
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting complexity to implement function in chemical systems
Jordi
Solà
 ,
Ciril
Jimeno
and
Ignacio
Alfonso
*
,
Ciril
Jimeno
and
Ignacio
Alfonso
*
Department of Biological Chemistry, Institute of Advanced Chemistry of Catalonia, IQAC-CSIC, Jordi Girona 18-26, 08034 Barcelona, Spain. E-mail: ignacio.alfonso@iqac.csic.es
First published on 17th September 2020
Abstract
Chemistry deals with complex molecular systems that can be further connected by supramolecular interactions and reaction networks. However, chemists have taken little advantage of the intrinsic complexity of chemical systems, probably due to the lack of appropriate tools to analyse and understand complexity. In the last few decades, the concept of complexity has grown appealing: it allows the design of networks and dynamic systems expressing emerging properties and functions, which would be difficult to achieve from the mere addition of the components of the ensemble. Here we describe a personal overview of the recent state-of-the-art in the field, mainly focused on complex systems providing molecular recognition and catalysis. Far from being a thorough revision of the recent literature, we intend to illustrate the topic to attract the chemical community for considering complexity as an additional parameter in their research.
Introduction
Complexity is a very appealing concept present in Nature, since it is ubiquitous to living matter,1 climate phenomena,2 astrophysics,3 and biological ecosystems,4 but also social networks,5 economical markets6 and, in general, every network where individual entities interact with each other. The realisation of complexity has crystallized in a growing interest to understand the laws and properties governing the evolution and adaptation of complex systems to different conditions, such as the action of stimuli or external perturbations. The deep understanding of complex systems will allow taking advantage of emerging properties that are absent in the components of the systems but appear upon the construction of the corresponding network.7 By definition, molecular systems are in general complex dynamic systems.8 Even a mere dissolution of a molecule in a solvent can be considered a complex network where the physicochemical properties of both components (solute and solvent) change from the isolated pure states to the corresponding solution. In this simple example, the solute–solute, solute–solvent and even solvent–solvent interactions dramatically change in the mixture and can be also different depending on the conditions at which this mixture is formed (molar fraction, temperature, pressure, presence of heterogeneous domains and phases, etc.). Moreover, if we include in the system a chemical reaction9 or a recognition process,10 an additional level of complexity starts where temporal and spatial constitutional changes can occur. Thus, in general, chemistry is a scientific discipline dealing with complex systems. In contrast, chemists have avoided to work with high levels of complexity in order to be able to understand the processes under study. However, chemists are recently designing and studying more complex systems thanks to the acquired knowledge and to the emergence of new analytical and computational technologies that now make possible to measure, analyse and examine several parameters and variables.In the last decades, the proposal of systems chemistry11 as a new paradigm to face dynamic chemical systems has emerged as a different viewpoint in chemical research. In parallel to the well-established field of systems biology,12 the chemical partner aims to set up complex chemical systems to exploit their ability to produce emerging properties that arise from the network and are absent in the individual components of the ensemble. To that, dynamic combinatorial chemistry, self-assembling, self-replication, adaptation, molecular evolution or far-from-equilibrium concepts have entered in the chemists’ jargon to explain processes and properties of complex systems.13 The beauty of complex chemical systems is especially remarkable when developing a given function that comes from the coordinated action of the different components of the network. Thus, we propose the implementation of complexity as a design vector in our chemical research, like an additional parameter to manipulate the outcome of a chemical reaction or device. Besides, complexity can be expressed at different levels (Fig. 1). The molecular complexity can be attributed to the actual properties of the molecules themselves, such as different functionalities, conformational flexibility or tautomerism. An additional level sources when molecules interact by non-covalent forces leading to supramolecular structures. In parallel, molecules can interact between them in a network of chemical reactions and processes, producing an additional level of complexity. Also consequently, we can imagine situations where any combination of two or the three levels can cooperate.
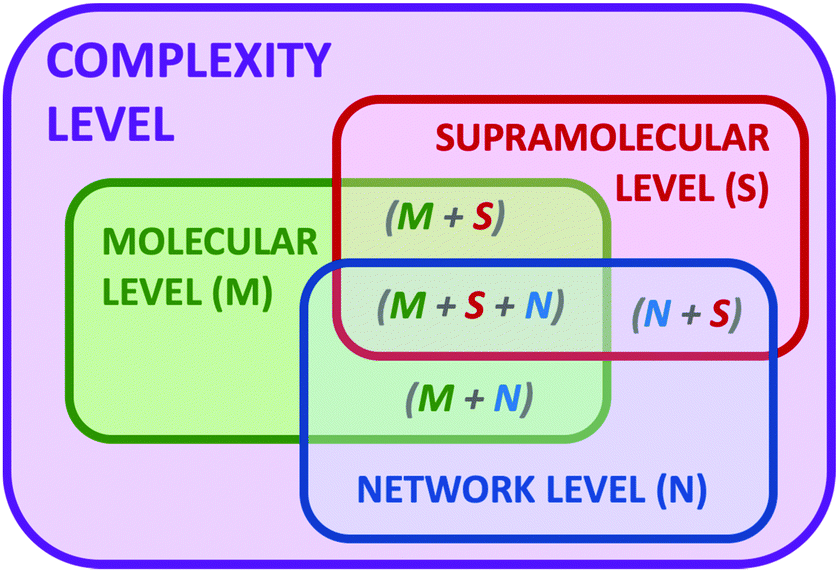 | ||
| Fig. 1 Schematic representation of the different levels within chemical systems, connecting molecular (M), supramolecular (S) and network (N) levels. Complexity can be expressed at all levels, as well as through their corresponding combinations. | ||
In this feature article, we will summarize our point of view in this area, specially by the implementation of complex chemical systems for a given function. We will illustrate the concept with selected examples that are not intended to deeply review the state of the art in the field, but to encourage the readership to think over the concept of complexity as an important additional variable to consider in future research planning.
Dynamic covalent chemistry in complex systems
The formation of chemical networks provides an excellent methodology for the generation and study of complex chemical systems.14 To this aim, Dynamic Covalent Chemistry (DCC) affords a valuable tool for the preparation of such networks.15 In the Dynamic Covalent Libraries (DCL) the members of the system are connected through the formation and exchange of reversible covalent bonds giving rise to combinatorial collections of compounds.16 Several dynamic bonds are suitable for that purpose being hydrazone and imine formation, and disulphide exchange maybe the most commonly employed.15a,e DCC was first envisioned for the preparation and study of ligand–receptor systems.17 As DCLs are intrinsically dynamic and subjected to thermodynamic control the receptor with a better affinity towards the guest should be stabilised and therefore its concentration should increase. This ideal concept, however, is not always perfect as the system will evolve towards the distribution that minimizes the energy of the whole system itself, which does not necessarily mean that the best binder is amplified the most.18 On the other hand, amplification of suitable receptors allows the emergence of compounds that were almost non-detectable in the absence of the guest.19 The size of the library is also an important parameter: Otto's research group addressed this issue by simulating a set of libraries leading to the conclusion that larger libraries offer the possibility to obtain stronger ligands and therefore complexity seems to deliver positive outcomes.20 The concentration of the template also plays an important role: if too much template is present it can hamper the emergence of the best receptors by the formation of a large number of lower affinity, worse, ligands.21In practical terms, DCC has been useful in the generation of efficient ligands, particularly in aqueous systems, which are especially relevant when pursuing biological applications because the binding and recognition of specific molecules by non-covalent, supramolecular interactions is competing with solvation interactions with the medium.22 Thus, several ligands with different structural features, from cyclic to linear scaffolds, have been described to target a large variety of compounds, from simple small molecules to bigger biomacromolecules.23 Maybe the most attractive characteristic of the DCC methodology is that a detailed structural knowledge of the host–guest interaction is not needed, even being possible to work with mixtures. This feature is especially useful when trying to recognise macromolecules with large conformational freedom or with heterogeneous composition. We took advantage of the intrinsic nature of DCC for the recognition of glycosaminoglycans (GAGs) such as heparin (1 in Fig. 2A) using a library of polyamines and aromatic aldehydes.24 We targeted two main binding motifs of this polysaccharide: the anionic sulphate and carboxylate functions on one side and the axial positions on the carbohydrates that are able to establish attractive CH–π interactions with aromatic rings. To do so, we used reductive amination reaction between spermine (2) as cationic platform at physiological pH and a mixture of five different aldehydes leading to a combinatorial output (Fig. 2A). When repeating the reaction in the presence of heparin we succeeded in identifying some amplified compounds. After preparative-scale synthesis of the potential best ligand 3AL we proceeded to study its interaction with heparin by means of NMR and fluorescence spectroscopies, as well as molecular modelling identifying a series of electrostatic, hydrogen bonding and C–H contacts as expected. Using Isothermal Titration Calorimetry (ITC, Fig. 2B) we were able to estimate a strong binding constant in aqueous medium between 3AL and heparin and, more importantly, this ligand was able to reverse the inhibitory effect of heparin in an enzymatic test related to human blood coagulation (Fig. 2C).
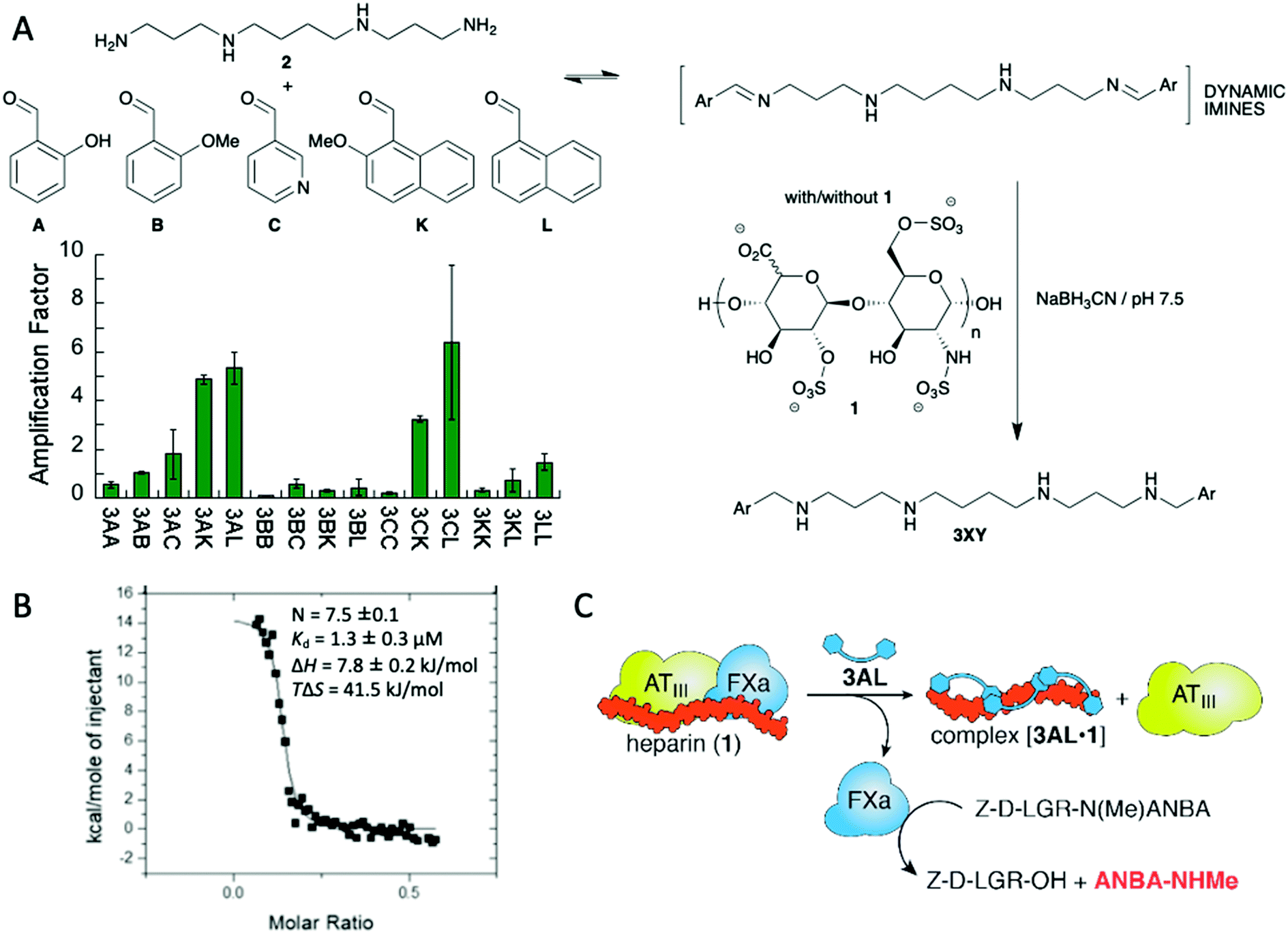 | ||
| Fig. 2 (A) Discovery of a ligand for heparin (1) using a library of dynamic imines. (B) ITC trace for the binding of 3AL to 1. (C) Reversal of the heparin inhibitory effect in an enzymatic assay related to blood coagulation. | ||
Encouraged by this success, we reasoned that an attractive and useful application to extend our research would be addressing the GAGs present in the cells. We anticipated that a suitable DCL against whole living cells should reflect the extracellular matrix composition. As we expected, using live A549 cancer cells as template and a dynamic library similar to that employed for the interaction with heparin, we could observe the amplification of a particular polyamine.25 Importantly, another cell line gave a different amplification pattern showing the dependence in the library behaviour towards the composition of the specific extracellular matrix. In this case, using a combination of on-cell NMR and SPR techniques we could prove an interaction between the amplified component and a GAG within the extracellular matrix. Our success highlights the power of dynamic combinatorial methodologies to address particularly challenging supramolecular processes specially when targeting not well-defined structures.
The adaptability of the chemical networks originated by DCC allows the sorting of their constituents in response to external stimuli or the presence of chemical effectors. Light,26 pH changes,27 temperature,27a pressure28 or salt concentration29 proved to be efficient in shifting the species' distribution. Studying these changes, it is possible to obtain individual fingerprints for each stimuli. Thus, the groups of Anslyn,30 Matile,31 Severin,32 Zonta33 and Lehn34 have used pattern-based techniques for the sensing of different analytes using the response of the system without the need to isolate, synthesise or study individual compounds. The analyte is monitored by the changes in the UV, fluorescence or even mass spectra and the corresponding chemometric analysis of the changes in the whole library leads to the identification of the analyte added. We started our research in this area analysing dynamic libraries of macrocyclic pseudopeptides and their response to several stimuli.29,35 We found out that these libraries responded to variations in the ionic strength leading to changes in the library composition that resemble the evolutionary patterns found in halophilic proteins.29a Moreover, the knowledge acquired allowed us to design de novo chemical systems with intended behaviour containing competing or cooperating species.29b In addition, chemometrics such as principal component analysis (PCA) or Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) allowed us to quantify the effect of multiple stimuli in a dynamic library (Fig. 3). These tools proved to be especially useful when two inter-connected stimuli were combined in the same dynamic system.36
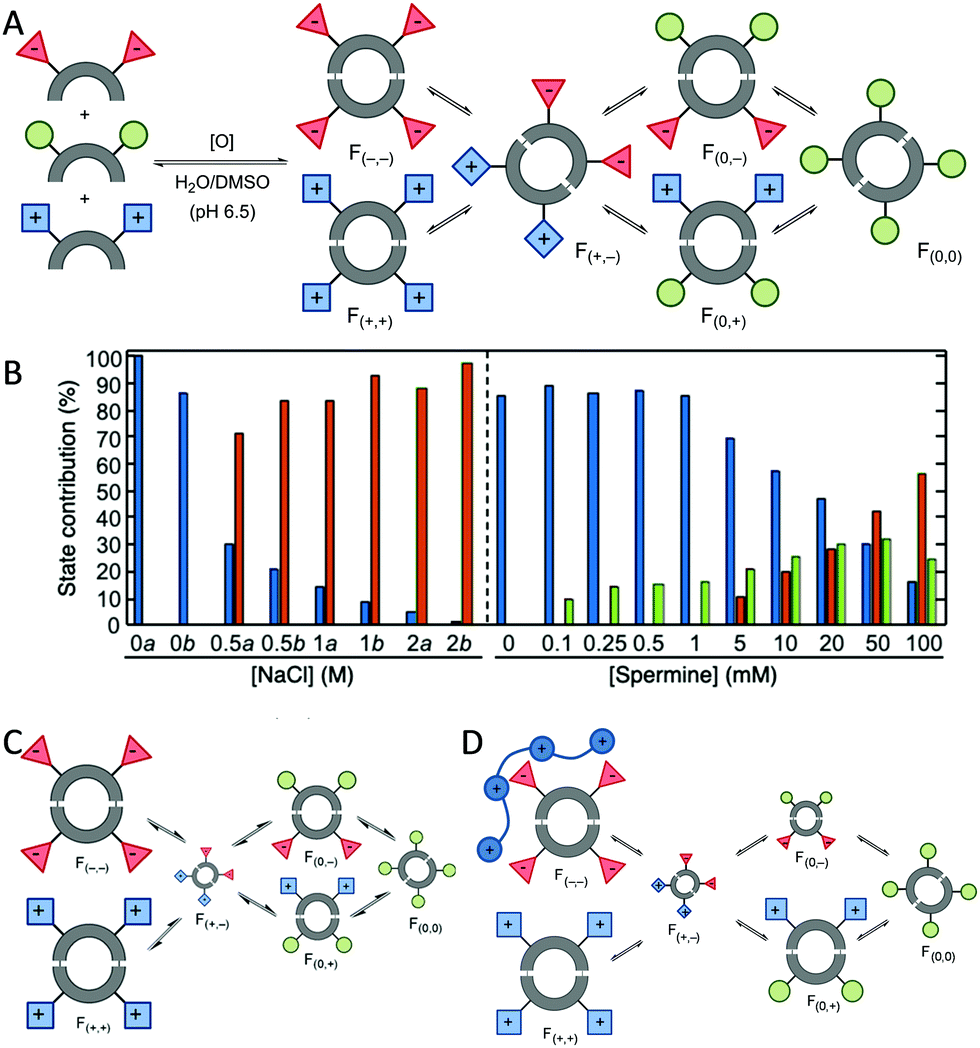 | ||
| Fig. 3 Use of chemometrics to unravel multi-stimuli response in DCL of macrocyclic pseudopeptides: (A) schematic representation of the DCL generated by disulphide formation in aqueous media giving yield to six different families classified by their charge at neutral pH. (B) Bar plots of the scores of the three states resolved by MCR-ALS analysis of the HPLC in the presence of salt or spermine data sets. (C) Schematic representation of the changes in concentration of the families of macrocycles due to the salt effect (larger/smaller size symbols represent an increase/decrease of the concentration) (D) cartoon representation of the effect of the spermine polycation binding on the DCL. | ||
As scientists have been able to deal with more complex systems thanks to the advanced analysis techniques, the complexity of the chemical networks has gained importance. Thus, despite that dynamic libraries usually contain just a dynamic bond, reaction networks containing more than one dynamic reaction have been described,37 with up to four orthogonal reactions operating at the same time and studied thanks to computational deconvolutions.38 Very interestingly, Furlan and co-corkers showed that combining sequential reversible reactions gives the chemical pathway a direction. Thus, the thermodynamic systems that result from combining these reactions keep memory of the previous equilibration processes leading to different layers of information.39
The group of Otto undertook a further step in the generation of complex systems by combining two different covalent chemistries operating simultaneously to generate diverse DCLs (Fig. 4).40 Thiol-disulphide and thio-Michael reactions were combined using the same building blocks (BBs) and, consequently, as the products of one reaction grow in concentration the products of the other have to decrease making the two processes antiparallel. External control over their ratios can be exerted by the use of reducing or oxidizing agents.
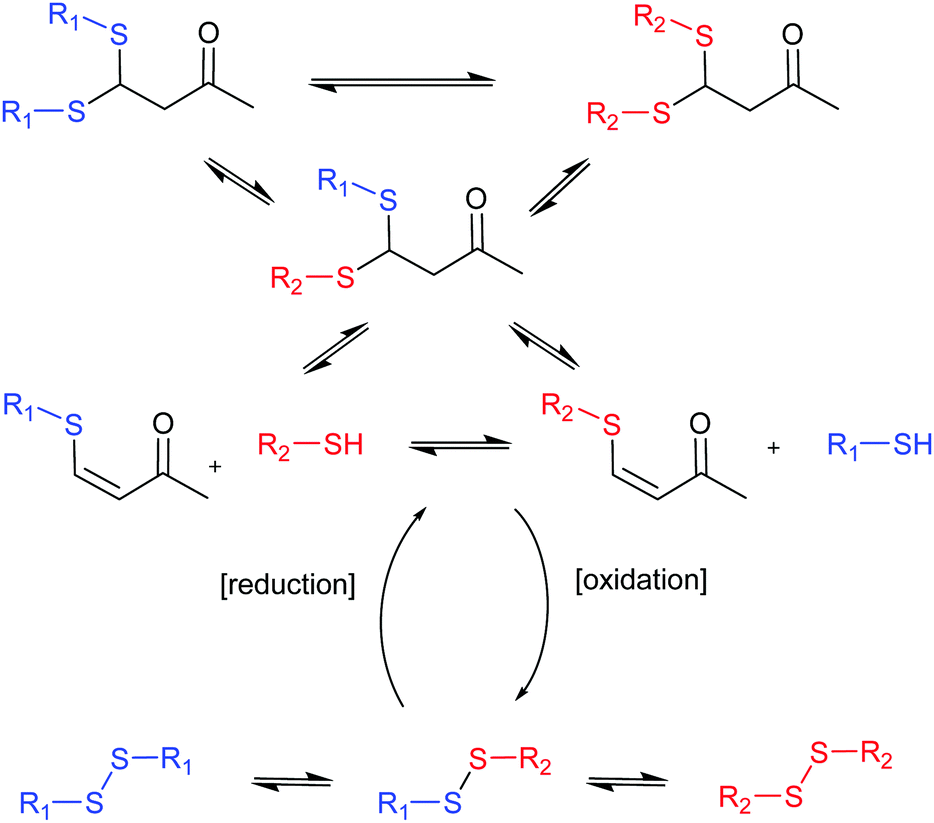 | ||
| Fig. 4 Antiparallel chemistries described by Otto.40 The final state of the system between thio-Michael chemistry (top) or disulphide chemistry (bottom) can be switched by addition of reducing or oxidising agents. | ||
This use of different chemistries was elegantly used by Withesides to build networks of reactions with autocatalytic, bistable and oscillatory behaviour. First, they developed a network based on amplification using thiols and thioesters. Addition of acrylamide delays the autocatalytic growth of the amplified species by the covalent capture of the thiols. Using a continuously stirred tank reaction that allowed a flux of species into and out of the system overtime they could design conditions in which the addition of acrylamide (in excess in the media) produced oscillations in the concentration of the amplified thiols.41 In a more recent study, the same authors showed that the oscillatory behaviour of thioester (AlaSEt) can be ‘trained’ to other non-oscillatory related compounds (GlySEt and AbuSEt) even increasing the period of oscillating behaviour and thus increasing its robustness. Finally, they showed that individual components that do not show oscillatory behaviour when isolated are able to oscillate when mixed together.42 Remarkably, the emergence of complex behaviour from simple molecules, such as those described in these papers, has been related to the pre-biotic origin of life.
The time-dependence of systems has also been studied by Lehn using DCL where its constituents can be switched from kinetic products (imines) to thermodynamic products (oximes or acylhydrazones) based on the reactivities and relative stabilities of the members.43
Another potential source of complexity arises from the number of functional groups involved in a given network and the combination of different geometries. In these scenarios one can envision the formation of large multimacrocycles or cage-like compounds among others. Building in the research performed by Sanders and Otto,44 we became particularly interested in this area as a manner to expand the geometrical possibilities of our dynamic libraries beyond the formation of cyclic compounds. By mixing BBs having a different number of thiol groups we were able to generate a variety of molecules with different topologies, some of them with degenerate mass. Importantly, we also observed self-selection mechanisms leading to a simplification of the libraries by the complementarity of charge and structure between BBs.45 Studying the molecular interactions that gave rise to these processes, we were able first to understand the mechanism leading to self-assembly and how the system evolved through time from complex kinetic mixtures to the thermodynamic final state. Strikingly, in some cases adding more components to the mixture resulted in the simplification in the number of the species formed (Fig. 5A). Mixing BB 4 having three thiol groups and dithiol 5 a complex library was formed. When cysteine 6 was added to the mixture, a single compound 7 containing all the BBs was formed which implies the covalent capture of cysteine in zwitterionic form.45a,b Including a chromophore (8) in the library that was released after the addition of cysteine (as the oxidized dimer 8-8) the system was able to act as a sensor for this amino acid.46
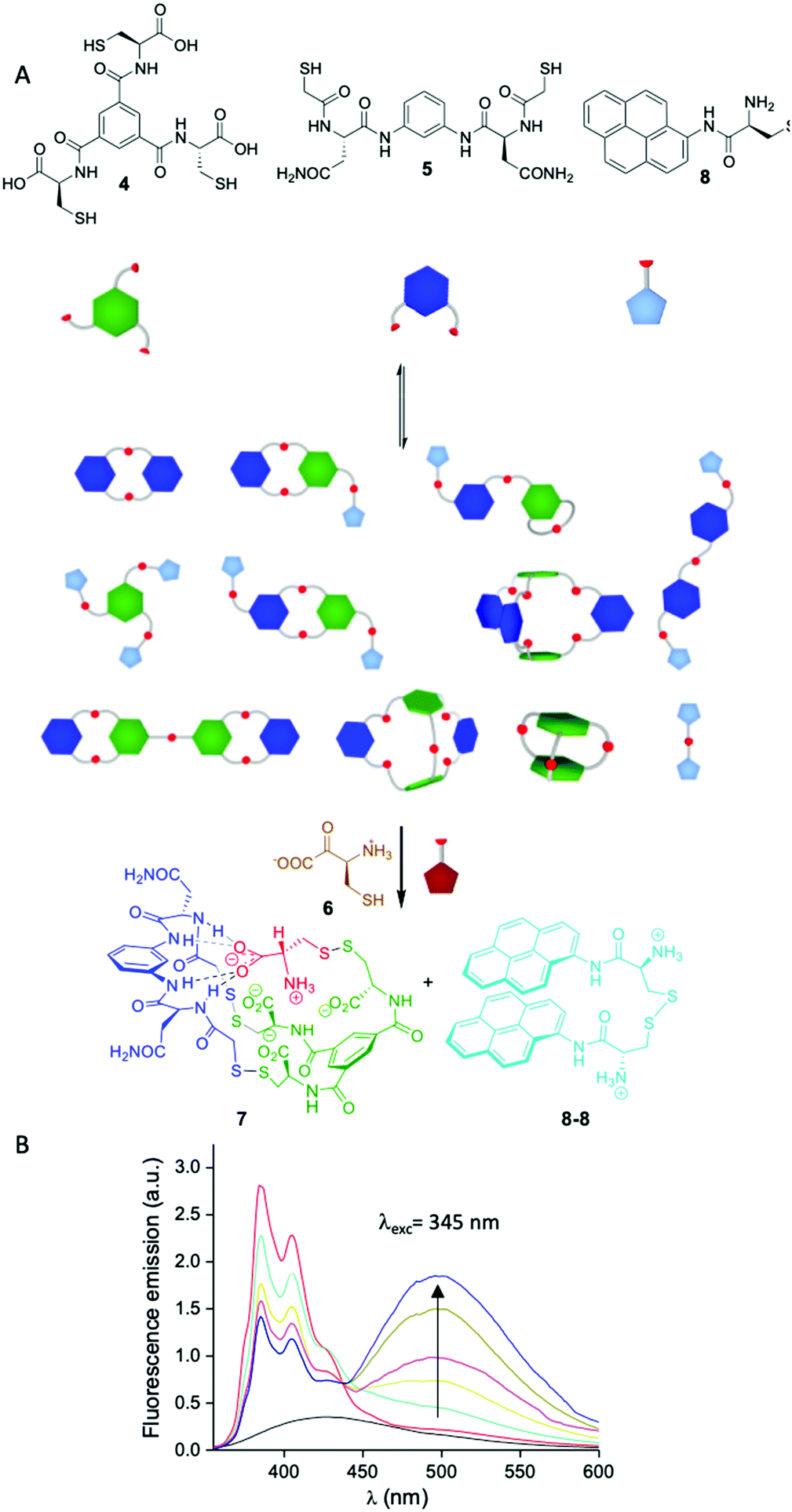 | ||
| Fig. 5 (A) Schematic representation of the sensing network for cysteine and cystine described by our group.46 (B) Fluorescence response of the dynamic network in response to increasing amounts of cysteine in human urine matrix (background urine fluorescence emission in black). | ||
The particularity of our strategy is that it is the library itself and not any of the individual compounds that form the sensor and the response is read by changes in fluorescence without the need to analyse the library composition once the system was optimised. This sensor was shown to be useful to detect cysteine in human urine being a potential tool for the detection and diagnosis of cystinuria (Fig. 5B).
Dynamic networks not only can react to external stimuli but also self-templation is an interesting phenomenon itself. Thus, unexpected complex structures such as catenanes,23c,47 knots48 or self-folding molecules have been described.49 In most cases their formation is driven by the hydrophobic effect as these compounds present a more compact structure with less areas exposed to the solvent. A particular case of study arises when one of the members of the library can aggregate with good specificity favouring its formation and making effectively a self-replicator.50 Minimal replicators are thought to be an essential requirement in the appearance of systems where life could emerge.51 Among other chemistries, dynamic covalent networks have been developed for the generation of replicating systems. Giuseppone using a dynamic equivalent of a replicator reported by Rebek52 described one of the first examples of the generation of replicators with DCC.53 Later, Philp designed a network of reactions leading to the amplification of a synthetic replicator by coupling a pre-existent nitrone-based replicator to an imine-nitrone exchange.54 In order to achieve new smart materials, Giuseppone and co-workers described the selection of a nanostructure resulting from phase separation (Fig. 6). They combined a kind of copolymers bearing a hydrophobic moiety (from 9) and hydrophilic ones (10–13) connected by a reversible imine linkage (dynablocks). Interestingly, the imime bond resulted stable in aqueous media thanks to the spontaneous formation of supramolecular structures. Moreover, the self-assembled structures made from those dynablocks were able to replicate by catalysing the formation from their BBs and selecting the most efficient replicator.55
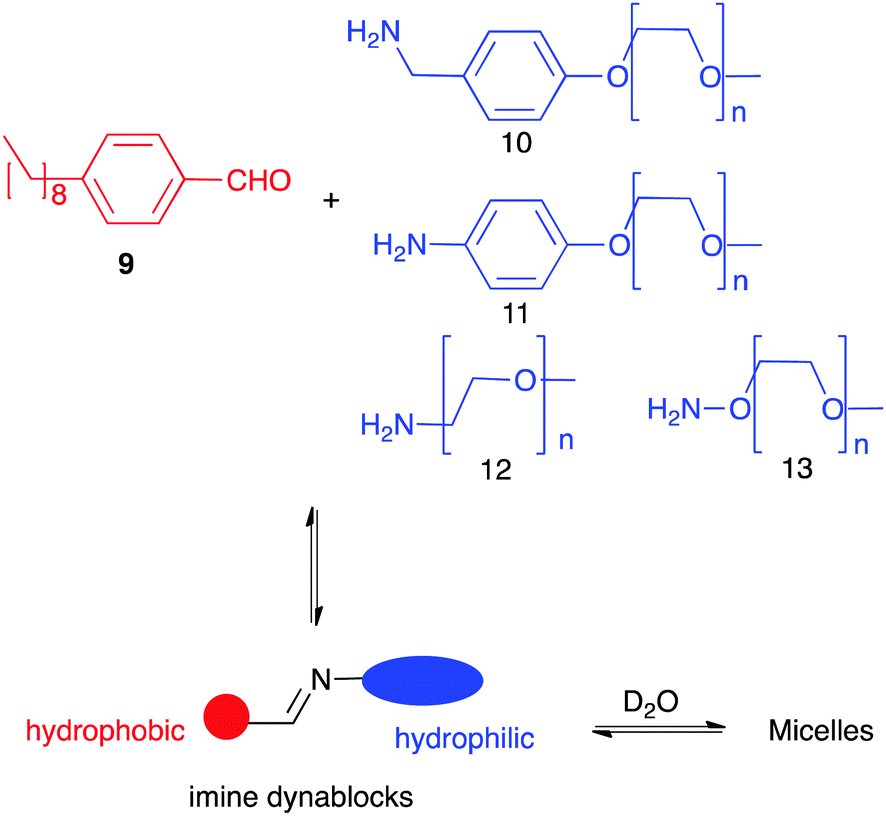 | ||
| Fig. 6 Formation of supramolecular aggregates using Imine dynablocks reported by Giuseppone.55 | ||
The use of peptide supramolecular structures such as coiled-coil motifs had been employed by Ghadiri56 for the generation of autocatalytic networks. More recently, Ashkenasy used the templating effect and folding properties of these structures to design a bistable network through the reversible thioester formation of a thiodepsipeptide. Depending on the initial concentration, the system leads to two different steady states which are switchable applying the appropriate stimuli.57 The same author reported the use of complex β-sheet peptidic networks from where the formation of a dominant functional polymer backbone can emerge.58
In the last years, the group of Otto has put a lot of effort in the preparation of several replicator systems based on libraries of dithiol pseudopeptidic BBs (14, Fig. 7). As the components are oxidised, several macrocycles arise with different size and the particularity is that some of those members are able to stack on top of themselves therefore promoting their own formation. As these stacks are prone to mechanical rupture the number of ends from where the replicator can grow increases enabling exponential growth.59
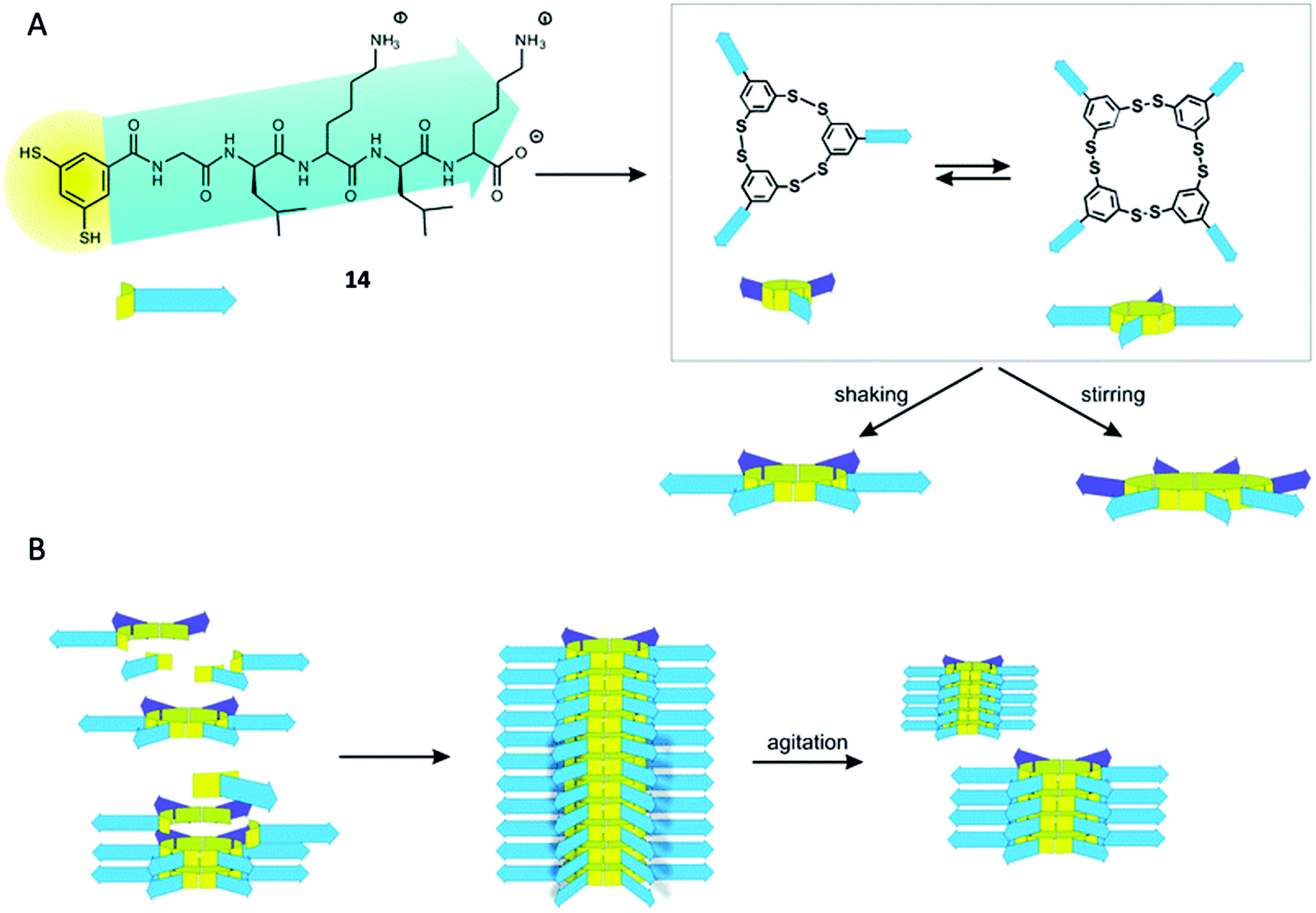 | ||
| Fig. 7 General replication process for the pseudopeptidic replicators described by Otto. (A) Oxidation of dithiol 14 gives rise to different macrocyclic compounds (mainly trimer and tetramer). After agitation the emergence of larger macrocycles takes place (hexamer and heptamer). (B) The hexamer can form stacks of compound held together by β-sheet formation of the peptidic moieties promoting its own formation. Mechanical rupture of the stacks due to agitation increases the number of ends leading to exponential growth. Reproduced from ref. 14b with permission from the Royal Society of Chemistry. | ||
However, a certain macrocycle size is needed to afford sufficiently strong interactions to drive the process.60 Thus, stacks of certain compound form fibres that can be obtained in controllable length and stabilized by covalent capture giving rise to self-synthesising materials. The dissimilar behaviour of s when they are alone or mixed with other species allowed the group to design a library that shows no replication until an effector is present.61 In this particular case, a BB capable of self-replication and a known to form a receptor in the presence of a template were combined together. In the absence of template, no self-catalysis is detected and the different components form mixed structures. Once the effector is introduced, the formation of a suitable receptor captures one of the BBs freeing the other one that is capable of self-templation, and autocatalytic behaviour is observed.
Remarkably, by studying different replicating systems, Otto's research group has been able to establish diverse relationships inside the chemical networks. On one hand, they described a system in which a diversification of the replicators that are competing for two common BBs into two different sets of replicators was observed.62 On the other hand, the appearance of some replicator molecules may require the presence of a pre-existing replicator, that is, in some cases the replicator itself does not form unless it is first templated by a previously formed oligomer.63 Moreover, the emergence of parasitic behaviours of replicators that consume those members to which they owe their own formation has also been witnessed.64 These dissimilar behaviours demonstrate the variety of possibilities that arise from studying complex systems. These replicators, however, are invariably either in thermodynamic or kinetic trapped states and no source of energy is required to maintain this state. A step forward in complexity was reported by the group of Fletcher. Building on the work of Giuseppone they described the formation of a metastable surfactant that is capable of forming micelles which increase the surfactant formation and therefore convert the process in self-catalytic.
However, the micelles are subjected to disassembly to reach the thermodynamic products. The addition of hydrogen peroxide as chemical fuel regenerates the BBs for further replicator formation.65 Following their work in phase separation autocatalysis, the same group reported the selection of components in a system of reproducing lipids forming micelles. In these systems, the replicators are metastable and selection can occur from a pool of competitors and out-of-equilibrium populations can be maintained by feeding the system with starting materials. Importantly, no selection is observed when the components are dissolved in a single phase.66
Controlled self-assembly and disassembly, coupled with functional molecular architectures is still a major challenge in the laboratory. One of the first examples of the use of dynamic libraries for the controlled formation of materials was described by Lehn using guanosine-5′-hydrazide and aldehydes to generate a library of gelators in the presence of metal cations. If a mixture of aldehydes was used, then the library evolved towards the formation of the most stable gel.67 Self-assembly of peptidic materials has been studied extensively by Ulijn group. In an early example, the use of reversible peptidic bond formation catalysed by a protease was reported to generate a DCL of dipeptides. As described above, gelation of one of the components drives the selection of a single product and allows for the discovery of stable self-assembled nanostructures. In these systems, the nature of the self-assembly can be modulated using changes in pH according the acid/base properties of the amino acid residues.68 In a more recent example, the group took a step further exploiting the molecular interactions resulting of incorporating amino acids into an organic semiconductor and thus electronic wires could be formed and degraded achieving temporary control over electroconductivity.69 As we described above, transient assembly of materials can be fuelled by a chemical reaction provided the self-assembled material can be switched between a non-associating and an associating states by a chemical reaction consuming a fuel, and reversed back to the first state by a different mechanistic pathway. The group of van Esch elegantly made use of ester formation and hydrolysis to control the gel properties of molecular gelators tuned by the kinetics of fuel consumption (Fig. 8).70
 | ||
| Fig. 8 Fuel-dependant modular pseudopeptidic gelators (15–17) reported by van Esch.70 | ||
The kinetic lability of dynamic bonds introduced in polymeric compounds gives them unique properties allowing for the formation of smart materials with self-healing properties, shape memory or degradability. The use of pH sensitive covalent bonds is especially useful when pursuing biological applications. The group of Matile reported the ring opening polymerization initiated by a substrate bearing a thiol group that allows the delivery of dyes, proteins or drugs inside the cells. These polymers were shown to depolymerise rapidly inside the cell due to the presence of endogenous glutathione, minimizing thus their toxicity.71 A different approach was reported by the group of Ulrich who prepared dynamic covalent polymers by condensation of hydrazone linkers. Thanks to the presence of cationic monomers these materials were capable of effectively complexing DNA. Moreover, the design was demonstrated to be pH sensitive, disassembling rapidly at acidic pHs.72 In an extension of this work, using amino acid BBs (18 in Fig. 9) they were able to complex DNA and siRNA and deliver the last one into live cells.73
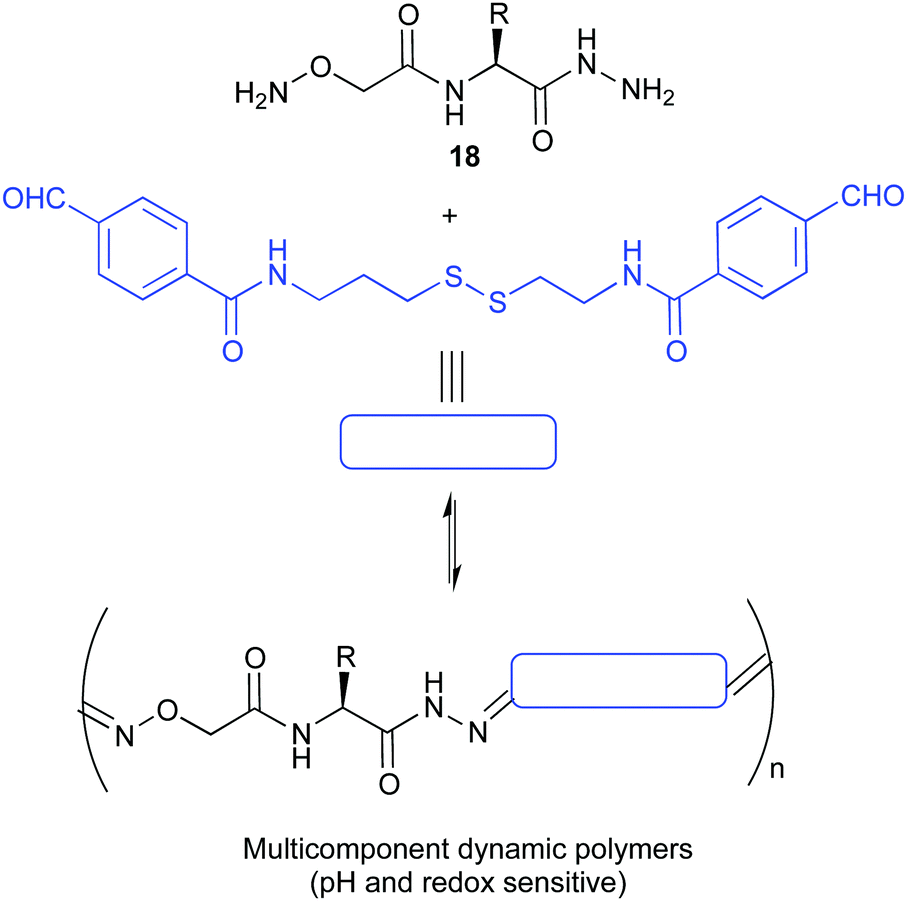 | ||
| Fig. 9 Multicomponent pseudopeptidic dynamic copolymers described by Ulrich for the delivery of siRNA.73 | ||
Overall, the use of dynamic covalent bonds represents a suitable approach to implement diversity in a chemical system leading to the potential emergence of interesting properties and functions.
Complex systems leading to efficient catalysis
Catalytic network and metabolism are synonymous in living organisms, likewise catalyst and enzyme. Indeed, a single enzymatic reaction in a cell is pointless without the context of a living organism. It is the overall combined network of catabolic and anabolic reactions that defines a living being. The whole metabolic network is a clear example of complex catalytic system that goes beyond simple reaction acceleration: substrate recognition, specificity and selectivity, enzyme inhibition or activation through allosteric regulation, the activation of cascade pathways, and self-regulation are emerging properties of complexity in catalytic networks. Artificial systems might depict some of these emerging properties as well, and there is a continuous flow of new and exciting discoveries in this field.74 Some illustrative examples from the last years that allow understanding the behaviour of complex catalytic networks are discussed below.A very interesting case of substrate activation of a catalytic assembly was reported by Lei and Ju.75 Mercury(II) was used as a co-factor to trigger the self-assembly of gold nanoparticles functionalized with hemin groups in the presence of a bridging pyrimidine-type ligand. The Hg(II) assembly showed three-times as much peroxidase activity as the mercury-free system. Ultimately, this activity was used to detect Hg(II) at a sub-attomolar level (Fig. 10).
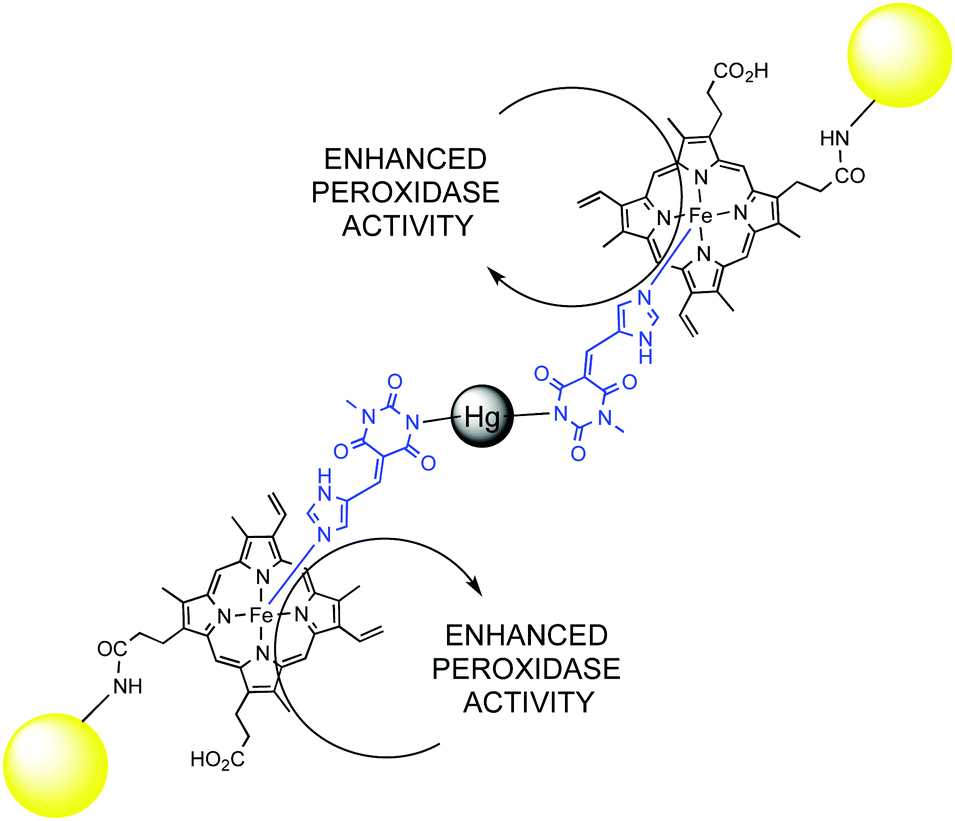 | ||
| Fig. 10 Enhanced peroxidase activity of Au-NP's functionalized with hemin groups in the presence of Hg(II). | ||
This is related to allosteric regulation as well, wherein a small molecule or metal regulates catalytic activity of an enzyme. Of course, supramolecular chemistry seems an ideal field for this approach. Remarkable examples have been developed in Mirkin's group.76 Two of them are discussed below. In a first experiment a P,S ligand containing a diphenylurea group was developed to coordinate Pt and form a tweezer complex. This is the catalytically active conformation of the complex, since the hydrogen bond donor properties of the adjacent urea groups enhance the Diels–Alder reaction. However, upon addition of chloride ions, the Pt–S bond breaks, destroying the tweezer conformation, promoting intermolecular aggregation of the urea groups, and ending catalytic activity in the DA reaction (Fig. 11A).77 In a second example, two different P,S ligands designed to form a triple decker complex with Pt contained a catalytic squaramide group (hydrogen bond donor) or a self-quenching ester groups. This complex was catalytically active in the Michael addition because the ester groups were positioned far away from the squaramide. Upon addition of chloride ions, the Pt–S bond is broken again and the structure of the triple decker complex changes allowing the quenching of the squaramide by ester groups from other complexes. These oligomers are catalytically inactive (Fig. 11B).78
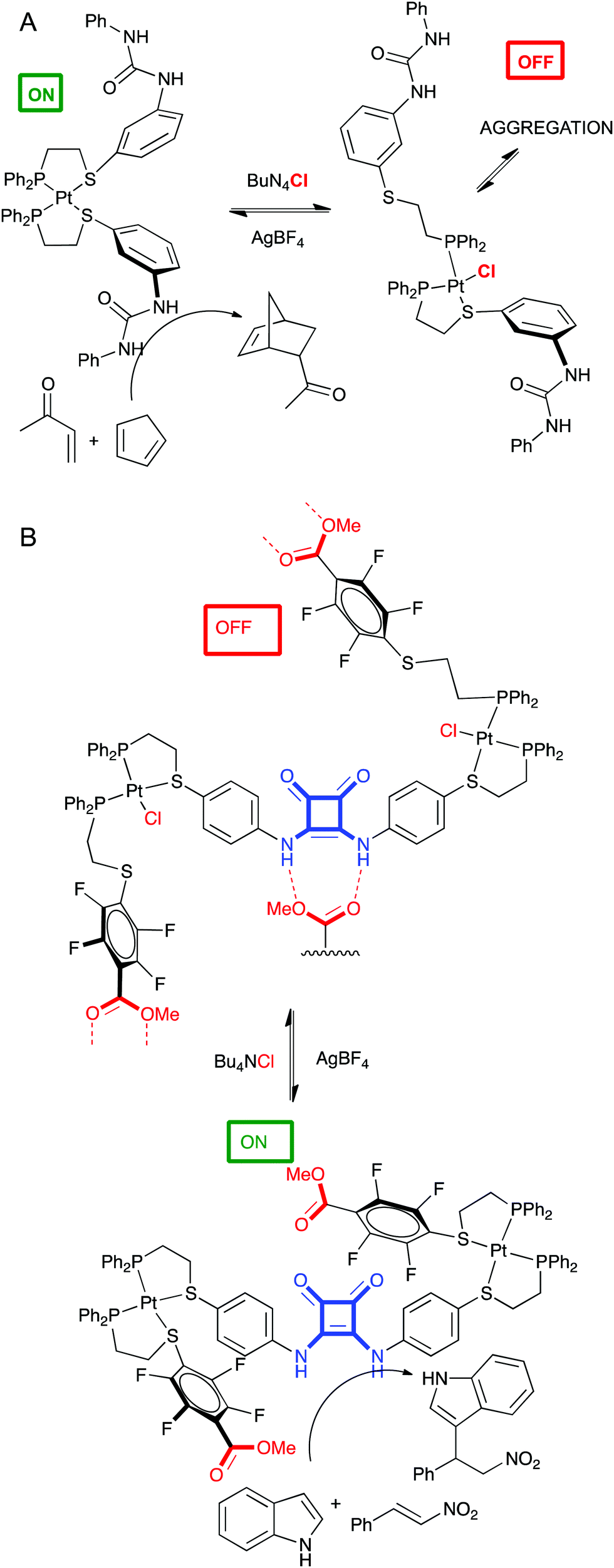 | ||
| Fig. 11 Allosteric control of catalytic activity of a tweezer complex (A) or a triple decker complex (B) by chloride. | ||
We will turn our attention now to some intrinsically dynamic catalytic systems, either in the substrates or the catalyst, and how chemists have learnt to deal with such complexity. Ramström and co-workers have developed several complex catalytic systems with unusual activities. Dynamic systemic resolutions79 of complex but reversible reaction networks were achieved through metal catalysis80 and biocatalysis.81 These are excellent examples of substrate specificity even in the presence of mixtures of compounds in equilibrium, which somehow relates to dynamic kinetic resolutions in asymmetric catalysis. For example, a mixture of α-iminonitriles in dynamic equilibrium obtained by mixing up to thirteen different aldehydes was subjected to azomethine ylide formation in the presence of a chiral Ag(I)–Taniaphos complex and base. After the addition of dimethyl fumarate as dipolarophile, two diastereomers derived from furfural (19 and 20) were isolated as the exclusive products (Fig. 12).80
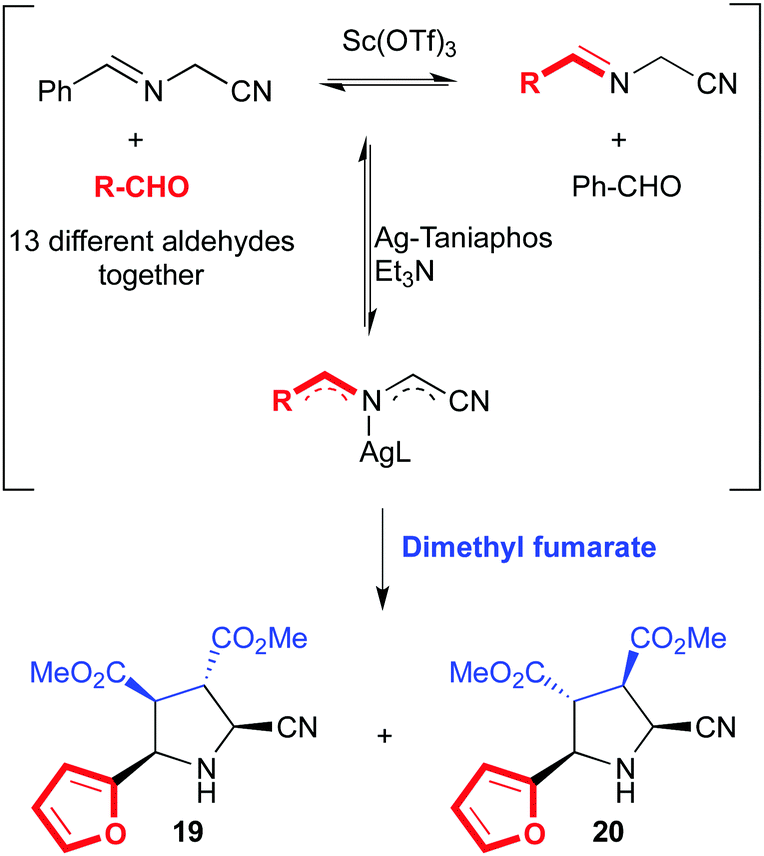 | ||
| Fig. 12 Dynamic systemic resolution of α-iminonitriles with silver(I)–Taniaphos. | ||
A similar process was developed using lipases as catalysts. A dynamic mixture was generated with two aromatic aldehydes (21a,b) and four related imines (22a–d). Then, 2-sulfanyl acetate (23) and 2-nitropropane (24) were added, which could lead to the reversible formation of multiple products arising from thioacetal or thioaminal formation, or Henry reactions. Indeed, Henry adducts were detected in the equilibrating mixture, but in the presence of CAL-B as lipase and phenyl acetate as acyl donor, the complex system was resolved into virtually two cyclized products (25a,b) out of eighteen possible products (Fig. 13).81
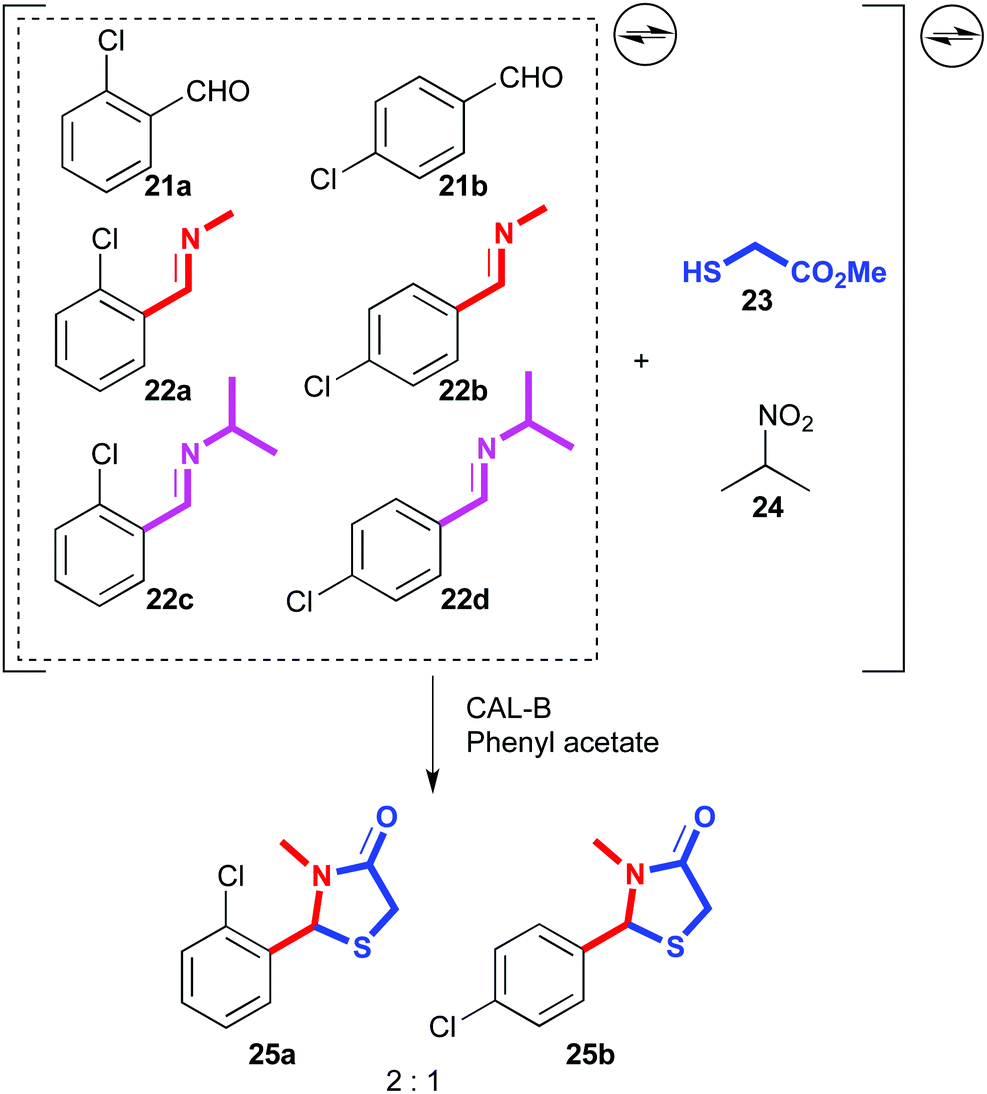 | ||
| Fig. 13 Lipase-catalysed dynamic systemic resolution of aldehydes and imines. | ||
Nevertheless, the use dynamic systems to generate new catalytic entities might be more common. In this sense, again in Ramström's group, a dynamic combinatorial approach to the discovery of a bifunctional organocatalyst for the Morita–Baylis–Hillman (MBH) reaction was undertaken (Fig. 14). A series of amines (26a–d, each with a different catalytic function) and aldehydes (27a–d) were subjected to reversible imine formation, and the resulting mixture tested in the MBH reaction. Significant catalytic activity was disclosed, which after deconvolution and independent preparation, led to the discovery of an efficient catalyst (28) for such reaction, although stereoselectivities were not reported.82
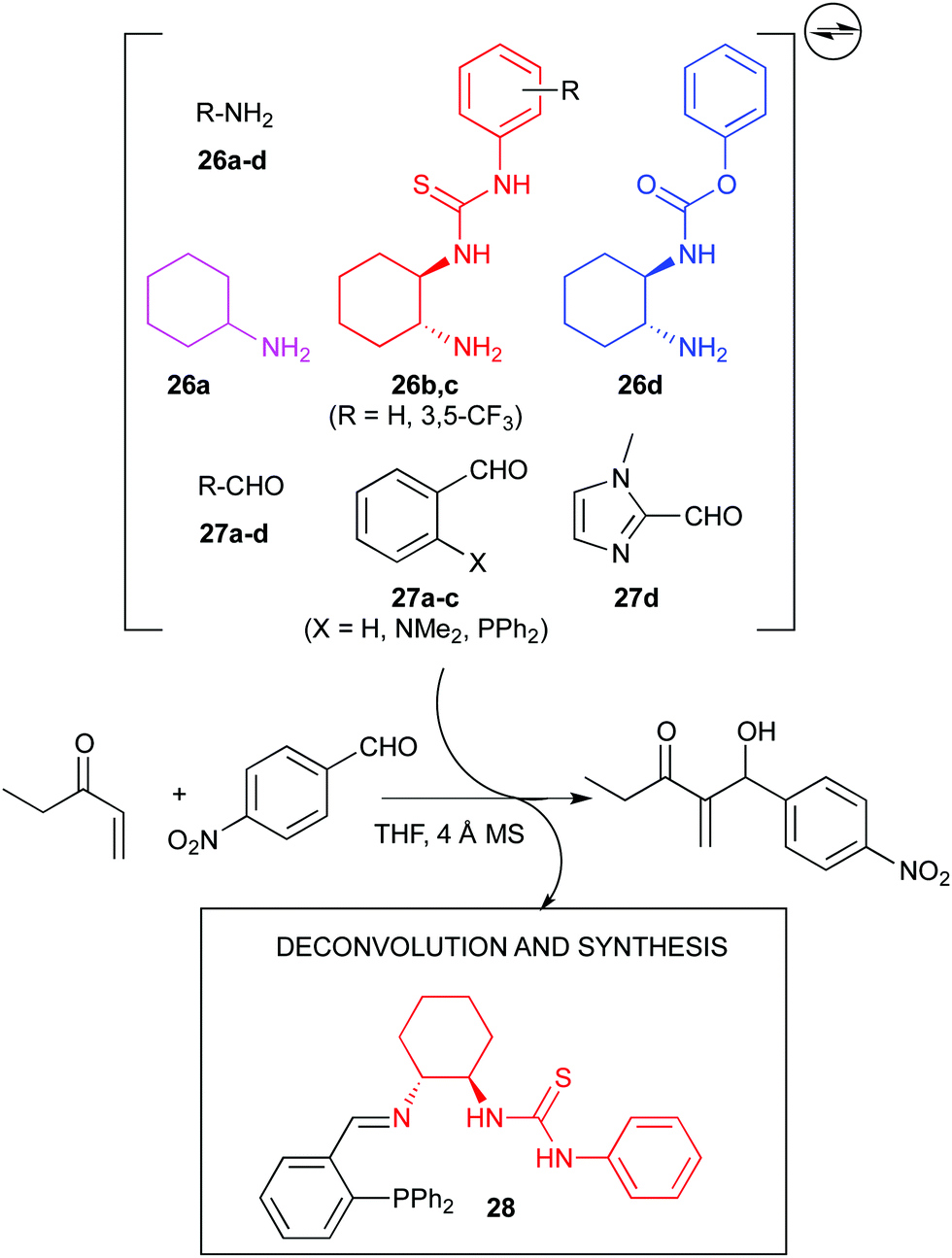 | ||
| Fig. 14 Dynamic combinatorial approach to the discovery of an organocatalyst for the MBH reaction. | ||
A more elaborated approach by the same group led to a kinetic self-sorting of a dynamic covalent catalyst for the MBH reaction, ultimately leading to feedback regulation. A family of quinuclidine catalysts with a reversible imine bond was tested in the above reaction showing a clear dependence of the reaction rate in the imine counterpart (arising from an aromatic aldehyde). Since an aromatic aldehyde was also used in the MBH reaction, this led to the development of a more or less active catalyst as a function of the aldehyde substrate chosen, thus being the substrate a key component to determine the catalyst structure and activity (Fig. 15).83
 | ||
| Fig. 15 Self-sorting of a dynamic covalent quinuclidine catalyst with substrate feedback regulation. | ||
Early examples of Dynamic Combinatorial Chemistry (DCC) applied to the discovery of new catalysts were actually pioneered by Otto and co-workers, in particular through the use of disulphide exchange chemistry. A significant example is provided next: their initial findings focused on the acceleration of the Diels–Alder (D–A) reaction through the adaptation of a library of disulphide ligands to a transition state analogue (TSA) as template. However, this catalytic reaction was hampered by product inhibition due to the similarities between the D–A adduct and the TSA.84 Later on, they focused on the aza-Cope rearrangement (Fig. 16) using an ammonium-based TSA (29). With this strategy, the properly designed ammonium-containing substrate would be recognized by the DCC library and trigger the formation of its own catalyst, a mixture of tetrameric isomers (30). After the rearrangement, the product would hydrolyse, losing the positive charge and avoiding product inhibition. Moreover, the catalyst also disassembled after the reaction was complete, which can be used to control catalysis in complex chemical systems. Still, a stoichiometric quantity of catalyst was used.85
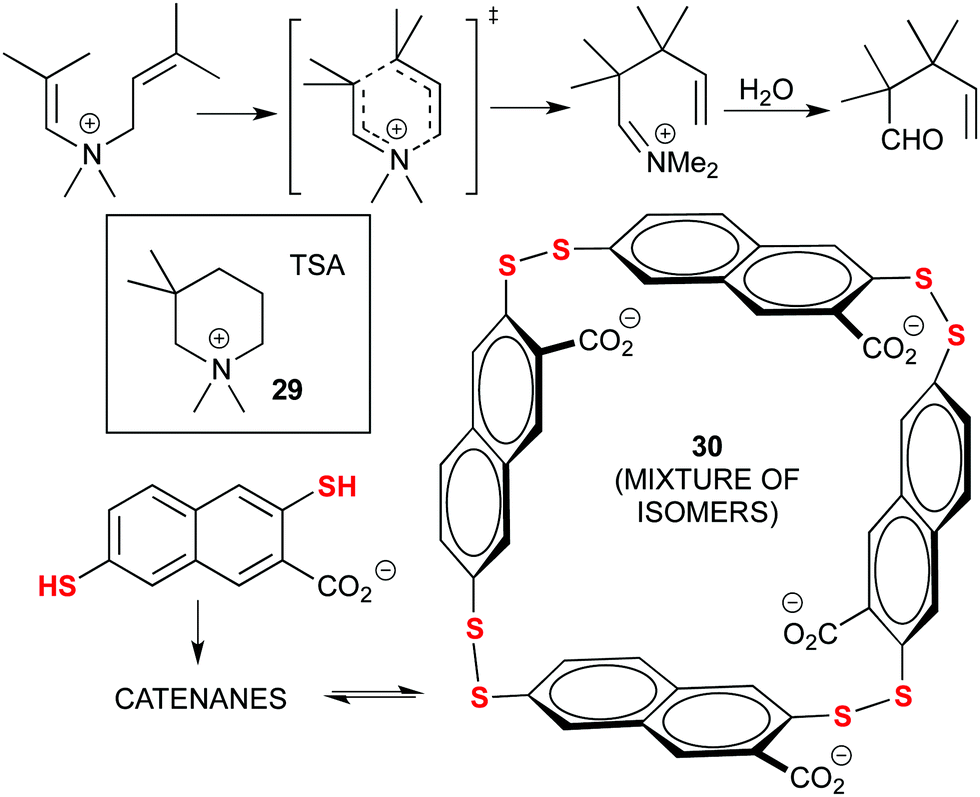 | ||
| Fig. 16 Amplification of a tetrameric catalyst (30) from a DCC library of sulphide ligands for the aza-Cope rearrangement, by using a TSA (29) as the template. | ||
Our own research group has been interested in complex catalytic systems, including the effect of water86 and other additives87 in reaction rate and stereoselectivity, as well as studying concentration effects.88 We eventually developed a dynamic catalytic network based on the binding of functional pyridine ligands to zinc(II), which thus behaves as a structural co-factor, in order to generate bifunctional catalysts for the asymmetric aldol reaction. This catalytic network showed fast exchange rate, and no single species could be identified. Nevertheless, high conversion and stereoselecivity were observed.89 Finally, in order to slow down the exchange process and favour the formation of more stable catalytic species, we developed the corresponding monofunctional bipyridine ligands 31 and 32 (Fig. 17).90 We demonstrated that the complex network thus generated, with several potential catalysts, actually simplified under the reaction conditions making the bifunctional species to predominate in a kind of thermodynamic self-sorting. As expected, this species also provided the highest reaction acceleration and stereoselectivity, improving the performance of the previous pyridine-based system at lower catalyst loading. Moreover, its reversible nature made impossible the synthesis and isolation of the catalytic complex, and the network generated with the three-components mixture was a convenient (and actually the only) manner of utilizing it.90 Thus, our approach illustrated how a dynamic system can cooperatively work for the formation of an active catalyst, even in the presence of other species in equilibrium, which are intrinsically needed for the function of the system.
 | ||
| Fig. 17 Dynamic network of organocatalytic bipyridine ligands templated by Zn(II). | ||
Other dynamic systems involve the use of vesicles or micelles due to the mobility of the self-assembled amphiphiles and therefore of the attached catalysts. Like in cell membranes, wherein enzymes and proteinic nanomachines are embedded in the lipidic bilayer, artificial vesicles or micelles provide a variety of new properties to catalysts or reactions different from the homogeneous systems. For instance, in our group we developed an amphiphilic catalyst for the asymmetric aldol reaction that featured an acylguanidinium group able to stablish a strong hydrogen bonding network. Indeed, intramolecular hydrogen bonding in the acylguanidine group was identified as the key structural motif. This catalyst worked nicely in the organic/aqueous interphase, outperforming other amphiphilic catalysts.91 A recent and more sophisticated example from Chen's group shows how an amphiphile containing an E-photoresponsive group and a catalytic Zn2+-triazacyclononane unit (33) self-assembles under visible light into a vesicle because of its E-configuration, which triggers catalytic activity towards transphosphorylation due to the cooperative action of the Zn2+-containing groups on the vesicle's surface. On the contrary, upon UV irradiation, the Z-configuration predominates, therefore the vesicle disassembles and the catalytic activity is lost (Fig. 18). This is an excellent example of catalyst activity control through an external stimulus in a fully reversible fashion, with concomitant control of both the molecular catalyst and the structure of the corresponding assembly.92
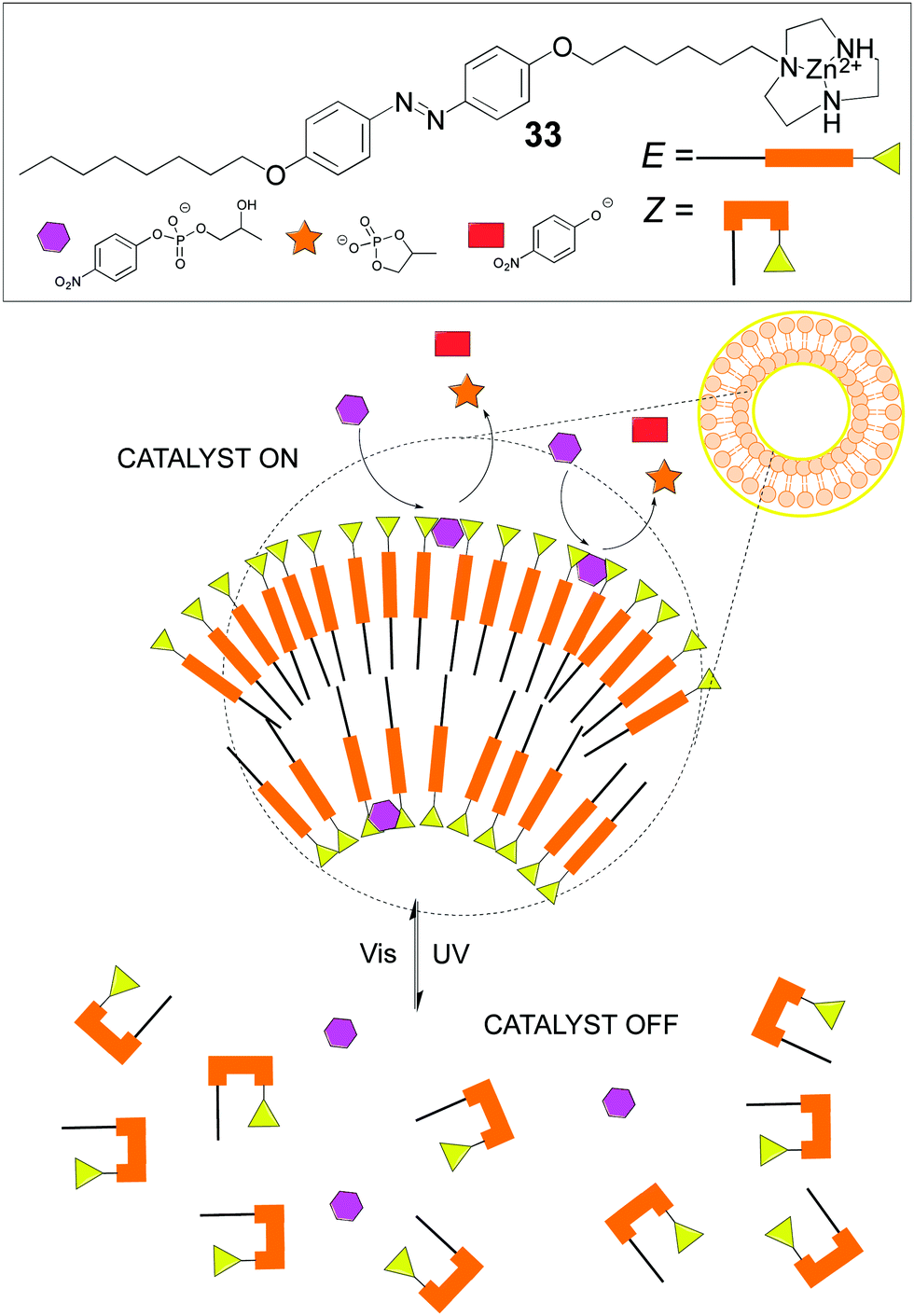 | ||
| Fig. 18 Photoactivation–deactivation in an amphiphilic catalytic system. | ||
This section beautifully reflects the power of using complexity in catalysis with selected examples. The main advantages of such an approach are the regulation of catalytic activity by simple means and the self-assembly of complex catalysts from simpler BBs, as well as product selectivity, even when dealing with complex mixtures in equilibrium (either for substrates or catalysts). We expect that further developments in the field will lead eventually to cost-effective and practical catalysts suitable for industrial applications, but meanwhile it is clear that Nature is an endless source of inspiration for the creativity in supramolecular catalysis.
Conclusions
Complexity has been traditionally avoided in chemical research as an unwanted complication and undesired side effect for achieving efficiency. The improvement of analytical techniques for the characterization of complex mixtures in addition to the development of elaborated mathematical tools have paved the way for the design and controlled implementation of complexity as an additional tool or parameter in molecular science. Following the growth of systems biology, the chemical counterpart systems chemistry has emerged as a subdiscipline with a bright future in different areas of research. Here, the use of dynamic combinatorial mixtures and stimuli-responsive networks for molecular recognition, sensing, chemical biology or catalysis has been illustrated with selected examples from the recent literature. Taking advantage of bioinspiration, several research groups have made this emergent field an appealing framework for the development of new functional materials and molecules. By constructing controlled mixtures of dynamic species, we and others have developed new chemical systems with potential applications in different fields. Although this can be considered a rather unconventional view to chemical design, it does not imply a very different methodological approach since complexity is inherent to molecular species. We think that complexity must be used in our benefit, instead of ignoring or avoiding its presence. We envision that the wise design of complex chemical systems will allow the emergence of new and even unexpected molecular properties and behaviours. With the progress of analytical instruments, we will be able to reduce the technical limitations of using complex systems in the near future, which will probably come from the capacity to rationalize and extract information from the data obtained. We have already shown that chemometric analysis may provide a solution to this bottleneck.36However, several challenges remain open for the future. The work in far-from-equilibrium systems is still in its infancy. The efficient implementation of fully synthetic systems able to replicate, merged with catalysis and compartmentalization would represent a big leap in the search for ‘artificial life’.93 Related to this, during the revision of the manuscript two important papers from the group of Otto have appeared. In a first study, the authors were able to combine a replicator and a cofactor that, when bound to the replicator, accelerates the formation of the components from which the replicator can emerge.94 In a second study, the replicator is able to catalyse a retro-aldol reaction due to the local increased basicity. Moreover, the same replicator promotes the deprotection of a N-Fmoc-protected amino acid. The particularity here is that the product of the reaction is able to catalyse the thiol oxidation necessary to form the replicator, thus exerting a positive feedback on replication.95
Last but not least, our final intention is not to create new terminology or to catalyse provocative discussion, but to encourage other chemists to reflect on the intrinsic complexity of molecules and their mixtures. This will hopefully create an opportunity to advance in basic knowledge, as well as on the impact of chemistry to solve the current societal challenges.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation and Spanish Research Agency (MCI/AEI/FEDER, RTI2018-096182-B-I00), as well as AGAUR (2017 SGR 208) is gratefully acknowledged. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- N. Pearcy, N. Chuzhanova and J. J. Crofts, J. Theor. Biol., 2016, 406, 99–104 Search PubMed.
- J. Bascompte, M. B. García, R. Ortega, E. L. Rezende and S. Pironon, Sci. Adv., 2019, 5, eaav2539 Search PubMed.
- M. J. Aschwanden, N. B. Crosby, M. Dimitropoulou, M. K. Georgoulis, S. Hergarten, J. McAteer, A. V. Milovanov, S. Mineshige, L. Morales, N. Nishizuka, G. Pruessner, R. Sánchez, A. S. Sharma, A. Strugarek and V. Uritsky, Space Sci. Rev., 2016, 198, 47–166 Search PubMed.
- (a) J. M. Montoya, S. L. Pimm and R. V. Solé, Nature, 2006, 442, 259–264 Search PubMed; (b) S. Azaele, S. Pigolotti, J. R. Banavar and A. Maritan, Nature, 2006, 444, 926–928 Search PubMed.
- S. P. Borgatti, A. Mehra, D. J. Brass and G. Labianca, Science, 2009, 323, 892–895 Search PubMed.
- F. Schweitzer, G. Fagiolo, D. Sornette, F. Vega-Redondo, A. Vespignani and D. R. White, Science, 2009, 325, 422–425 Search PubMed.
- M. Gosak, R. Markovič, J. Dolenšek, M. Slak Rupnik, M. Marhl, A. Stožer and M. Perc, Phys. Life Rev., 2018, 24, 118–135 Search PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 Search PubMed.
- A. S. Y. Wong and W. T. S. Huck, Beilstein J. Org. Chem., 2017, 13, 1486–1497 Search PubMed.
- (a) E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 Search PubMed; (b) I. Alfonso, Chem. Commun., 2016, 52, 239–250 Search PubMed.
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 Search PubMed.
- (a) A. Aderem, Cell, 2005, 121, 511–513 Search PubMed; (b) Special issue devoted to systems biology: L. Chong and L. B. Ray, Science, 2002, 295, 1661–1682 Search PubMed.
- G. Ashkenasy, T. Hermans, S. Otto and A. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 Search PubMed.
- (a) J. R. Nitschke, Nature, 2009, 462, 736–738 Search PubMed; (b) S. Otto and A. R. Hunt, Chem. Commun., 2011, 47, 847–858 Search PubMed; (c) J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 520–5126 Search PubMed.
- (a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 Search PubMed; (b) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 Search PubMed; (c) J. N. H. Reek and S. Otto, Dynamic Combinatorial Chemistry, Wiley-VCH, Weinheim, 2010 Search PubMed; (d) F. B. L. Cougnon and J. K. M. Sanders, Acc. Chem. Res., 2012, 45(12), 2211–2221 Search PubMed; (e) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 Search PubMed; (f) P. Frei, R. Hevey and B. Ernst, Chem. – Eur. J., 2019, 25, 60–73 Search PubMed.
- (a) J.-M. Lehn, Chem. – Eur. J., 1999, 5, 2455–2463 Search PubMed; (b) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 Search PubMed.
- (a) R. L. E. Furlan, S. Otto and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4801–4804 Search PubMed; (b) S. Otto, Curr. Opin. Drug Discovery Dev., 2003, 6, 509–520 Search PubMed.
- (a) P. T. Corbett, S. Otto and J. K. M. Sanders, Chem. – Eur. J., 2004, 10, 3139–3143 Search PubMed; (b) I. Saur and K. Severin, Chem. Commun., 2005, 1471–1473 Search PubMed.
- P. T. Corbett, S. Otto and J. K. M. Sanders, Org. Lett., 2004, 6, 1825–1827 Search PubMed.
- R. F. Ludlow and S. Otto, J. Am. Chem. Soc., 2010, 132, 5984–5986 Search PubMed.
- (a) I. Saur and K. Severin, Chem. Commun., 2005, 1471–1473 Search PubMed; (b) P. T. Corbett, J. K. M. Sanders and S. Otto, J. Am. Chem. Soc., 2005, 127, 9390–9392 Search PubMed.
- (a) S. Otto and S. Kubik, J. Am. Chem. Soc., 2003, 125, 7804–7805 Search PubMed; (b) L. Vial, R. F. Ludlow, J. Leclaire, R. Pérez-Fernández and S. Otto, J. Am. Chem. Soc., 2006, 128, 10253–10257 Search PubMed.
- Selected examples: (a) S. Otto, R. L. E. Furlán and J. K. M. Sanders, Science, 2002, 297, 590–593 Search PubMed; (b) A. González-Álvarez, I. Alfonso, F. López-Ortiz, S. Aguirre, S. García-Granda and V. Gotor, Eur. J. Org. Chem., 2004, 1117–1127 Search PubMed; (c) T. S. R. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667–669 Search PubMed; (d) S. Ladame, A. Whitney and S. Balasubramanian, Angew. Chem., Int. Ed., 2005, 44, 5736–5739 Search PubMed; (e) A. Bugaut, K. Jantos, J. L. Wietor, R. Rodriguez, J. K. M. Sanders and S. Balasubramanian, Angew. Chem., Int. Ed., 2008, 47, 2677–2680 Search PubMed; (f) S. Hamieh, V. Saggiomo, P. Nowak, E. Mattia, R. F. Ludlow and S. Otto, Angew. Chem., Int. Ed., 2013, 52, 12368–12372 Search PubMed; (g) R. Brachvogel, F. Hampel and M. von Delius, Nat. Commun., 2015, 6, 7129 Search PubMed.
- M. Corredor, D. Carbajo, C. Domingo, Y. Pérez, J. Bujons, A. Messeguer and I. Alfonso, Angew. Chem., Int. Ed., 2018, 57, 11973–11977 Search PubMed.
- D. Carbajo, Y. Pérez, J. Bujons and I. Alfonso, Angew. Chem., Int. Ed., 2020, 59, 17202–17206 Search PubMed.
- (a) M. Samiappan, Z. Dadon and G. Ashkenasy, Chem. Commun., 2011, 47, 710–712 Search PubMed; (b) X. Dadon, M. Samiappan, N. Wagner and G. Ashkenasy, Chem. Commun., 2012, 48, 1419–1421 Search PubMed.
- (a) N. Giuseppone and J.-M. Lehn, Chem. – Eur. J., 2006, 12, 1715–1722 Search PubMed; (b) L. Tauk, A. P. Schröder, G. Decher and N. Giuseppone, Nat. Chem., 2009, 1, 649–656 Search PubMed.
- S. Sobczak, W. Drozḋz, G. I. Lampronti, A. M. Belenguer, A. Katrusiak and A. R. Stefankiewicz, Chem. – Eur. J., 2018, 24, 8769–8773 Search PubMed.
- (a) J. Atcher, A. Moure and I. Alfonso, Chem. Commun., 2013, 49, 487–489 Search PubMed; (b) J. Atcher, A. Moure, J. Bujons and I. Alfonso, Chem. – Eur. J., 2015, 21, 6869–6878 Search PubMed.
- (a) J. Zhang, L. You, E. V. Anslyn and X. Qian, Chem. – Eur. J., 2012, 18, 1102–1110 Search PubMed; (b) D. Zamora-Olivares, T. S. Kaoud, K. N. Dalby and E. V. Anslyn, J. Am. Chem. Soc., 2013, 135, 14814–14820 Search PubMed; (c) L. You, D. Zhaa and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 Search PubMed.
- (a) J. Montenegro, P. Bonvin, T. Takeuchi and S. Matile, Chem. – Eur. J., 2010, 16, 14159–14166 Search PubMed; (b) T. Takeuchi, J. Montenegro, A. Hennig and S. Matile, Chem. Sci., 2011, 2, 303–307 Search PubMed; (c) T. Takeuchi and S. Matile, Chem. Commun., 2013, 49, 19–29 Search PubMed.
- (a) A. Buryak, A. Pozdnoukhov and K. Severin, Chem. Commun., 2007, 2366–2368 Search PubMed; (b) S. Rochat, J. Gao, X. Qian, F. Zaubitzer and K. Severin, Chem. – Eur. J., 2010, 16, 104–113 Search PubMed; (c) Z. Köstereli, R. Scopelliti and K. Severin, Chem. Sci., 2014, 5, 2456–2460 Search PubMed.
- (a) F. A. Scaramuzzo, G. Licini and C. Zonta, Chem. – Eur. J., 2013, 19, 16809–16813 Search PubMed; (b) C. Bravin, A. Guidetti, G. Licini and C. Zonta, Chem. Sci., 2019, 10, 3523–3528 Search PubMed.
- (a) A. Osypenko, S. Dhers and J.-M. Lehn, J. Am. Chem. Soc., 2019, 141, 12724–12737 Search PubMed; (b) G. Men and J.-M. Lehn, Chem. Sci., 2019, 10, 90–98 Search PubMed.
- J. Atcher, J. Solà and I. Alfonso, Org. Biomol. Chem., 2017, 15, 213–217 Search PubMed.
- A. M. Valdivielso, F. Puig-Castellví, J. Atcher, J. Solà, R. Taulera and I. Alfonso, Chem. – Eur. J., 2017, 23, 10789–10799 Search PubMed.
- K.-D. Zhang and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 8980–8983 Search PubMed.
- H. M. Seifert, K. R. Trejo and E. V. Anslyn, J. Am. Chem. Soc., 2016, 138, 10916–10924 Search PubMed.
- M. Martínez-Amezaga, A. G. Orrillo and R. L. E. Furlan, Chem. Sci., 2019, 10, 8338–8347 Search PubMed.
- B. M. Matysiak, P. Nowak, I. Cvrtila, C. G. Pappas, B. Liu, D. Komáromy and S. Otto, J. Am. Chem. Soc., 2017, 139, 6744–6751 Search PubMed.
- S. N. Semenov, L. J. Kraft, A. Ainla, M. Zhao, M. Baghbanzadeh, V. E. Campbell, K. Kang, J. M. Fox and G. M. Wihtesides, Nature, 2016, 537, 656–660 Search PubMed.
- B. J. Cafferty, A. S. Y. Wong, S. N. Semenov, L. Belding, S. Gmür, W. T. S. Huck and G. M. Whitesides, J. Am. Chem. Soc., 2019, 141, 8289–8295 Search PubMed.
- M. He and J.-M. Lehn, J. Am. Chem. Soc., 2019, 141, 18560–18569 Search PubMed.
- (a) K. West, K. Bake and S. Otto, Org. Lett., 2005, 7, 2615–2618 Search PubMed; (b) A. R. Stefankiewicz and J. K. M. Sanders, Chem. Commun., 2013, 49, 5820–5822 Search PubMed.
- (a) J. Solà, M. Lafuente, J. Atcher and I. Alfonso, Chem. Commun., 2014, 50, 4564–4566 Search PubMed; (b) M. Lafuente, J. Atcher, J. Solà and I. Alfonso, Chem. – Eur. J., 2015, 21, 17002–17009 Search PubMed; (c) M. Lafuente, I. Alfonso and J. Solà, ChemSystemsChem, 2019, 1, 25–31 Search PubMed.
- M. Lafuente, J. Solà and I. Alfonso, Angew. Chem., Int. Ed., 2018, 57, 1–5 Search PubMed.
- (a) K. R. West, R. F. Ludlow, P. T. Corbett, P. Besenius, F. M. Mansfeld, P. A. G. Cormack, D. C. Sherrington, J. M. Goodman, M. C. A. Stuart and S. Otto, J. Am. Chem. Soc., 2008, 130, 10834–10835 Search PubMed; (b) M.-K. Chung, S. J. Lee, M. L. Waters and M. R. Gagne, J. Am. Chem. Soc., 2012, 134, 11430–11443 Search PubMed; (c) F. B. L. Cougnon, N. Ponnuswamy, N. A. Jenkins, G. D. Pantos and J. K. M. Sanders, J. Am. Chem. Soc., 2012, 134, 19129–19135 Search PubMed.
- (a) N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantoş and J. K. M. Sanders, Science, 2012, 338, 783–785 Search PubMed; (b) N. Ponnuswamy, F. B. L. Cougnon, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2014, 136, 8243–8251 Search PubMed.
- B. Liu, C. G. Pappas, E. Zangrando, N. Demitri, P. J. Chmielewski and S. Otto, J. Am. Chem. Soc., 2019, 141, 1685–1689 Search PubMed.
- (a) A. J. Bissette and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 12800–12826 Search PubMed; (b) T. Kosikova and D. Philp, Chem. Soc. Rev., 2017, 46, 7274–7305 Search PubMed.
- (a) K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 114, 285–366 Search PubMed; (b) J. D. Sutherland, Angew. Chem., Int. Ed., 2016, 55, 104–121 Search PubMed.
- T. Tjivikua, P. Ballester and J. Rebek, J. Am. Chem. Soc., 1990, 112, 1249–1250 Search PubMed.
- S. Xu and N. Giuseppone, J. Am. Chem. Soc., 2008, 130, 1826–1827 Search PubMed.
- W. Sadownik and D. Philp, Angew. Chem., Int. Ed., 2008, 47, 9965–9970 Search PubMed.
- R. Nguyen, L. Allouche, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2009, 48, 1093–1096 Search PubMed.
- K. Severin, D. H. Lee, J. A. Martinez, M. Vieth and M. R. Ghadiri, Angew. Chem., Int. Ed., 1998, 37, 126–128 Search PubMed.
- R. Mukherjee, R. Cohen-Luria, N. Wagner and G. Ashkenasy, Angew. Chem., Int. Ed., 2015, 54, 12452–12456 Search PubMed.
- J. Nanda, B. Rubinov, D. Ivnitski, R. Mukherjee, E. Shtelman, Y. Motro, Y. Miller, N. Wagner, R. Cohen-Luria and G. Ashkenasy, Nat. Commun., 2017, 8, 434 Search PubMed.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 Search PubMed.
- (a) M. Malakoutikhah, J. J.-P. Peyralans, M. Colomb-Delsuc, H. Fanlo-Virgos, M. C. A. Stuart and S. Otto, J. Am. Chem. Soc., 2013, 135(49), 18406–18417 Search PubMed; (b) M. Colomb-Delsuc, E. Mattia, J. W. Sadownik and S. Otto, Nat. Commun., 2015, 6, 7427 Search PubMed.
- D. Komáromy, M. Tezcan, G. Schaeffer, I. Marić and S. Otto, Angew. Chem., Int. Ed., 2017, 56, 14658–14662 Search PubMed.
- J. W. Sadownik, E. Mattia, P. Nowak and S. Otto, Nat. Chem., 2016, 8, 264–269 Search PubMed.
- Y. Altay, M. Tezcan and S. Otto, J. Am. Chem. Soc., 2017, 139, 13612–13615 Search PubMed.
- M. Altay, Y. Altay and S. Otto, Angew. Chem., Int. Ed., 2018, 139, 10564–10568 Search PubMed.
- S. M. Morrow, I. Colomer and S. P. Fletcher, Nat. Commun., 2019, 10, 1011 Search PubMed.
- I. Colomer, A. Borissov and S. P. Fletcher, Nat. Commun., 2020, 11, 176 Search PubMed.
- J. Sreenivasachary and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938–5943 Search PubMed.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 Search PubMed.
- M. Kumar, N. L. Ing, V. Narang, N. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696–703 Search PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 Search PubMed.
- (a) E.-K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 Search PubMed; (b) G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 Search PubMed.
- C. Bouillon, D. Paolantoni, J. C. Rote, Y. Bessin, L. W. Peterson, P. Dumy and S. Ulrich, Chem. – Eur. J., 2014, 20, 14705–14714 Search PubMed.
- C. Bouillon, Y. Bessin, F. Poncet, M. Gary-Bobo, P. Dumy, M. Barboiu, N. Bettache and S. Ulrich, J. Mater. Chem. B, 2018, 6, 7239–7246 Search PubMed.
- (a) M. Vlatkovic, B. S. L. Collins and B. L. Feringa, Chem. – Eur. J., 2016, 22, 17080–17111 Search PubMed; (b) B. A. Grzybowski, K. Fitzner, J. Paczesny and S. Granick, Chem. Soc. Rev., 2017, 46, 5647–5678 Search PubMed; (c) A. M. Lifschitz, M. S. Rosen, C. M. McGuirk and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 7252–7261 Search PubMed; (d) N. Kumagai and M. Shibasaki, Catal. Sci. Technol., 2013, 3, 41–57 Search PubMed.
- L. Zhang, F. Ma, J. Lei, J. Liu and H. Ju, Chem. Sci., 2017, 8, 4833–4839 Search PubMed.
- (a) A. I. d’Aquino, H. F. Cheng, J. Barroso-Flores, Z. S. Kean, J. Mendez-Arroyo, C. M. McGuirk and C. A. Mirkin, Inorg. Chem., 2018, 57, 3568–3578 Search PubMed; (b) N. C. Gianneschi, S.-H. Cho, S. T. Nguyen and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 5503–5507 Search PubMed.
- C. M. McGuirk, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 4689–4696 Search PubMed.
- C. M. McGuirk, J. Méndez-Arroyo, A. M. Lifschitz and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 16594–16601 Search PubMed.
- M. Sakulsombat, Y. Zhang and O. Ramström, Top. Curr. Chem., 2012, 322, 55–86 Search PubMed.
- L. Hu and O. Ramström, Chem. Commun., 2014, 50, 3792–3794 Search PubMed.
- (a) Y. Zhang and O. Ramström, Chem. – Eur. J., 2014, 20, 3288–3291 Search PubMed; (b) Y. Zhang, P. Vongvilai, M. Sakulsombat, A. Fischer and O. Ramström, Adv. Synth. Catal., 2014, 356, 987–992 Search PubMed.
- F. Schaufelberger and O. Ramström, Chem. – Eur. J., 2015, 21, 12735–12740 Search PubMed.
- F. Schaufelberger and O. Ramström, J. Am. Chem. Soc., 2016, 138, 7836–7839 Search PubMed.
- B. Brisig, J. K. M. Sanders and S. Otto, Angew. Chem., Int. Ed., 2003, 42, 1270–1273 Search PubMed.
- H. Fanlo-Virg, A.-N. R. Alba, S. Hamieh, M. Colomb-Delsuc and S. Otto, Angew. Chem., Int. Ed., 2014, 53, 11346–11350 Search PubMed.
- C. Jimeno, Org. Biomol. Chem., 2016, 14, 6147–6164 Search PubMed.
- Z. I. Günler, X. Companyó, I. Alfonso, J. Burés, C. Jimeno and M. A. Pericàs, Chem. Commun., 2016, 52, 6821–6824 Search PubMed.
- Z. Inci Günler, I. Alfonso, C. Jimeno and M. A. Pericàs, Synthesis, 2017, 319–325 Search PubMed.
- (a) A. Serra-Pont, I. Alfonso, C. Jimeno and J. Solà, Chem. Commun., 2015, 51, 17386–17389 Search PubMed; (b) A. Serra-Pont, I. Alfonso, J. Solà and C. Jimeno, Org. Biomol. Chem., 2017, 15, 6584–6591 Search PubMed.
- A. Serra-Pont, I. Alfonso, J. Solà and C. Jimeno, Chem. Commun., 2019, 55, 7970–7973 Search PubMed.
- A. M. Valdivielso, A. Catot, I. Alfonso and C. Jimeno, RSC Adv., 2015, 5, 62331–62335 Search PubMed.
- C. Z.-J. Ren, P. Solís Muñana, J. Dupont, S. S. Zhou and J. L.-Y. Chen, Angew. Chem., Int. Ed., 2019, 58, 15254–15258 Search PubMed.
- (a) P. Adamski, M. Eleveld, A. Sood, Á. Kun, A. Szilágyi, T. Czárán, E. Szathmáry and S. Otto, Nat. Rev. Chem., 2020, 4, 386–403 Search PubMed; (b) E. A. J. Post and S. P. Fletcher, Chem. Sci., 2020, 11, 9434–9442 Search PubMed.
- G. M. Santiago, K. Liu, W. R. Browne and S. Otto, Nat. Chem., 2020, 12, 603–607 Search PubMed.
- J. Ottelé, A. S. Hussein, C. Mayer and S. Otto, Nat. Catal., 2020, 3, 547–553 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |