
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrocatalytic synthesis of organic carbonates
Dimitra
Anastasiadou
,
Emiel J. M.
Hensen
 and
Marta C.
Figueiredo
*
and
Marta C.
Figueiredo
*
Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, Eindhoven, The Netherlands. E-mail: m.c.costa.figueiredo@tue.nl
First published on 29th September 2020
Abstract
Organic carbonates are considered environmentally benign alternatives for various fossil-derived compounds used in the chemical industry. Replacing current costly and toxic production methods by greener alternatives offers opportunities to cover the increasing demand for these intermediates in a more sustainable manner. In this feature article, the prospect of electrochemical synthesis of organic carbonates is presented as an approach to use carbon dioxide and green electricity to arrive at such compounds. We explore the strengths and limitations of the different methods by looking into the electrode and electrolyte composition effects and operating conditions with a focus on the synthesis of dimethylcarbonate from methanol and either carbon monoxide or carbon dioxide. The proposed mechanisms are discussed in an effort to understand the underlying steps and their challenges. This review concludes with a perspective on the broader developments needed to turn the basic chemistry into a practical application.
 Dimitra Anastasiadou | Dimitra Anastasiadou is working as a PhD researcher in the Department of Chemical Engineering and Chemistry at Eindhoven University of Technology (Netherlands) since May 2019, under the supervision of Prof. Emiel Hensen and Dr Marta Costa Figueiredo. Her doctoral research is focused on the electrochemical conversion of CO2 and NOx towards high value chemicals. Prior to Eindhoven University, she worked in industry as an assistant researcher on the electrochemical reduction of CO2. |
 Emiel J. M. Hensen | Emiel Hensen (Geleen, The Netherlands 1971), professor of Inorganic Materials and Catalysis at Eindhoven University of Technology, has been carrying out research in heterogeneous catalysis for nearly 2 decades. He focuses on fundamental aspects of reaction mechanism, combining operando characterization with theoretical modelling aiming at understanding the nature of active sites. He has published more than 500 research papers, more than 10 book chapters and is inventor of several patents. Hensen earned his MSc and PhD at Eindhoven University of Technology under direction of Prof. Rutger van Santen and worked also at the University of Amsterdam, Hokkaido University and Shell. |
 Marta C. Figueiredo | Marta Costa Figueiredo is Assistant Professor of Electrocatalysis at Eindhoven University of Technology since April 2019. She obtained her PhD in electrocatalysis, science and technology in 2012 at the University of Alicante, Spain under the supervision of Prof. Juan Feliu. After that, she was a postdoctoral researcher at different Universities in Europe such as Aalto University (Finland), Leiden University and University of Copenhagen. Before joining TU/e, Marta worked in the industry as Jr Scientist at Avantium (Amsterdam). In Eindhoven, her research is devoted to electrocatalysis and electro(catalytic) synthesis for sustainable processes and production of high value chemicals. |
Introduction
Our way of living is strongly dependent on Earth's natural resources to fulfil our needs for energy and materials. There is growing concern that the emissions of greenhouse gases, most notably carbon dioxide (CO2), associated with the use of fossil resources will lead to climate change. Therefore, we need to substantially increase the share of renewable energy resources to cover our immediate energy demands. The production of chemicals is also mostly based on fossil resources, and their manufacture typically requires large amounts of energy. Thus, the chemical industry can also contribute to lower CO2 emissions by introducing renewable feedstock and energy.1Many different technologies are being explored to enable the envisioned transition to renewable energy with a focus on renewable electricity derived from solar and wind.2 In the long term, carbon capture and sequestration and use (CCS and CCU) combined with renewable energy can form the basis for the sustainable production of chemicals.3 Based on the expected availability and low pricing of renewable electricity, electrochemistry can become a key enabling technology for such a sustainable chemical industry.4,5 Electrochemical conversion routes are attractive for many reasons. The technology can in principle be scaled up relatively easily, and it provides opportunities to use a variety of feedstock including renewable biobased waste products and even CO2, which can in principle be captured from the air. This strategy of turning “waste” into valuable products can reduce the carbon footprint of the chemical industry to zero, if not negative.
Organic carbonates are an important class of chemical intermediates, for which a transition of a fossil-based process to an electrochemical alternative involving renewable energy and the use of CO2 is considered. Organic carbonates find widespread application as aprotic polar solvents, monomers in the manufacture of plastics and intermediates in the production of pharmaceutical, cosmetics and fine chemicals (e.g., glycols, purines, carbamates). They can also be used as fuel additives6 and electrolyte solvents in lithium-ion batteries.7 Moreover, their low toxicity and biodegradability make them useful intermediates for green chemistry applications.8 Currently, the industrial synthesis of organic carbonates is environmentally stressing due to the use of toxic reactants (e.g. phosgene and carbon monoxide), harsh operative conditions and large amount of waste.9 Thus new approaches to manufacture these chemicals sustainably, such as electrosynthesis, are desired. By using electrosynthesis, not only renewable energy is directly used to create bonds, but also electrons are used instead of highly toxic reactants. Moreover, when designing a novel electrochemical process for organic carbonate synthesis, the use of CO2 instead of carbon monoxide (CO) can be considered.10,11
The formation of organic carbonates from CO2 can be achieved via two pathways. The first pathway involves the direct reduction of CO2, while the second one first reduces CO2 to CO followed by organic carbonate synthesis.12 Technology for the electrochemical CO2 reduction to CO is advancing very fast.13–15 In this review, we will discuss both electrochemical pathways. We finalise with an outlook by discussing the challenges in this area.
Electrocatalytic synthesis
As a general introduction, Fig. 1 depicts several electrochemical cells involving three electrodes, used for lab-scale testing. The simplest electrochemical cell configuration is an undivided three-electrode cell, in which a cathode and an anode are immersed into a conductive solution containing the substrate(s). At the cathode, electrons are removed from the surface and transferred to the substrate, which results in its reduction. At the same time at the anode, an electron from the substrate is removed, resulting in its oxidation. The electrochemical reaction is driven by a power supply that applies a potential difference between the cathode and the anode (Fig. 1a). In electrochemical synthesis, typically only one of the reactions, anodic or cathodic, is investigated. For example, the synthesis of dimethyl carbonate (DMC) from CO2 and methanol (CH3OH), which requires reduction, takes place in the cathodic compartment (Fig. 1b). On the other hand, to synthesise DMC from CO and CH3OH, electrooxidation is required, which takes place at the anode (Fig. 1c).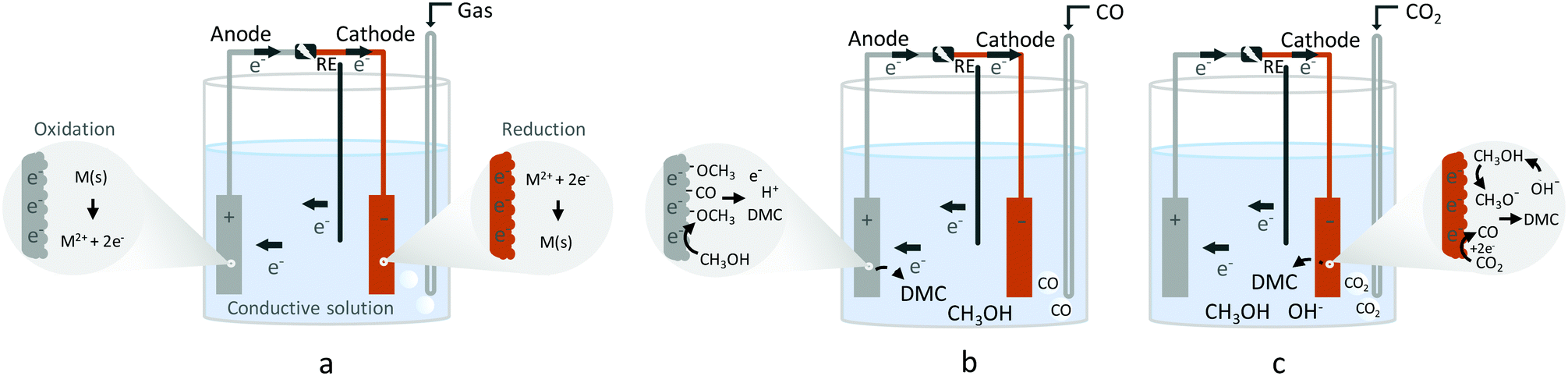 | ||
| Fig. 1 Electrochemical three-electrode cells. (a) General electrochemical cell, (b) electrocatalytic synthesis of DMC with CO2, (c) electrocatalytic synthesis of DMC with CO. | ||
Electrocatalytic synthesis of organic carbonates from CO2
Several researchers have reported electrocatalytic synthesis methods to obtain organic carbonates,16–25 mainly targeting DMC. DMC is an environmentally benign alkyl carbonate, which is suitable for a wide range of applications. For example, it can serve as a methoxycarbonylation and methylation agent, replacing toxic and hazardous compounds such as phosgene, methyl halides, and dimethyl sulphate.26 DMC can also replace tert-butyl methyl ether as an oxygenated fuel additive in gasoline or diesel. Another application of growing importance is as electrolyte solvent in lithium-ion batteries.7Industrially, DMC is produced via various routes, all of which are hazardous and energy-intensive due to the high temperatures and pressures required to drive the reactions. The most important commercial process to manufacture DMC is oxidative carbonylation of CH3OH with oxygen (R1).27
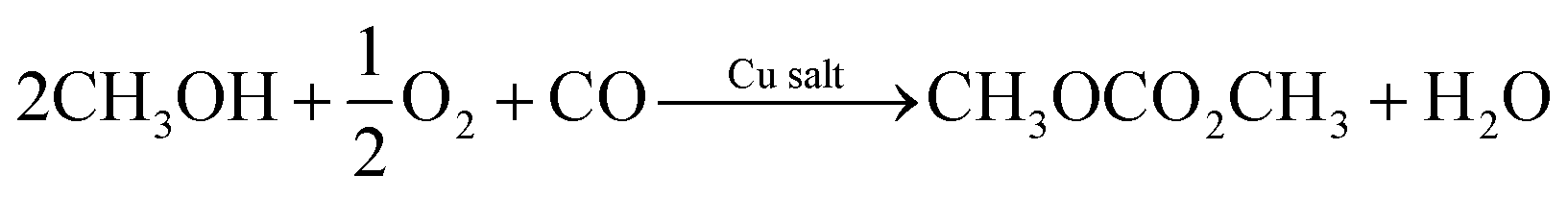 | (R1) |
Given the interest in DMC and the environmental concerns of current production methods, substantial research is directed towards cheaper and less toxic alternatives.28,29 Electrochemical approaches have been explored for this purpose with most efforts made for the reaction of CO2 with CH3OH (R2). This process involves different electrochemical and chemical reactions. The main electrochemical reactions are the reduction of CO2 (to CO) and water (R3) and (R4). The OH− generated from water reduction will facilitate the deprotonation CH3OH to form the methoxy intermediate (R5). DMC is then produced by reaction of the electrochemical formed CO and the methoxy species (R6).
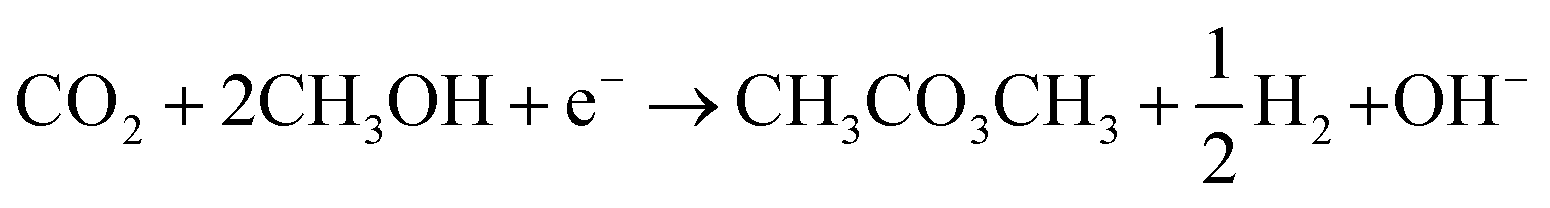 | (R2) |
| CO2 + H2O + 2e− → CO + 2OH− | (R3) |
| 2H2O + 2e− → H2 + 2OH− | (R4) |
| CH3OH + OH− ↔ CH3O− + H2O | (R5) |
| CO + 2CH3O− → CH3CO3CH3 + 2e− | (R6) |
The majority of studies involving the direct electrochemical conversion of CO2 to DMC are performed in imidazolium-based ionic liquids (ILs)30 like 1-butyl,3-methyl-imidazolium tetrafluoroborate and 1-ethyl-3-methylimidazolium tetrafluoroborate. These solvents are attractive because of the high CO2 solubility compared to conventional solvents.16 The solubility of CO2, expressed as the mole fraction of CO2 in these imidazolium-based ILs are around 0.14 and 0.10, respectively, at ∼300 K and 10 bar.31,32 The corresponding solubilities in acetonitrile (<0.09) and methanol and ethanol (<0.07) are significantly lower under similar conditions.
Literature shows that DMC synthesis in ILs can be carried out with good reaction yields (ratio of the obtained product to the starting amount of alcohol). For example, yields around 75% were reported at In, Cu, and Ag based electrodes (Table 1, entries 9 and 7),23,24 while a yield of 81% was mentioned for a nanoporous Cu–Pt based catalyst (Table 1, entry 10).21 Despite these promising DMC yields, these systems use methyl iodide as an alkylating agent, which is a carcinogenic compound. Another study on Pt electrodes reported a high yield of 76%, using (carcinogenic) propylene oxide (Table 1, entry 8).22 Various efforts to exclude the use of such harmful compounds and reduce the production steps have been reported,10,25,33–36 which however negatively affect the DMC yield in general. For example, the synthesis of DMC using Pt or graphite working electrodes reported low yields, below 5%.33 The authors proposed the reaction mechanism shown in Fig. 2a and suggested that the electrochemical synthesis of DMC involves activation of CH3OH on the electrode surface as an initial step leading to the methoxide anion. Methoxide then reacts with CO2 to form DMC. These findings are contradictory to other reports that consider that CO2 is adsorbed on the electrode surface and interacts with CH3OH.21,22 Even though no additives were used in this study,33 benzyl chloride is used to prepare the IL solvent, which is also classified as a carcinogenic compound and brings unnecessary risks and waste to the process.
| Entry | Catalyst | Electrochemical cell | Electrolyte | Temperature (K) | Potential | Current | Time | DMC yielda | DMC selectivity | By-products |
|---|---|---|---|---|---|---|---|---|---|---|
| a The reported yield is based on the starting alcohol unless stated otherwise. | ||||||||||
| 133 | Graphite | Divided electrolytic cell | CH3OH & BzmimCl | 293 | 4.0 V (cell voltage) | 164 to 68 mA cm−2 | 18.5 to 80 h | 3.8% | 51.8% | Dimethoxymethane (DMM), 2,2-dimethoxylpropane, 1,1,2,2-tetramethoxylethane, methyl 2-methoxylpropionate |
| CH3OH & BmimCl | 3.7% | 12.9% | ||||||||
| CH3OH & BmimOH | 3.1% | 42.5% | ||||||||
| CH3OH & BmimBr | 2.1% | 39.7% | ||||||||
| CH3OH & BmimBF4 | 3.0% | 15.5% | ||||||||
| CH3OH & EmimBF4 | 1.3% | 37.6% | ||||||||
| CH3OH & EmimBr | 6.0% | 24.3% | ||||||||
| Pt | CH3OH & BzmimCl | 1.9% | 8.8% | |||||||
| Glassy carbon | No detection | |||||||||
| Cu | ||||||||||
| 234 | Pt/Nb plates | Divided electrochemical flow cell | CH3OK & BmimBr | 279 | 3.5 V (cell voltage) | — | 48 h | 12.5% (based on CO2) | — | Dimethyl ether (DME), DMM and tetramethyl orthocarbonate (OC) |
| 335 | Undivided electrochemical flow cell | 5.5 V (cell voltage) | 70.7% (based on CO2) | — | OC, DMM, bromomethane | |||||
| 436 | Graphite | Divided electrolytic cell | CH3OH & BmimOH | 293 | 3.5 V (cell voltage) | — | 40 h | 0.9% | 76.7% | DMM |
| CH3OH & ApmimBr | 1.1% | 94.3% | ||||||||
| CH3OH & BmimBr | 0.4% | 67.3% | ||||||||
| CH3OH & BmimBF4 | 0.2% | 53.1% | ||||||||
| Pt | CH3OH & ApmimBr | 0.3% | 84.7% | |||||||
| Glassy carbon | 0.3% | 90.7% | ||||||||
| Cu | No detection | |||||||||
| 510 | Cu disc | Undivided electrolytic cell | Tetraethylammonium tetrafluoroborate & CH3OH | 298 | −1.2 and −2.4 V vs. Ag/AgCl | — | — | No detection | — | Alkylammonium methyl carbonate |
| Pt disc | ||||||||||
| Pd disc | ||||||||||
| 625 | Pt | Undivided electrolytic cell | CH3OH, CH3OK & bmimBr | 303 | 5.5 V (cell voltage) | — | 48 h | 3.9% | 88.4% | Trimethoxy methane, OC, H2, CO |
| 724 | Cu | Undivided electrolytic cell | BmimBF4, CH3I, (alcohols added after electrolysis) | 328 | −1.8 V vs. Ag/AgI | — | Stirring for 6 h after electrolysis | 74% | 100% | No by-products |
| Ni | 18% | 100% | ||||||||
| Ag | 73% | 100% | ||||||||
| Stainless steel | 67% | 100% | ||||||||
| Ti | −2.5 V vs. Ag/AgI | 58% | 100% | |||||||
| 822 | Pt | Undivided electrolytic cell | BmimBr, CH3OH & PO | 303 | 5.5 V (cell voltage) | — | 48 h | 75.5% | 77.8% | Not reported |
| Glassy carbon | 49.7% | 53.1% | ||||||||
| Graphite | 30.3% | 37.1% | ||||||||
| 923 | In | Undivided electrolytic cell | BmimBF4, CH3OH & CH3I | 313 | −1.7 V vs. Ag | — | DMC detected 6 h after electrolysis | 76% | — | Not reported |
| 1021 | Cu skeletons/Pt shells | Undivided electrolytic cell | BmimBF4, CH3OH & CH3I | 298 | −2.2 V vs. Ag | 3.2 mA cm−2 | Stirring for 6 h after electrolysis | 81% | — | Not reported |
| Pt | 1.7 mA cm−2 | 64% | ||||||||
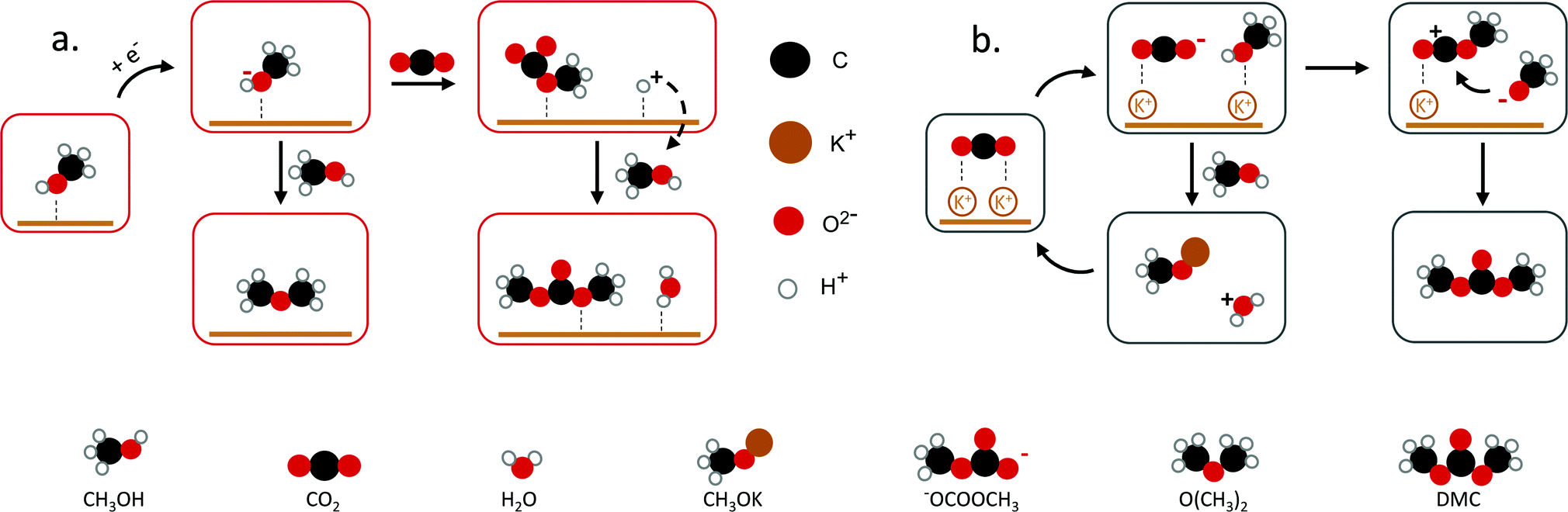 | ||
| Fig. 2 Proposed mechanisms towards DMC. (a) DMC formation through methoxy and CO2, (b) K+ interaction with CO2 to DMC. Figures adapted from ref. 25 and 33. | ||
Yuan et al. reported the electrochemical synthesis of DMC on Pt electrodes in a dialkyl imidazolium IL–potassium methoxide (CH3OK)–CH3OH system, where CH3OK acted as a co-catalyst.25 Amongst the basic compounds investigated (CH3ONa, NaOH, KOH, K2CO3, CH3OK), CH3OK led to the highest DMC yield (3.9%) (Table 1, entry 6). The authors suggested that the K+ ion interacts with adsorbed CO2 stabilising the CO2− anion (Fig. 2b). This system was further studied in a filter-press electrochemical reactor with a divided anodic and cathodic compartment, resulting in an increased DMC yield of 12.5% (Table 1, entry 2).34,35 By applying a higher potential, the authors achieved the highest efficiency for the electrosynthesis of DMC from CO2 without the use of harmful compounds (Table 1, entry 3). Another study by Lu et al. reported a yield of 2.5% with 94.5% selectivity on a graphite electrode using an amino-functionalised, imidazolium-based IL (Table 1, entry 4).36 The basicity of the amino-functionalised IL is beneficial for capturing and activating the CO2 molecule. These findings are in agreement with another work25 and support that basic groups are advantageous for CO2 activation. Even though the use of carcinogenic compounds can be avoided, the use of ILs has some limitations. These include, besides the low yield, their high cost and viscosity.37 Hence, a more cost-efficient and feasible system for the electrochemical conversion of CO2 to DMC needs to be developed. To achieve this, understanding the underlying mechanism and identifying the reaction intermediates is essential.
A recent investigation of the synthesis of organic carbonates from CO2 and CH3OH in acetonitrile provides better insight into the reaction mechanism and the associated intermediates (Table 1, entry 5).10 The proposed reaction pathway (Fig. 3b) involves the electrochemical reduction of CO2 to CO and the simultaneous activation of CH3OH. The CH3OH is activated via dehydrogenation/deprotonation by OH− species derived from residual water oxidation. The CH3OH activation leads to the formation of methoxy species, which react with CO and form DMC and monomethyl carbonate (Fig. 1c). Under the applied conditions, the researchers did not detect DMC by Fourier transformed infrared spectroscopy (FTIR), as it quickly decomposes below −1.4 V vs. Ag/Ag+ to the monomethyl carbonate as shown experimentally.10Fig. 3a shows the FTIR spectrum for a Cu electrode in the presence of CH3OH and CO2. The authors explained that the bands at 1673 and 1281 cm−1 can be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–OCH3 stretching vibrations of monomethyl carbonate, respectively, and not to functional groups of DMC. The formation of this alkyl carbonyl intermediate was observed on different electrodes like Cu, Pt and Pb. The results suggest that the overall reaction requires methoxy species and the ability of the electrode to reduce CO2 to CO or CO2−. Further experiments with isotopically labelled compounds showed that CO is responsible for the carbonylation step and not the CO2 anion radical.25,36
O and C–OCH3 stretching vibrations of monomethyl carbonate, respectively, and not to functional groups of DMC. The formation of this alkyl carbonyl intermediate was observed on different electrodes like Cu, Pt and Pb. The results suggest that the overall reaction requires methoxy species and the ability of the electrode to reduce CO2 to CO or CO2−. Further experiments with isotopically labelled compounds showed that CO is responsible for the carbonylation step and not the CO2 anion radical.25,36
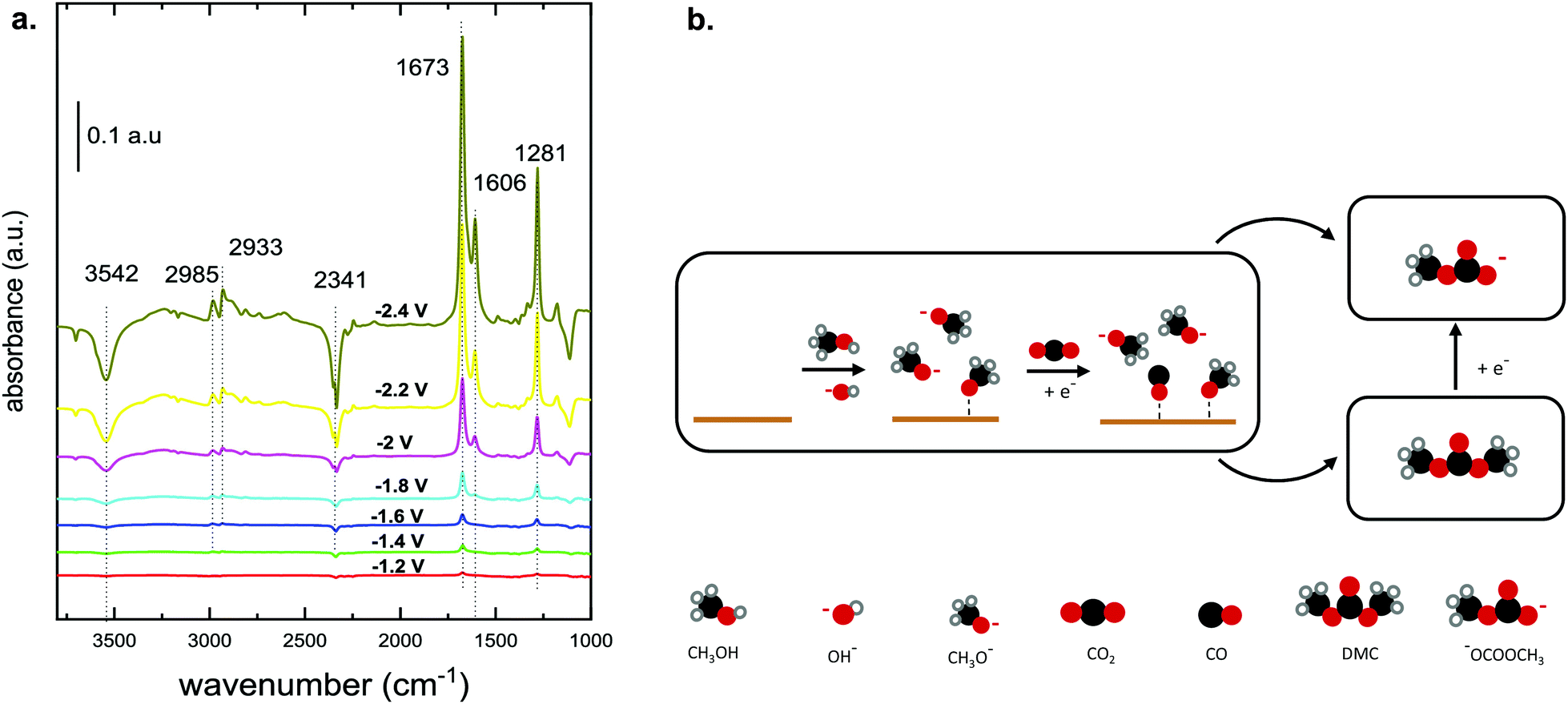 | ||
| Fig. 3 (a) FTIR spectrum of CO2 reduction in CH3OH on a Cu electrode at potentials between −1.2 and −2.4 V vs. Ag/AgCl.10 (Reprinted with permission from American Chemical Society. Copyright©2019, American Chemical Society.) (b) Proposed reaction mechanism that involves the electrochemical reduction of CO2 to CO with a simultaneous activation of CH3OH. Scheme adapted from ref. 10. | ||
So far, the studies discussed target the formation of DMC using CO2 directly as feedstock. As mentioned, higher yields were achieved with the use of ILs.21–24 Investigations into avoiding the use of such media have succeeded in producing DMC, but with low yields.25,34–36 A study from Figueiredo et al.10 examined the formation of DMC in organic solvents. Despite the mechanistic insights obtained, DMC was not detected under the applied reaction conditions due to its low stability at negative potentials. As DMC can also be obtained under oxidative conditions using CO as a reactant, this approach will be discussed in the next section.
Electrochemical synthesis of organic carbonates from CO
An alternative route to synthesise DMC electrochemically is by using CO under oxidative conditions. Through this route, CH3OH and CO are converted to DMC (R7), similar to the current industrial process. Other products often reported in the electrochemical equivalent of this process are dimethyl oxalate (DMO), dimethyl ether (DME), dimethoxymethane (DMM), and methyl formate (MF). CO2 has also been observed, which points to the undesired direct oxidation of CH3OH.38| 2CH3OH + CO ↔ CH3OCO2CH3 + 2H+ + 2e− | (R7) |
An interesting approach of CH3OH carbonylation with CO involved a solution-phase Br2/Br− redox couple in an indirect electrochemical system (Table 2, entry 2).39 Br− is oxidised at the Pd anode to Br2 and Br2 acts as an oxidant for the oxidative carbonylation of CH3OH and CO. This does not constitute an electrochemical reaction. Even though the system is very selective, it is not scalable due to the low yield (7.2%) and the use of Br2, a highly toxic compound.
| Entry | Catalyst | Electrochemical cell | Electrolyte | Temperature (K) | Potential | Current (mA cm−2) | Time | DMC formation | DMC faradaic/current efficiency | DMC selectivitya | By-products |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a The selectivity reported is based on the conversion of starting alcohols. | |||||||||||
| 138 | Cu | Divided H-cell | NaClO4/MeOH | 298 | 0.2 V vs. SCE | 4.5 | 70 days post electrolysis | 24 μM | 6% | 100% | No other products detected |
| 239 | Pd/AC + VGCF | Divided H-cell | LiBr & MeOH | 298 | 1.0 V vs. Ag/AgCl | 9–10 | 60 min | 150 μmol h−1 cm−2 | 80% | 95% | DMO, DMM, MF, and CO2 |
| 340 | CuCl2-graphite | Divided flow cell | H3PO4 & MeOH | 343 | 1.4 V vs. Ag/AgCl | 3.2 | 120 min | 0.1 μmol min−1 cm−2 | 11% | Not reported | DMM, MF, CO2 |
| 441 | PdCl2-graphite | Divided flow cell | H3PO4/MeOH | 343 | 1.3 V vs. Ag/AgCl | 3.2 | 30 min | 0.1 μmol min−1 cm−2 | 10% | 30% | DMM, MF, CO2 |
| 542 | PdCl2/VGCF | Divided flow cell | MeOH/NaClO4 or HClO4 | 298 | 1.8 V vs. Ag/AgCl | 8 | 60 min | 80 μmol h−1 cm−2 | 67% | 82% | DMO, DMM, MF, CO2 |
| 643 | CuCl(bipy) | Divided H-cell | MeOH/NaOCH3 | 293 | 0.3 V vs. SCE | 5 | — | 18 mmol | 64% | Not reported | No other products reported |
| 744 | CuCl(bipy) | Divided H-cell | MeOH/NaOCH3 | 293 | 0.3 V vs. SCE | — | — | — | 85% | Not reported | No other products reported |
| PdCl2(bipy) | Divided H-cell | MeOH/NaOCH3 | 293 | 0.9 V vs. SCE | — | — | — | 64% | Not reported | ||
| 845 | CuCl2(bipy) | Divided electrolytic cell | MeOH/KOH | 298 | 0.3 V vs. Ag/Ag2O | 2.3 | 120 min | 0.3161 mmol | 76% | 100% | No other products detected |
| 946 | Cu(bipy) MOF | Divided electrolytic cell | MeOH/KOH | 298 | 0.3 V vs. Ag/Ag2O | — | — | 110.33 μmol | — | Not reported | No other products reported |
| 1047 | PdCl2 + VGCF | Divided H-cell | NaClO4/MeOH | 298 | 1.0 V vs. Ag/AgCl | 4.6 | — | 58.6 μmol cm−2 h−1 | 68% | Not reported | DMO, DMM, MF, CO2 |
| 1147 | HAuCl4/AC + VGCF | Divided H-cell | NaClO4/MeOH | 298 | 1.0 V vs. Ag/AgCl | 2.4 | — | 2.3 μmol cm−2 h−1 | 5% | Not reported | DMO, DMM, MF, CO2 |
| 1248 | HAuCl4/AC | Divided H-cell | NaClO4/MeOH | 298 | 1.5 V vs. Ag/AgCl | 8 | 250 min | 300 μmol | 20% | 32% | DMO, DMM, MF, CO2 |
| 1349 | Au | Divided H-cell | NaClO4/MeOH | 298 | 1.2 V vs. SCE | 6 | 20 min | 430 μM | 30% | — | MF, DMM, DME and CO2 |
| 1450 | Au and Pd | Three electrode spectro-electrochemical cell | LiClO4/MeOH | 298 | 0 to 1 V vs. Ag/AgCl | — | — | — | Not reported | Not reported | MF |
| Ag and Pt | — | — | — | Not active | — | — | |||||
| 1553 | Pd | Divided H-cell | NaClO4/MeOH | 298 | 1.2 V vs. SCE | 0.4 | 30 min | 30 μM | 15% | — | DMO, DMM, MF, CO2 |
| 1655 | Au | Three electrode spectro-electrochemical cell | LiClO4/PhOH, MeOH | 298 | 0.5 to 1 vs. Ag/AgCl | — | — | — | Not reported | Not reported | DPC, Methyl phenyl carbonate (MPC) |
Otsuka et al. tested the activity of PdCl2 and CuCl2 anodes for the electrochemical carbonylation of CH3OH to DMC in a gas phase system (Table 2, entries 3 and 4).40,41 A higher applied voltage was required for CuCl2 in comparison with PdCl2. This was explained by the involvement of Cu(II) in the carbonylation of CH3OH. Subsequent studies of the electrosynthesis of DMC over a PdCl2/vapor-grown carbon fibre (VGCF) anode and a CuCl2/VGCF anode were done by Yamanaka et al.42 in a three-phase-boundary system (gas phase: CO, liquid phase CH3OH and solid phase: anode material). The results suggested that reduced Pd0 species are responsible for catalysing CH3OH carbonylation from CO, whereas oxidised Pd species catalyse direct CH3OH electrochemical carbonylation to the DMO by-product. The CuCl2/VGCF anode displayed no activity when tested in the same system. The authors could not further explain this difference between Pd and Cu. The three-phase-boundary system method showed promising results with the PdCl2/VGCF anode with a selectivity of 82% and 67% current efficiency (Table 2, entry 5), obtained in CH3OH by applying a potential of +1.8 V vs. Ag/AgCl and feeding CO at 101 kPa.
Metal–ligand complexes were also studied for this reaction. CuCl-2,2′-bipyridine (bipy) complex was found to be the most efficient for the electrochemical carbonylation of CH3OH to DMC at room temperature and atmospheric pressure (Table 2, entries 6 and 7).43,44 The drawbacks of this system are that the CuCl(bipy) complex has a low solubility in CH3OH and is unstable in air. Other Cu-based materials such as CuCl2(bipy)45 and Cu-based metal–organic frameworks (MOF)46 (e.g., Cu-2,2-bipyridine-trimesic acid) have also been studied in a two-compartment electrolytic cell (Table 2, entries 8 and 9). Based on the results, the Cu2+/Cu+ couple is thought to be solely responsible for catalysing the carbonylation reaction of CH3OH and 2,2′-bipyridine is only serving as a non-participating ligand. According to the proposed reaction mechanism ((R8)–(R11)), DMC is formed in solution, and the OH− groups from the electrolyte serve as co-catalyst by dissociating CH3OH.
Anode:
| 2Cu+ → 2Cu2+ + 2e− | (R8) |
Cathode:
| 2CH3OH + 2e− → 2CH3O− + H2 | (R9) |
Anolyte:
| 2CH3OH + 2Cu2+ + CO → CH3OCO2CH3 + 2Cu+ + 2H+ | (R10) |
| 2CH3O− + 2H+ → 2CH3OH | (R11) |
A corollary of the work mentioned above is that DMC selectivity depends strongly on both the reaction conditions and the presence of additives. Funakawa et al.47,48 studied CH3OH carbonylation on a Au anode, in the form of Au/activated carbon (AC) with vapor-grown carbon fibre (VGCF) and reported a strong dependence of selectivity on the applied potential (Table 2, entries 11 and 12). The reaction products were mainly DMO and DMC with DMM and MF as by-products. According to the authors, the DMO and DMC selectivity is controlled by the oxidation state of Au and hence the applied potential. However, the formation of DMC is through indirect electrochemical carbonylation of CH3OH, mediated by the Au3+/Au0 (or Au+) redox reaction on AC at high potential (>+1.3 V vs. Ag/AgCl). On the other hand, Au0 was found to be the active species for the formation of DMO by direct electrochemical carbonylation at low potential (<+1.2 V vs. Ag/AgCl). Both suggested reaction pathways proceed via methoxycarbonyl intermediates. These observations agree with results of a density functional theory (DFT) study that showed that DMO is formed at lower potentials due to the presence of MF at the catalyst surface, whereas with increasing potential methoxy starts to adsorb favouring the formation of DMC.49
Figueiredo et al. reported for different electrode materials, including Au, Pd, Pt, and Ag, that DMC can be formed without any additives or oxidised metals (i.e., formed at potentials below +0.4 V vs. Ag/AgCl) (Table 2, entry 14).50In situ FTIR was applied to detect intermediate products formed at different electrode potentials. The results revealed that DMC was only formed on electrodes that are capable of oxidising CH3OH to (adsorbed) methoxy groups. Another requirement was that CO does not adsorb too strongly, because it would inhibit any further reaction. Ag and Pt were found to be inactive towards the formation of DMC. Pt suffers from CO poisoning, while the reaction on Ag is inhibited by CH3OH adsorption.
On the other hand, Au and Pd, which bind CO weaker than the other metals, allow CO to act as a carbonylation agent to CH3OH (Fig. 4). The importance of CH3OH decomposition on the surface and the fact that DMC is formed through the methoxy intermediate was supported by FTIR spectroscopy and the formation of MF. The observation of MF as a by-product highlights the involvement of methoxy groups at the electrode surface in the overall reaction.51
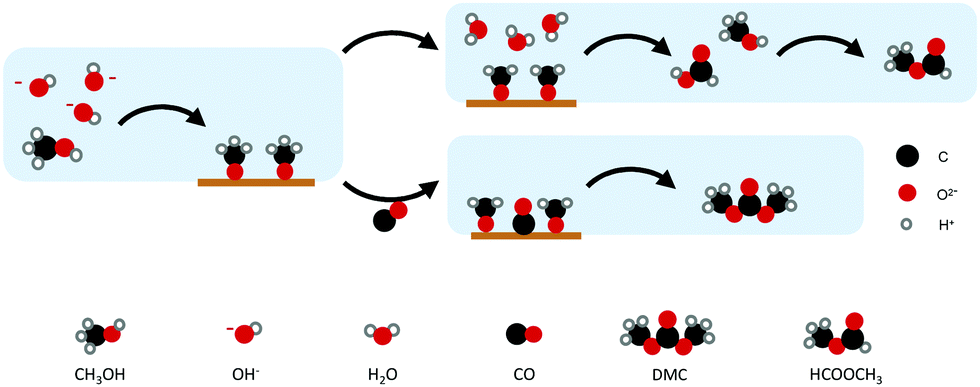 | ||
| Fig. 4 Proposed mechanism for DMC production on Au and Pd. (Scheme adapted from ref. 50.) | ||
Due to the technical limitations of in situ spectro-electrochemical FTIR, quantitative analysis cannot be easily performed. Thus, techniques such as gas or liquid chromatography are necessary52 to analyse the product distribution in more detail. However, the application of these analytic techniques is limited by experimental aspects like clogging of separation columns by salts used as electrolytes to enhance conductivity.
Davies et al.53 combined FTIR spectroscopy with headspace (HS) gas chromatography (GC) and mass spectrometry (MS) to achieve a quantitative analysis of the CH3OH carbonylation with a Pd electrocatalyst (Table 2, entry 15). This HS-GC-MS approach was applied in order to avoid salts entering the analysis system, as only gas-phase products enter the GC system.54 The electrolyte solution was collected and analysed offline within 48 hours. The analysis time plays a crucial role in the analysis since it has been previously shown that in some systems DMC can be formed in the solution via an indirect electrochemical, homogeneous reaction.38,45,46,49 Therefore, a shorter analysis time can improve the analysis of the results. The drawback of this analytical technique is that it can only detect products that are volatile below the boiling temperature of the sample matrix.
DFT studies on metallic surfaces38,49 showed that Cu, different from Au and Pd, is an interesting candidate for the electrosynthesis of DMC, as it is predicted to require lower overpotentials. A good catalytic material would bind the methoxy group strongly enough in order to lower the overpotential needed for CH3OH oxidation and, at the same time, allow for an optimal coverage of CO. However, the binding of CO and methoxy intermediates should not be too strong, otherwise the surface will be poisoned by one of these intermediates. With the help of FTIR, ICP-MS and HS-GS-MS, the authors38,49 found that Cu does not act as a direct electrocatalyst for CH3OH carbonylation to DMC (Table 2, entries 1 and 13). According to spectro-electrochemical measurements, adsorbed methoxy, which is an intermediate for the direct electrochemical synthesis of DMC, was not observed.50 On the other hand, a Cu carbonyl (Cu–CO) species was observed in the same potential range, in which DMC formation was seen. Besides, analysis of the electrolyte solution after the experiments did not show any DMC. However, after 70 days DMC was observed in the solution and its concentration increased over time. (Fig. 5) The authors proposed that the Cu(I) species act as the initiator of a post-electrolysis homogeneous reaction towards DMC, according to the mechanism ((R12)–(R14)), shown below.
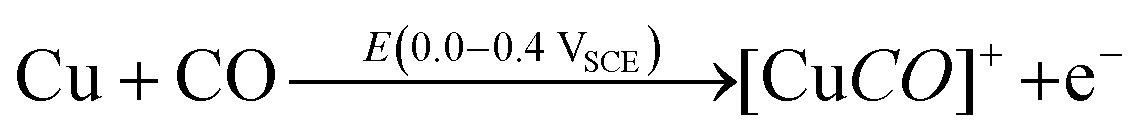 | (R12) |
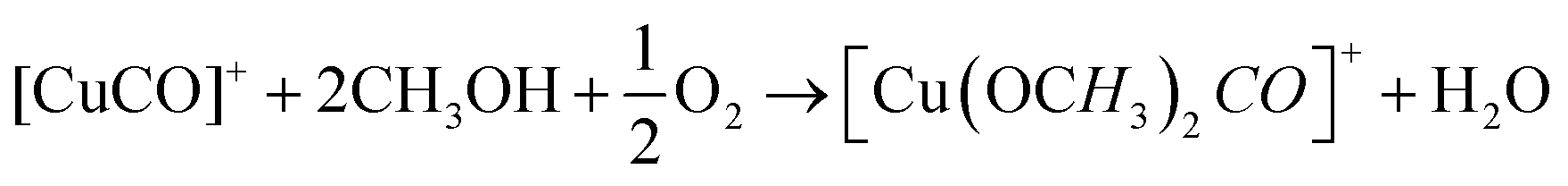 | (R13) |
| [Cu(OCH3)2CO]+ → Cu+ + CH3OCO2CH3 | (R14) |
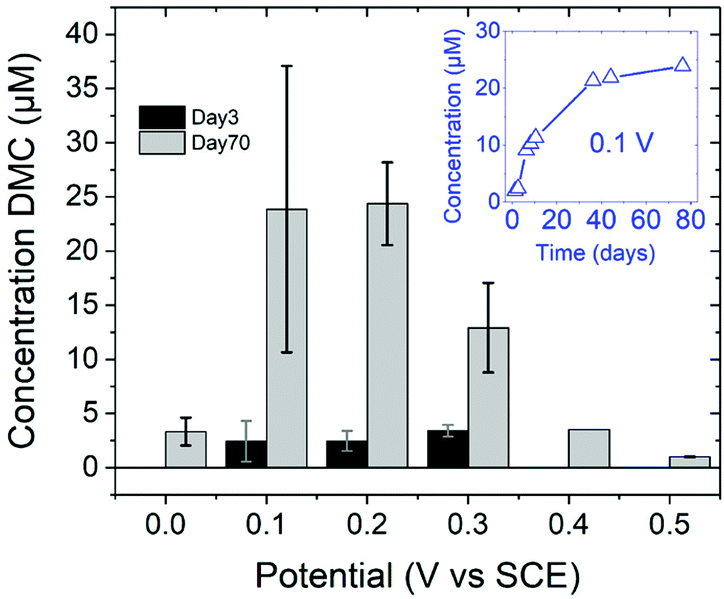 | ||
| Fig. 5 DMC concentration at 3 and 70 days after electrolysis. (inset) DMC concentration change over time since the day of electrolysis, shown for 0.1 V vs. SCE.38 Reprinted with permission from American Chemical Society. Copyright©2019, American Chemical Society. | ||
Based on the evidence that this reaction does not require an oxidised surface to obtain DMC,50 the electrosynthesis of diphenyl carbonate (DPC) was investigated on Au under similar conditions (Table 2, entry 16).55 DPC is also an important organic carbonate, because it has versatile chemical properties and can be used in synthetic chemistry as a phenylation and carbonylation agent.56 Most importantly it is used for the solvent-free production of polycarbonates. Similar to DMC, DPC manufacturing procedures involve several steps and a large number of reactants.8,57
Employing in situ FTIR, DPC was identified at low overpotential (+0.5 V vs. Ag/AgCl) on Au electrodes via an indirect electrochemical path. This pathway involves DMC conversion to DPC through an “electro-transesterification” step.55 The results suggest that DPC formation occurs similarly as DMC formation in which phenoxy groups take the role of the methoxy groups instead of a direct mechanism via phenol (PhOH) carbonylation. DMC is regarded as an electrophile and has three active centres for reaction with a nucleophile, i.e., the carbonyl group and the two methyl groups. According to the HSAB (hard–soft acid–base) principle, hard nucleophiles prefer to react with hard electrophiles and soft nucleophiles with soft electrophiles. The carbonyl group is considered to be the harder electrophile due to the partial positive charge on the carbon atom and its sp2 hybridisation. On the other hand, the methyl groups are softer electrophiles due to their sp3 hybridised orbital and saturated carbon atom.27 Although alkoxides are considered hard nucleophiles, PhOH has a decreased nucleophilic nature due to the conjugation of the oxygen lone pair electrons with the aromatic ring when deprotonated, thereby rendering a softer base/nucleophile and facilitating the reaction with the methyl groups.
The first step in the proposed mechanism is the formation of DMC via carbonylation of the methoxy groups from CH3OH and CO, which are then replaced by the phenoxy groups. The application of an electrochemical potential was found to be essential for the nucleophilic attack to occur. FTIR spectra showed that the addition of PhOH does not change the reaction pathway of CH3OH oxidation to CO2, and no evidence of PhOH decomposition or oxidation products were observed. In Fig. 6A, the spectra under Ar saturation are characterised by bands at 2341 cm−1 (CO2), 2047 cm−1 (CH3OH), and 1737 cm−1 (MF) related with the adsorption and oxidation of CH3OH. In spectra obtained after saturation with CO (Fig. 6B), two new positive bands are observed at potentials higher than +0.4 V vs. Ag/AgCl. These bands at 1757 cm−1 and 1780 cm−1 are attributed to DMC and DPC, respectively. These findings are relevant because they show that the direct DPC production by carbonylation of PhOH can be achieved with non Pd-based catalysts.
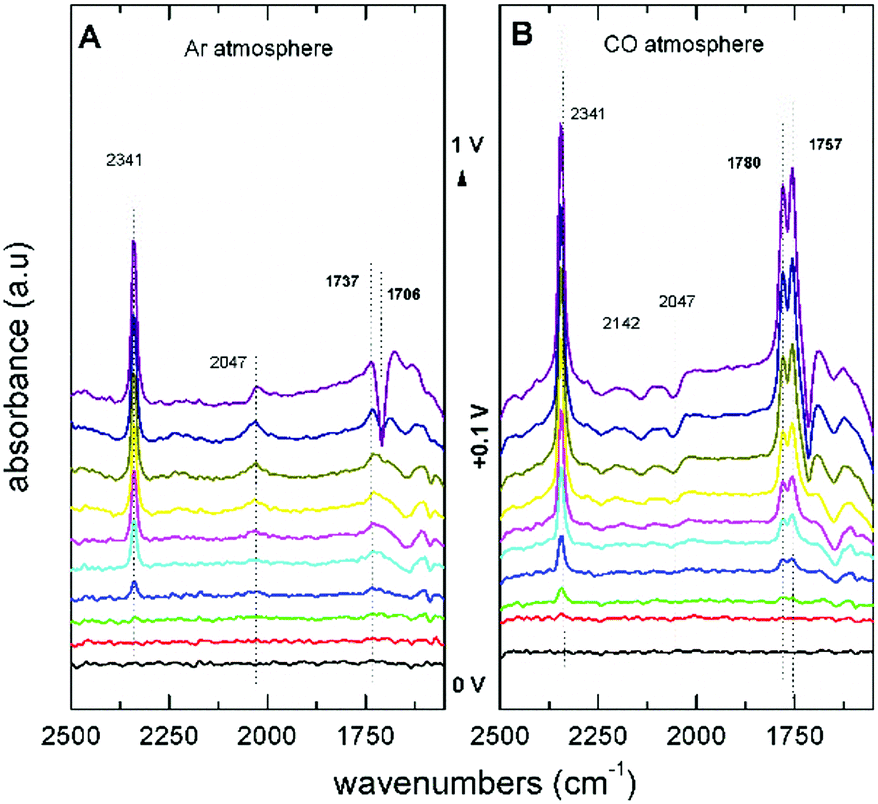 | ||
| Fig. 6 FTIR spectra of an Au electrode in 0.1 M LiClO4 in CH3OH with 0.1 M PhOH saturated with (A) Ar and (B) CO.55 Reprinted with permission from American Chemical Society. Copyright©2019, American Chemical Society. | ||
This research provides evidence that the transesterification of DMC with PhOH can occur under electrochemical conditions, avoiding thermocatalytic processes, and using the same catalyst as used to obtain DMC by electrochemical methods. Yet, more in-depth research into the nature of the “electro-transesterification” step is required.
Future directions and outlook
A summary of the suggested reactions pathways and intermediates towards DMC and DPC (and the other major products) from CH3OH and CO/CO2 is shown in Fig. 7.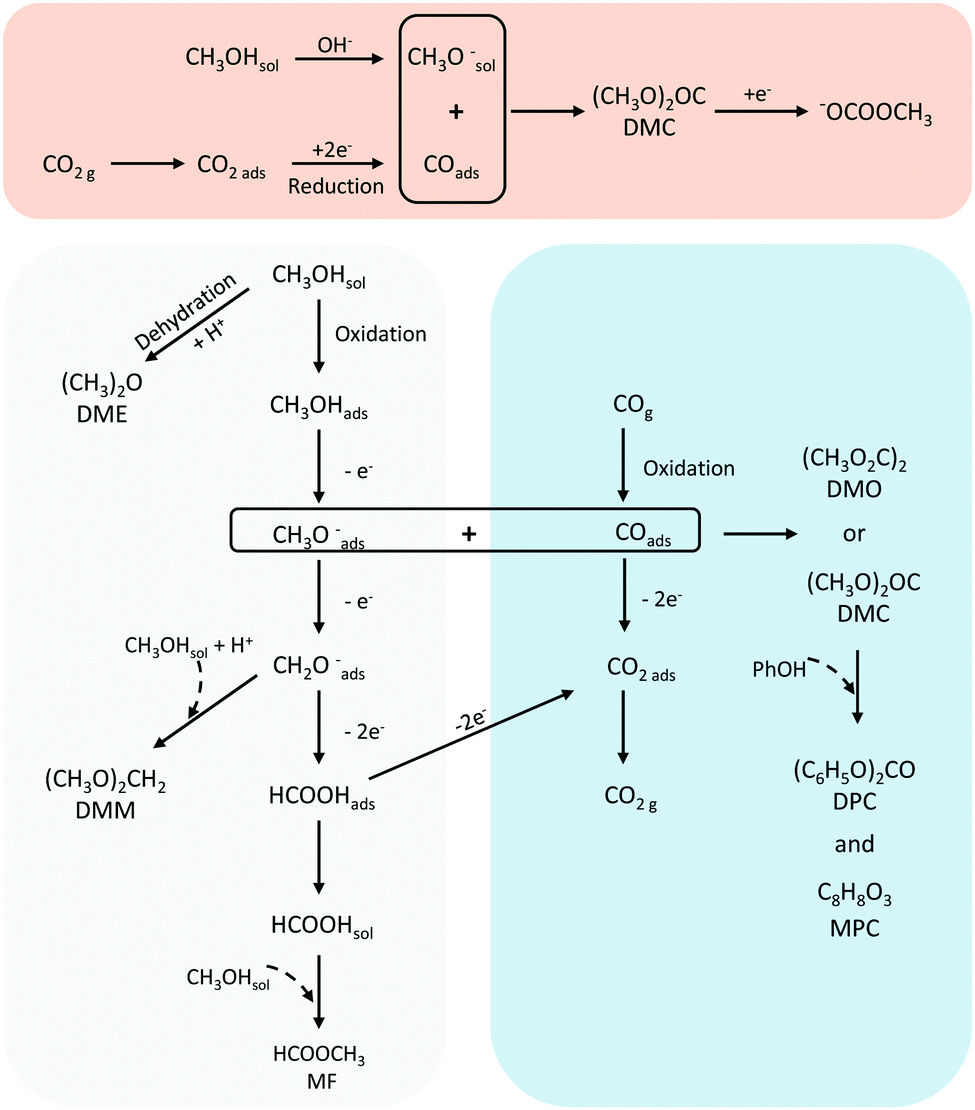 | ||
| Fig. 7 Suggested reaction pathways and intermediates from CO2 or CO and CH3OH. | ||
The works cited demonstrate the substantial efforts made by the scientific community to achieve efficient electrosynthesis methods for organic carbonates, in particular those involving the electrochemical conversion of CO and CO2. While the understanding of the processes associated with CO2 reduction reaction (CO2RR) has come a long way,58–62 we need to understand this reaction much better as such and certainly in the context of the formation of organic carbonates. A major drawback remains the low yields of the desired organic carbonates, which requires high energy costs in downstream processing. Garcia-Herrero et al.63 showed that the DMC yield in electrochemical synthesis from CH3OH and CO2 must be at least 20% in order to compete with the commercial process. For these methods to become viable in the long term and to produce the targeted products on a global scale, different aspects such as thermodynamics, kinetics and infrastructure (reactors and process units), must be understood and optimised. In this review, we focused primarily on the electrocatalysts-solvent-conditions conducive to organic carbonate formation, which remains a key aspect to be improved before a process can be realized. However, also other aspects need to be addressed, such as stable membranes and electrodes, optimised electrolyser and environmental conditions (local pH, electrolytes). In the following, we briefly touch upon such aspects.
In terms of catalyst development, there are more than a handful of examples that can be obtained from other research areas as a source of inspiration. Catalyst design approaches that include alloying, dopants, tuning oxidation state, ligand and facet control can be used. For example, the addition of ligands has been implemented in previously mentioned works with the use of CuCl-2,2′-bipyridine (bipy) complexes,43–46 producing DMC with high selectivity showing that this strategy can be used for improved results. Catalyst morphology and structure are additional factors that can be adjusted to improve catalytic activity and selectivity towards the desired product.61 So far, to the best of our knowledge, no research has been done on the impact of the particle size, shape, exposed facets and defects of the nanomaterials (e.g. atom vacancies) or catalyst dopants for the electrochemical production of carbonates. One potential method is tuning the catalyst's oxidation state as it affects its local electronic structure. Oxide-derived materials, for example, can be used to tune the oxidation state of the catalyst. Enhanced performance of such electrocatalysts is attributed to the formation of active grain boundaries, edges, and steps that can have significantly different adsorption energies and interactions with the reactants and intermediates.64 The use of bimetallic electrocatalysts is another viable strategy. Such catalysts have been widely investigated in the CO2 reduction towards C1 and C2 products.62,65,66 The surface interaction between two different materials results in altered electronic states and chemical properties, while at the same time, the local atomic arrangement can affect the binding strength of the reaction intermediates.
The lack of in situ analytical and catalyst characterisation studies for the electrochemical production of organic carbonates from CO2 or CO limits the fundamental understanding of the reaction mechanism. This hinders improving the overall system and optimising the design of the catalyst. Furthermore, there is a lack of fundamental theoretical studies, which can help understand structure–performance relationships and design better catalysts.38,49,67 For instance, modelling of the electrode–electrolyte interface can provide insight into the reaction kinetics and mass transport phenomena, which are of particular importance for continuous cell applications and porous electrode materials. Porous materials, usually prepared by catalyst deposition on a gas diffusion layer (GDL), have the advantage of supplying enough CO and CO2 to the reaction zone, while enhancing a rapid electrolyte diffusion rate at the catalyst surface minimising mass transport limitations. Besides, GDLs act as low-resistance transportation media for protons, electrons, and reduction products from the catalyst to the electrolyte.68 Catalysts assembled with GDLs have been widely used in CO2 electroreduction to C2+ products, and even though they are linked to high performances, stability is often problematic.69 The reports of the use of GDLs for electrosynthesis of organic carbonates are, as far as we are aware, absent in the literature despite their proved advantages in reactions involving gaseous reactants. Their applicability, optimisation and stability should be explored in order to accelerate electrosynthesis as an industrial solution for the synthesis of organic carbonates.
Another aspect that should be further explored is the influence of the electrolyte. It is known that the use of ILs and organic solvents provide a higher CO2 solubility and the absence of protons improves the current density while lowering the overpotential required for the CO2 conversion.70 The utilisation of mixed electrolytes, containing both organic and aqueous solvents, allows tuning the proton concentrations and consequently controlling the selectivity.71 Additionally, mixing ILs with an organic or aqueous solution can reduce their viscosity37,72 and therefore enhance the CO2 diffusion rate, leading to better performance and cost-effectiveness. The used concentrations of ILs reported in literature vary from pure ILs to millimolar, but the optimum concentration has not been determined. Another alternative for making the overall process more cost-efficient, while maintaining the advantages of ILs, is the use of deep eutectic solvents (DES).73,74 DES have not been investigated in electrosynthesis of organic carbonates, but they can be used either as additives or as the electrolyte. These solvents are typically not consumed in the reaction, making them easier to recycle, reducing the operational costs of the process. However, their recycling might prove costly due to their viscosity and volatility.
The engineering aspects of the reactions should also be evaluated. Development of electrochemical processes requires significant capital investments, which even affects the scale-up of electrochemical reactions to a larger scale than that of the lab. Continuous flow microfluid cells are an emerging class of electrolysers that allow more effective scale up by numbering.75 Such reactors can also have a positive impact on the overall efficiency of the reaction by improving mass transfer and reducing cell resistance, resulting in higher current densities.76 The evaluation and optimisation of electrosynthesis of organic carbonates in continuous flow cells is a very promising research direction in our view.
Overall, this review sketched the basic electrochemistry of organic carbonate synthesis with an emphasis on potential electrocatalysts and electrolytes for obtaining dimethyl carbonate. Clearly, significant improvements are needed in terms of product yield to make a step towards implementing such technology at a larger scale. The main research questions to arrive at this stage were formulated in this work and can hopefully be an inspiration to others working in this field.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. P. Windisch, The European Chemical Industry, 1959, vol. 15 Search PubMed.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y. M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B. M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C. J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef.
- S. Pacala and R. Socolow, Science, 2004, 305, 968–972 CrossRef CAS.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS.
- M. A. Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2–29 CrossRef CAS.
- R. H. Heyn, Carbon Dioxide Util. Closing Carbon Cycle, 1st edn, 2015, pp. 97–113 Search PubMed.
- A. A. G. Shaikh and S. Sivaram, Organic carbonates, 1996, vol. 96 Search PubMed.
- T. Tabanelli, D. Bonincontro, S. Albonetti and F. Cavani, Studies in Surface Science and Catalysis, Elsevier, 2019, vol. 178, pp. 125–144 Search PubMed.
- M. C. Figueiredo, V. Trieu and M. T. M. Koper, ACS Sustainable Chem. Eng., 2019, 7, 10716–10723 CrossRef CAS.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- V. Trieu, S. Eiden, D. Kaubitzsch, J. R. Alvarez, M. Koper, M. Figueiredo, J. Heijl and N. Meine, Electrochemical method for producing arylalkyl carbonates or diaryl carbonates, WIPO PCT, WO 2019/007828 AI, 2019 Search PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS.
- D. U. Nielsen, X. M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244–254 CrossRef CAS.
- M. Kathiresan and D. Velayutham, Chem. Commun., 2015, 51, 17499–17516 RSC.
- H. Yang, Y. Gu, Y. Deng and F. Shi, Chem. Commun., 2002, 274–275 RSC.
- H. Wang, L. X. Wu, Y. C. Lan, J. Q. Zhao and J. X. Lu, Electrosynthesis of cyclic carbonates from CO2 and diols in ionic liquids under mild conditions, 2011, vol. 6 Search PubMed.
- M. A. Casadei, S. Cesa and L. Rossi, Eur. J. Org. Chem., 2000, 2445–2448 CrossRef CAS.
- L. X. Wu, H. Wang, Z. Y. Tu, B. Bin Ding, Y. Xiao and J. X. Lu, Synthesis of cyclic carbonates from CO2 and diols via electrogenerated N-heterocyclic carbenes, 2012, vol. 7 Search PubMed.
- Q. Feng, S. Liu, X. Wang and G. Jin, Appl. Surf. Sci., 2012, 258, 5005–5009 CrossRef CAS.
- C. Yan, B. Lu, X. Wang, J. Zhao and Q. Cai, J. Chem. Technol. Biotechnol., 2011, 86, 1413–1417 CrossRef CAS.
- F. Liu, S. Liu, Q. Feng, S. Z. Zhang and P. Bu, Electrochemical synthesis of dimethyl carbonate with carbon dioxide in 1-butyl-3-methylimidazoliumtetrafluoborate on indium electrode, 2012, vol. 7 Search PubMed.
- L. Zhang, D. Niu, K. Zhang, G. Zhang, Y. Luo and J. Lu, Green Chem., 2008, 10, 202–206 RSC.
- D. Yuan, C. Yan, B. Lu, H. Wang, C. Zhong and Q. Cai, Electrochim. Acta, 2009, 54, 2912–2915 CrossRef CAS.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS.
- F. Aricò and P. Tundo, Russ. Chem. Rev., 2010, 79, 479–489 CrossRef.
- H. Z. Tan, Z. Q. Wang, Z. N. Xu, J. Sun, Y. P. Xu, Q. S. Chen, Y. Chen and G. C. Guo, Catal. Today, 2018, 316, 2–12 CrossRef CAS.
- A. A. Olajire, J. CO2 Util., 2013, 3–4, 74–92 CrossRef CAS.
- M. Alvarez-Guerra, J. Albo, E. Alvarez-Guerra and A. Irabien, Energy Environ. Sci., 2015, 8, 2574–2599 RSC.
- E. Torralba-Calleja, J. Skinner and D. Gutiérrez-Tauste, J. Chem., 2013, 2013, 16 Search PubMed.
- X. Zhang, X. Zhang, H. Dong, Z. Zhao, S. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 RSC.
- X. Yuan, B. Lu, J. Liu, X. You, J. Zhao and Q. Cai, J. Electrochem. Soc., 2012, 159, E183–E186 CrossRef CAS.
- I. Garcia-Herrero, M. Alvarez-Guerra and A. Irabien, J. Chem. Technol. Biotechnol., 2015, 90, 1433–1438 CrossRef CAS.
- I. Garcia-Herrero, M. Alvarez-Guerra and A. Irabien, J. Chem. Technol. Biotechnol., 2016, 91, 507–513 CrossRef CAS.
- B. Lu, X. Wang, Y. Li, J. Sun, J. Zhao and Q. Cai, J. CO2 Util., 2013, 3–4, 98–101 CrossRef CAS.
- M. Aghaie, N. Rezaei and S. Zendehboudi, Renewable Sustainable Energy Rev., 2018, 96, 502–525 CrossRef CAS.
- B. J. V. Davies, M. Šarić, M. C. Figueiredo, N. C. Schjødt, S. Dahl, P. G. Moses, M. Escudero-Escribano, M. Arenz and J. Rossmeisl, ACS Catal., 2019, 9, 859–866 CrossRef CAS.
- A. Funakawa, I. Yamanaka and K. Otsuka, J. Electrochem. Soc., 2006, 153, D68 CrossRef CAS.
- K. Otsuka, Electrolytic Carbonylation of Methanol over the CuCl[sub 2] Anode in the Gas Phase, 1995, vol. 142 Search PubMed.
- K. Otsuka, T. Yagi and I. Yamanaka, Electrochim. Acta, 1994, 39, 2109–2115 CrossRef CAS.
- I. Yamanaka, A. Funakawa and K. Otsuka, J. Catal., 2004, 221, 110–118 CrossRef CAS.
- A. Galia, G. Filardo, S. Gambino, R. Mascolino, F. Rivetti and G. Silvestri, Electrochim. Acta, 1996, 41, 2893–2896 CrossRef CAS.
- G. Filardo, A. Galia, F. Rivetti, O. Scialdone and G. Silvestri, Electrochim. Acta, 1997, 42, 1961–1965 CrossRef CAS.
- Y. Yu, X. Liu, W. Zhang, Y. Zhang, L. Li, Z. Cao, Z. Guo, H. Wang, G. Jia, Y. Pan and Y. Gao, Ind. Eng. Chem. Res., 2013, 52, 6901–6907 CrossRef CAS.
- G. Jia, W. Zhang, Z. Jin, W. An, Y. Gao, X. Zhang and J. Liu, Electrochim. Acta, 2014, 144, 1–6 CrossRef CAS.
- A. Funakawa, I. Yamanaka, S. Takenaka and K. Otsuka, J. Am. Chem. Soc., 2004, 126, 5346–5347 CrossRef CAS.
- A. Funakawa, I. Yamanaka and K. Otsuka, J. Phys. Chem. B, 2005, 109, 9140–9147 CrossRef CAS.
- M. Šarić, B. J. V. Davies, N. C. Schjødt, S. Dahl, P. G. Moses, M. Escudero-Escribano, M. Arenz and J. Rossmeisl, Green Chem., 2019, 21, 6200–6209 RSC.
- M. C. Figueiredo, V. Trieu, S. Eiden and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 14693–14698 CrossRef CAS.
- T. H. M. Housmans, A. H. Wonders and M. T. M. Koper, J. Phys. Chem. B, 2006, 110, 10021–10031 CrossRef CAS.
- C. Schultz, V. Kraft, M. Pyschik, S. Weber, F. Schappacher, M. Winter and S. Nowak, J. Electrochem. Soc., 2015, 162, A629–A634 CrossRef CAS.
- B. J. V. Davies, M. Arenz, J. Rossmeisl and M. Escudero-Escribano, J. Phys. Chem. C, 2019, 123, 12762–12772 CrossRef CAS.
- E. Bertheussen, Y. Abghoui, Z. P. Jovanov, A. S. Varela, I. E. L. Stephens and I. Chorkendorff, Catal. Today, 2017, 288, 54–62 CrossRef CAS.
- M. C. Figueiredo, V. Trieu, S. Eiden, J. Heijl and M. T. M. Koper, ACS Catal., 2018, 8, 3087–3090 CrossRef CAS.
- E. R. Baral, J. H. Lee and J. G. Kim, 2018, 83, 11768–11776.
- J. Gong, X. Ma and S. Wang, Appl. Catal., A, 2007, 316, 1–21 CrossRef CAS.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- D. Anastasiadou, M. Schellekens, M. de Heer, S. Verma and E. Negro, ChemElectroChem, 2019, 6, 3928–3932 CrossRef CAS.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Strategies in catalysts and electrolyzer design for electrochemical CO2 reduction toward C2+ products, 2020, vol. 6 Search PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- I. Garcia-Herrero, R. M. Cuéllar-Franca, V. M. Enríquez-Gutiérrez, M. Alvarez-Guerra, A. Irabien and A. Azapagic, ACS Sustainable Chem. Eng., 2016, 4, 2088–2097 CrossRef CAS.
- Y. Lum and J. W. Ager, Nat. Catal., 2019, 2, 86–93 CrossRef CAS.
- Z. P. Jovanov, H. A. Hansen, A. S. Varela, P. Malacrida, A. A. Peterson, J. K. Nørskov, I. E. L. Stephens and I. Chorkendorff, J. Catal., 2016, 343, 215–231 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, 2017, 139, 47–50.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- K. Liu, W. A. Smith and T. Burdyny, ACS Energy Lett., 2019, 4, 639–643 CrossRef CAS.
- U. O. Nwabara, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, ChemSusChem, 2020, 13, 855–875 CrossRef CAS.
- P. Sebastián-Pascual, S. Mezzavilla, I. E. L. Stephens and M. Escudero-Escribano, ChemCatChem, 2019, 11, 3626–3645 CrossRef.
- M. Moura de Salles Pupo and R. Kortlever, ChemPhysChem, 2019, 20, 2926–2935 CrossRef CAS.
- M. Hasib-ur-Rahman, M. Siaj and F. Larachi, Chem. Eng. Process., 2010, 49, 313–322 CrossRef CAS.
- D. V. Vasilyev, A. V. Rudnev, P. Broekmann and P. J. Dyson, ChemSusChem, 2019, 12, 1635–1639 CrossRef CAS.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
| This journal is © The Royal Society of Chemistry 2020 |