hodges and J. R. Rubin, Tetrahedron, 1999, 55, 1647 CrossRef CAS; (b) V. Lupi, M. Penso, F. Foschi, F. Gassa, V. Mihali and A. Tagliabue, Chem. Commun., 2009, 5012 RSC; (c) S. Johnson, E. Kovacs and M. F. Greaney, Chem. Commun., 2020, 56, 3222 RSC.
- Recent non-desulfonylative Smiles examples: (a) X. Chang, Q. Zhang and C. Guo, Org. Lett., 2019, 21, 4915 CrossRef CAS PubMed; (b) J. Li, Z. Liu, S. Wu and Y. Chen, Org. Lett., 2019, 21, 2077 CrossRef CAS PubMed; (c) C. Faderl, S. Budde, G. Kachkovskyi, D. Rackl and O. Reiser, J. Org. Chem., 2018, 83, 12192 CrossRef CAS PubMed; (d) D. Alpers, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2018, 12167 CrossRef CAS PubMed; (e) D. L. Leonard, J. W. Ward and J. Clayden, Nature, 2018, 562, 105 CrossRef CAS PubMed; (f) R. Costil, Q. Lefebvre and J. Clayden, Angew. Chem., Int. Ed., 2017, 56, 14602 CrossRef CAS PubMed; (g) S.-F. Wang, X.-P. Cao and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 13809 CrossRef CAS PubMed; (h) R. Costil, H. J. A. Dale, N. Fey, G. Whitcombe, J. V. Matlock and J. Clayden, Angew. Chem., Int. Ed., 2017, 56, 12533 CrossRef CAS PubMed; (i) D. Janssen-Mueller, S. Singha, F. Lied, K. Gottschalk and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 6276 CrossRef CAS PubMed; (j) S. S. Bhojgude, T. Roy, R. G. Gonnade and A. T. Biju, Org. Lett., 2016, 18, 5424 CrossRef CAS PubMed.
- S. Freeman and J. F. Adler, Eur. J. Med. Chem., 2002, 37, 527 CrossRef CAS PubMed.
- H. J. Köhler and W. N. Speckamp, J. Chem. Soc., Chem. Commun., 1980, 142 RSC ; and references therein.
- A. Gheorghe, B. Quiclet-Sire, X. Vila and S. Zard, Org. Lett., 2005, 7, 1653 CrossRef CAS PubMed.
- D. Whalley, H. Duong and M. Greaney, Chem. – Eur. J., 2019, 25, 1927 CrossRef CAS PubMed.
- T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514 CrossRef CAS PubMed.
- T. Patra and D. Maiti, Chem. – Eur. J., 2017, 23, 7382 CrossRef CAS PubMed.
- S. Pathania, R. K. Narang and R. K. Rawal, Eur. J. Med. Chem., 2019, 180, 486 CrossRef CAS PubMed.
- M. M. Goodman, G. W. Kabalka, J. Lee, G. W. Kabalka, R. C. Marks, Y. Liang and F. F. Knapp, J. Med. Chem., 1992, 35, 280 CrossRef CAS PubMed.
- R. Schmidt, J. H. Oh, Y. Sen Sun, M. Deppisch, A. M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk and Z. Bao, et al. , J. Am. Chem. Soc., 2009, 131, 6215 CrossRef CAS PubMed.
- K. Lee, Z. Lei, C. Morales-Rivera, P. Liu and M. Ngai, Org. Biomol. Chem., 2016, 14, 5599 RSC.
- M. Dukat, C. Smith, K. Herrick-Davis, M. Teitler and R. A. Glennon, Bioorg. Med. Chem., 2004, 12, 2545 CrossRef CAS PubMed.
- M. L. E. N. Da Mata, W. B. Motherwell and F. Ujjainwalla, Tetrahedron Lett., 1997, 38, 137 CrossRef CAS.
- C. Krips and D. R. Lines, J. Paediatr. Child Health, 1972, 8, 318 CrossRef CAS PubMed.
- B. Mohar and M. Stephan, Adv. Synth. Catal., 2013, 355, 594 CAS.
- C. Paizs, A. Katona and J. Rétey, Chem. – Eur. J., 2006, 12, 2739 CrossRef CAS PubMed.
- L. Chen, L. Guo, Z. Ma, Y. Gu, J. Zhang and X. Duan, J. Org. Chem., 2019, 84, 6475 CrossRef CAS PubMed.
- C. Meyer, S. Hell, A. Misale, A. Trabanco and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 8829–8833 CrossRef CAS PubMed.
- (a) T. Patra, P. Bellotti, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 3172 CrossRef CAS PubMed; (b) V. Soni, S. Lee, J. Kang, Y. Moon, H. Hwang, Y. You and E. Cho, ACS Catal., 2019, 9, 10454 CrossRef CAS.
- Reiser and co-workers reported an example of acetone-sensitised, UV-mediated Smiles reaction in their decarbonylative system, see ref. 5c.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05049k |
| This journal is © The Royal Society of Chemistry 2020 |