
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetrazine metallation boosts rate and regioselectivity of inverse electron demand Diels–Alder (iEDDA) addition of dienophiles†
Marc
Schnierle
 a,
Svenja
Blickle
a,
Vasileios
Filippou
*ab and
Mark R.
Ringenberg
*a
a,
Svenja
Blickle
a,
Vasileios
Filippou
*ab and
Mark R.
Ringenberg
*a
aUniversität Stuttgart, Institut für Anorganische Chemie, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: mark.ringenberg@iac.uni-stuttgart.de
bVarimol TGU, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: filippou@varimol.de
First published on 9th September 2020
Abstract
Reported herein is the coordination of rhenium complexes to tetrazine ligand in [ReCl(CO)3(TzPy)] [1] (TzPy = 3-(2-pyridyl)-1,2,4,5-tetrazine) and the rates of addition of different dienophiles to the tetrazine. Tetrazine coordiation lowers the ΔS‡ contribution to ΔG‡ for iEDDA addition.
The inverse electron demand Diels–Alder (iEDDA) reaction between 1,2,4,5-tetrazines and olefins is a substrate controlled click-reaction1,2 and does not require the addition of a catalyst (CuAAC)3 or light (thiol–ene).4,5 The modification of the 1,(4)-positions on the tetrazine can be synthetically arduous but rational design of the tetrazine diene and dienophile has resulted in very fast iEDDA reaction rates, where rates >106 M s−1 have been reported.6,7 These factors have made the iEDDA addition a useful reaction in several applications, chiefly among them in biological labelling experiments.1,5,8 The iEDDA addition is, however, not regioselective9 and it produces a mixture of regioisomers, e.g., 1,4- and 1,5-isomers (Scheme 1).
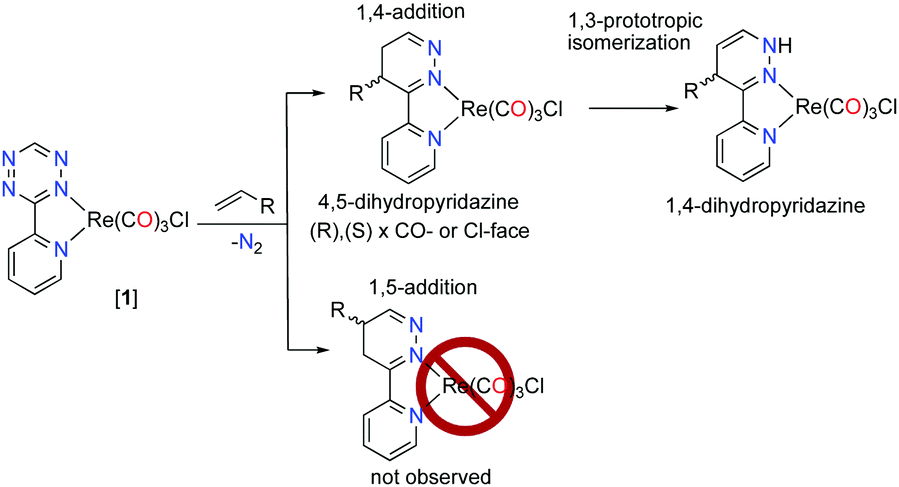 | ||
| Scheme 1 Addition of dienophile to [1]. | ||
Transition metal(s) are known to coordinate tetrazines and these complexes can exhibit ligand non-innocence.10,11 Coordinated tetrazines show anodically shifted reduction potentials due to metal back-bonding, i.e., the tetrazine moiety is more electrophilic.10,12 The metal back-bonding would also lower the activation energy of the addition of a dienophile to the tetrazine diene, however, the often transoid bridging geometry prevents the approach of a dienophile to the tetrazine.10,11 Unlike the majority of the reported symmetric tetrazines,10 2-pyridyltetrazine (TzPy), isoelectronic and isostructural to 2,2′-bipyridine, can be used as bidentate ligand, and the tetrazine diene is free to add dienophiles. For example, addition of cyclooctyne to the fluorescent iridium complex [Ir(PhPy)2(TzPy)]+ (PhPy = 2-phenylpyridine) have been described and the rate of the iEDDA addition was between 2.5 and 60 times faster than with the free TzPy ligand.13 The addition of cyclooctyne to [Ir(PhPy)2(TzPy)]+ generates the aromatic 1,2-diazine and thus no stereochemical information was generated from this reaction.
Herein we report the synthesis and rate of addition of three dienophiles to the metallotetrazine [ReCl(CO)3(TzPy)] [1], see ESI,† for synthetic and kinetic details. The ReCl(CO)3 moiety was chosen to coordinate TzPy because tricarbonylrhenium(I) complexes with pyridine donor ligands have found uses as imaging reagents in cells and they have also shown cytotoxic activity for cancer treatment.14 The water soluble variants [Re(OH2)(CO)3(L^L)]+ (L^L = bidentate amine donor ligands) have also been described by replacing the Cl-ligand for the aquo ligand.15 Additionally, tricarbonylrhenium(I) coordinated to a bidentate ligand, e.g., 2,2′-bipyridine, are electrocatalysts for CO2 reduction.16
The tetrazine moiety in [1] can add dienophiles and the rate of addition of vinylferrocene (ViFc), styrene (Ci), and trans-cyclooctene (TCO) to [1] were measured and are reported in Table 1. Different dienophiles were also tested for their ability to add to [1], such as vinyl and allyl functionality (e.g. vinyltrimethoxysilane and allyltrimethylsilane), a bulky olefin (e.g. quinine) and phenylacetylene to [1] was also demonstrated (see ESI†) indicating coordination of TzPy does not inhibit addition of electron rich olefins. The addition of Ci and TCO to [1], respectively, showed enhanced rates compared to the reported rates between the symmetric 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine (Py2Tz) and the self-similar dieneophiles.7,17,18 The work herein was performed in 1,2-dichloroethane (DCE) due to the poor solubility of [1] in H2O, although [1] does show moderate solubility in MeOH, the comparison is used to demonstrate the enhanced rate compared to the reported k2 values.
| Reaction | k 2 /M−1 s−1 | ΔH‡/kJ mol−1 | ΔS‡/J mol−1 K−1 | ΔG‡![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (DFTc)/kJ mol−1 b (DFTc)/kJ mol−1 |
|---|---|---|---|---|
| a Rate at 25 °C in C2H4Cl2. b At 25 °C. c Gas phase reaction TPSS/def2-TZVP/J level, see ESI of details. d MeOH ref. 18. e 9:1 MeOH/H2O ref. 7 and CH2Cl2 see ESI figure S. f DFT M06L/6(311)+G(d,p) level ref. 7. | ||||
| [1] + ViFc | 2.80 ± 0.1 | 23 | −150 | 68 (40) |
| TzPy + ViFc | 1.80 × 10−2 | 27 | −192 | 84 (54) |
| [1] + Ci | 6.03 ± 0.02 × 10−2 | 55 | −125 | 92 (62) |
| Py2Tz + Ci | 3.0 ± 0.3 × 10−3d |
— | — | — |
| [1] + TCO | 4.06 ± 0.52 × 105 | 26 | −50 | 41 (28) |
| Py2Tz +TCO | 2.0 × 103e |
— | — | (37f) |
The rate of the addition of ViFc, Ci, or TCO to [1] was measured using time-resolved variable-temperature UV vis spectroscopy in C2H4Cl2, respectively and the rates and thermodynamic values are reported in Table 1. The rate for [1] + ViFc k2 = 2.80 ± 0.1 M−1 s−1 at 22 °C was 160 times faster than the control reaction TzPy + ViFc k2 = 0.0180 M−1 s−1 at 22 °C. The Eyring analysis19 of [1] + ViFc found ΔH‡ = 22.6 kJ mol−1 and ΔS‡ = −150 J mol−1 K−1, with ΔG‡(25 °C) = 68 kJ mol−1. The Eyring analysis of TzPy + ViFc showed a small increase in the ΔH‡ = 27 kJ mol−1, however the ΔS‡ = −192 J mol−1 K−1 contributed more to the ΔG‡(25 °C) = 84 kJ mol−1. Coordination of TzPy lowers the ΔΔG‡ = 16 kJ, and the ΔΔG‡(calc.) = 14 kJ was in good agreement with experimental value (see ESI†). The discrepancy between DFT and experimental values can be attributed to solvent effects.
TzPy shows resonance stabilization of δ+ at the C4 of the tetrazine, and coordination of TzPy enhances this resonance structure due to back-bonding of the δ− on the ReCl(CO)3 moiety (Fig. 2). The addition of ViFc to TzPy shows a larger contribution of ΔS‡ to the transition state ΔG‡. One rational is that the molecular structure of TzPy (see Fig. S27, ESI†) is planar while the TzPy in [1] (Fig. 1) tilts towards the Cl-ligand. This distortion may approximate the dien-dienophile transition state (Scheme 2), another contribution could be that back-bonding affords a weakening of the double bonds in the tetrazine. Additionally, coordination of TzPy restricts its motion, which may also contribute to a lower transition state energy. The effect as to why coordination of the tetrazine lowers the ΔG‡ is currently under investigation.
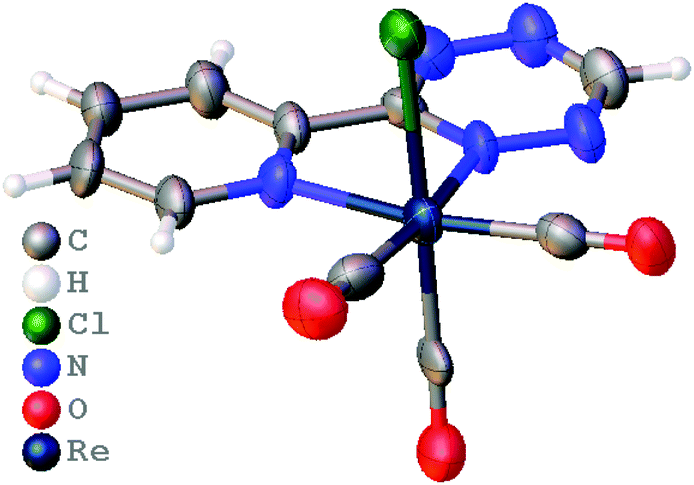 | ||
| Fig. 1 Molecular structure of [1] determined crystallographically, co-crystallized CH2Cl2 was omitted for clarity. | ||
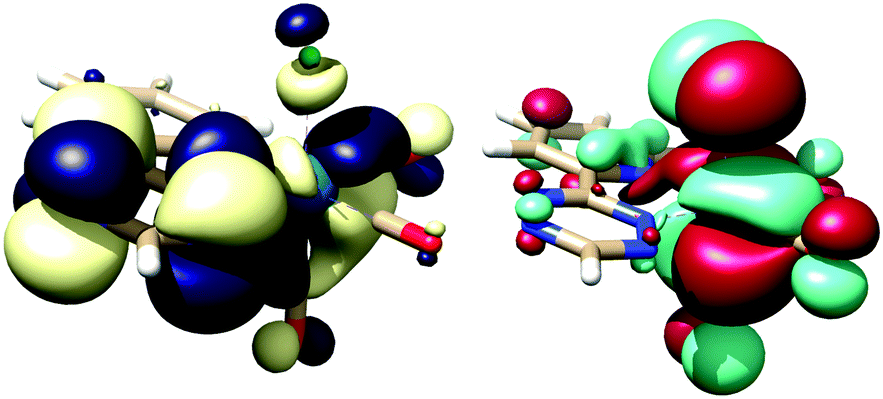 | ||
| Fig. 2 Lowest unoccupied molecular orbital (LUMO, yellow/blue) and highest occupied MO (HOMO, red/green) of [1], isosurface at 0.02, TPSS basis set at def2-TZVP/J level of theory (see ESI,† for full details). | ||
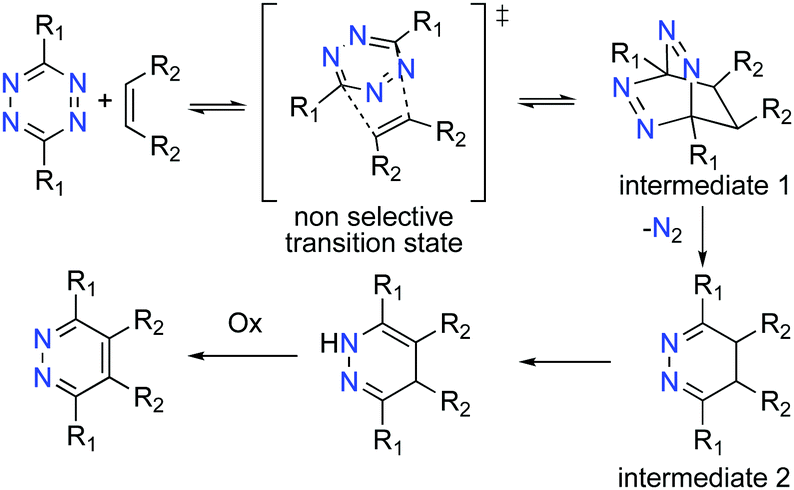 | ||
| Scheme 2 Reaction mechanism of olefin with tetrazine. | ||
ViFc is an electron rich dienophile (δ− on α-carbon, fulvene resonance with δ+ on Fe atom),20 therefore the addition of the unactivated styrene (Ci) to [1] was also studied. The rate of addition of Ci to [1], k2 = 6.03 ± 0.02 × 10−2 M−1 s−1 was nearly 20 times faster than the addition of Ci to Py2Tz (k2 = 3.0 × 10−3 M−1 s−118). The Eyring analysis of the addition of Ci to [1] found a larger ΔH‡ = 55 kJ mol−1 was more than double the ΔH‡ for the addition of ViFc to [1]. The lower contribution of ΔS‡ = −125 J mol−1 K−l to the transition state (Table 1) can be attributed to the size of the Ph verse Fc. The rate of the addition of TCO to [1] was k2 = 4.06 ± 0.52 × 105 M−1 s−1 which is 200 time faster than the addition of TCO to Py2Tz.7 The ΔH‡ = 26 kJ mol−1 is on the order of the addition of ViFc to [1], however there is a significantly lower contribution of the ΔS‡ = −50 J mol−1 K−1 to ΔG‡ = 41 kJ mol−1, as should be expected with the strained TCO dienophile. The difference in rates between the addition of ViFc and TCO respectively to [1], shows that coordination lowers the entropic barrier (ΔS‡) as the enthalpic barriers (ΔH‡) are nearly isoenergetic (Table 1).
The addition of dienophiles to the tetrazine diene is generally unselective (Scheme 2), and the 1,3-prototropic isomerization is rapid which prevents the 4,5-dihydropyridazine (Scheme 2, intermediate 2) from being isolated.11 The Alder–Stein principle (the relative stereochemistry of dienes and dienophiles is conserved), and Alder's endo-rule (the endo-adduct is the kinetically preferred product), apply in the iEDDA addition, but stereochemical information is often lost due to rapid isomerization and rearomatisation of the 1,4-dhp to the 1,2-pyrazine, especially under aerobic conditions (Scheme 2).
The addition of ViFc, TCO, or Ci to [1] at room temperature generated the species [1Fc], [1TCO], and [1Ci] and crystals were obtained directly from the reaction mixtures where the molecular structures were determined crystallographically. The molecular structure of [1Fc] showed the 4,5-dhp isomer, while [1TCO] and [1Ci] were found as the 1,4-dhp isomer (Fig. 3). DFT analysis for the 1,3-prototropic isomerization for all three complexes showed the 1,4-dhp was energetically favorable, however, the 1,4-dhp isomer of [1Fc] was only observed as a minor product in the 1H-NMR (see ESI†). DFT analysis found rearomatisation for both [1Fc] and [1Ci] were both endergonic (see ESI†).
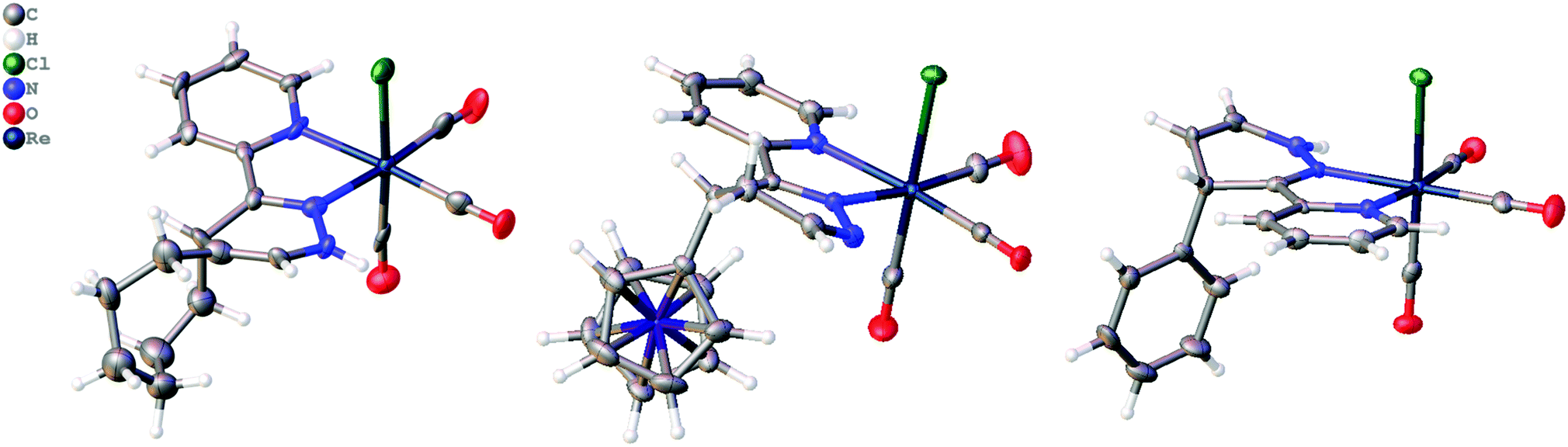 | ||
| Fig. 3 Molecular structure of [1TCO], [1Fc] and [1Ci] determined crystallographically, thermal ellipsoids are shown at 50% probability. | ||
The 4,5-dhp isomer of [1Fc] is, to the best of our knowledge, the first molecular structure of this intermediate. Based on the structure of [1Fc] the endo approach of the dienophile to [1] at CO-face of the molecule (Chart 1, Fig. 1) is favorable. The 1H NMR21 of the reaction product [1Fc] showed a mixture of two major 4,5-dhp products in a ratio of 80:12, and a minor product (∼8%) that appears to be the 1,4-dhp isomer due to slow 1,3-prototropic isomerization. The major 4,5-dhp product is the same found in the crystal state, however, based purely on these data the dienophile's exo approach at the Cl-face cannot be differentiated from the endo approach. However, DFT calculations found that exo approach of any dienophile to [1] at either Cl- or CO-face produced unrealistic activation energy (>50 kcal mol−1) and was therefore not considered. The endo approach of ViFc to [1] was found to be favorable at the CO-face versus the Cl-face of [1] based on DFT calculations (Fig. S5, ESI†).
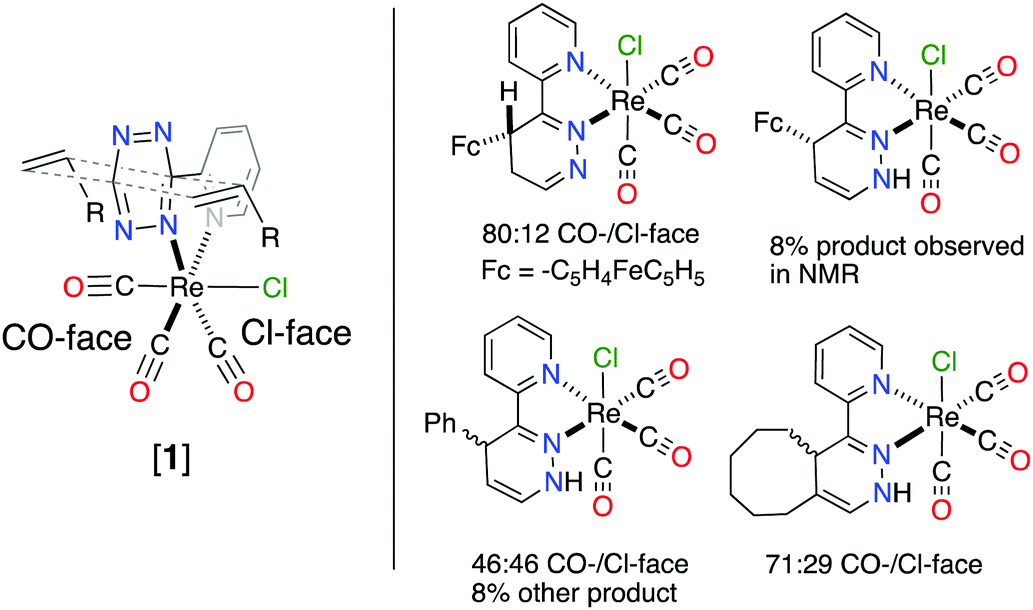 | ||
| Chart 1 Different facial approach of dienophile (left) and products from the reaction between [1] and olefin with observed product ratio from 1H NMR of reaction mixture (right). | ||
The crystal structure of [1TCO] and [1Ci] revealed that the 1,3-prototropic isomerization had occurred. The 1H NMR of the reaction mixture for [1Ci] confirmed the 1,4-dhp isomer in a ratio of 46:46 with 8% other products (Fig. S22, ESI†). According to DFT analysis the endo approach of Ci to [1] is nearly isoenergetic at both the faces, however, only the product of the endo CO-face addition was found in the crystal state (Fig. 3). Analysis of the reaction mixture from [1] + TCO also showed two similar products in a ratio of 71:29, and the 1,4-dhp isomer was assigned based on 1H NMR analysis, the molecular structure, and DFT calculations (Fig. S25, ESI†). These data show that the endo approach of dienophiles is favored to occur and only the 1,4-additon and not the 1,5-addition are observed.
We report the increased rate of the iEDDA addition of three dienophiles to the metallotetrazine [1]. The combination of the strong endo effect and the back-bonding from the tetrazine to the metal is thought to increase the rate of this reaction. Coordination of the rhenium(I) moiety to the tetrazine lowers the ΔS‡, while the nature of the dienophile shows the larger influence on the contribution of ΔH‡ to the transition state ΔG‡. The metallotetrazine [1] also allows for the facial approach of the dienophile to be prejudiced, albeit the Cl- and CO-face of [1] only imparts a small influence. Currently we are exploring this complex for its biological activity, and for immobilization of the complex onto solid supports to generate new electrocatalysts.
M. Schnierle and this project funded by Deutsche Forschungsgesellschaft SFB1333 C2. Calculations supported by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through Grant INST 40/467-1 FUGG for access to the Justus cluster. The authors gratefully acknowledge Dr Wolfgang Frey and Dr Ingo Hartenbach for advice and crystallographic measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- G. Linden, L. Zhang, F. Pieck, U. Linne, D. Kosenkov, R. Tonner and O. Vázquez, Angew. Chem., Int. Ed., 2019, 58, 12868–12873 CrossRef CAS; A. Rondon and F. Degoul, Bioconjugate Chem., 2020, 31, 159–173 CrossRef; T. Cañeque, S. Müller and R. Rodriguez, Nat. Rev. Chem., 2018, 2, 202–215 CrossRef; E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851 RSC.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC; L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS.
- W. Chen, D. Wang, C. Dai, D. Hamelberg and B. Wang, Chem. Commun., 2012, 48, 1736–1738 RSC; J. Sauer, D. K. Heldmann, J. Hetzenegger, J. Krauthan, H. Sichert and J. Schuster, Eur. J. Org. Chem., 1998, 2885–2896 CrossRef CAS.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646–9649 CrossRef CAS.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS; H. Wu and N. K. Devaraj, Top. Curr. Chem., 2015, 374, 3 CrossRef; A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770–3776 RSC.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS; K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298–304 CrossRef.
- W. Kaim, Coord. Chem. Rev., 2002, 230, 127–139 CrossRef CAS.
- M. Maekawa, T. Miyazaki, K. Sugimoto, T. Okubo, T. Kuroda-Sowa, M. Munakata and S. Kitagawa, Inorg. Chim. Acta, 2014, 410, 46–53 CrossRef CAS.
- M. Glöckle and W. Kaim, Angew. Chem., Int. Ed., 1999, 38, 3072–3074 CrossRef CAS; S. Kraft, E. Hanuschek, R. Beckhaus, D. Haase and W. Saak, Chem. – Eur. J., 2005, 11, 969–978 CrossRef.
- T. S.-M. Tang, H.-W. Liu and K. K.-W. Lo, Chem. Commun., 2017, 53, 3299–3302 RSC.
- K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS; E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef; M. S. Capper, H. Packman and M. Rehkämper, ChemBioChem, 2020, 21, 2111–2115 CrossRef.
- K. M. Knopf, B. L. Murphy, S. N. MacMillan, J. M. Baskin, M. P. Barr, E. Boros and J. J. Wilson, J. Am. Chem. Soc., 2017, 139, 14302–14314 CrossRef CAS.
- L. Suntrup, F. Stein, J. Klein, A. Wilting, F. G. L. Parlane, C. M. Brown, J. Fiedler, C. P. Berlinguette, I. Siewert and B. Sarkar, Inorg. Chem., 2020, 59(7), 4215–4227 CrossRef CAS; S. Sung, D. Kumar, M. Gil-Sepulcre and M. Nippe, J. Am. Chem. Soc., 2017, 139, 13993–13996 CrossRef; B. P. Sullivan, C. M. Bolinger, D. Conrad, W. J. Vining and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 1414–1416, 10.1039/C39850001414.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- A.-C. Knall, M. Hollauf and C. Slugovc, Tetrahedron Lett., 2014, 55, 4763–4766 CrossRef CAS.
- S. Choi, M. L. Personick, J. A. Bogart, D. Ryu, R. M. Redman and E. Laryea-Walker, Dalton Trans., 2011, 40, 2888–2897 RSC.
- I. Martínez-Montero, S. Bruña, A. M. González-Vadillo and I. Cuadrado, Macromolecules, 2014, 47, 1301–1315 CrossRef.
- J. A. Bravo, F. M. Raymo, J. F. Stoddart, A. J. P. White and D. J. Williams, Eur. J. Inorg. Chem., 1998, 2565–2571 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Containing experimental section, DFT calculations, crystallographic tables, and molecule coordinates determined by DFT description. CCDC 1991846–1991849 and 2008845. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03805a |
| This journal is © The Royal Society of Chemistry 2020 |