
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolated zirconium centres captured from aqueous solution: the structure of zirconium mandelate revealed from NMR crystallography†
James M.
Crosland
a,
Emily K.
Corlett
b,
Dave
Scapens
c,
Nathalie
Guillou
d,
Steven P.
Brown
 b and
Richard I.
Walton
*a
b and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: r.i.walton@warwick.ac.uk
bDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
cLuxfer MEL Technologies, Swinton, Manchester, M27 8LN, UK
dInstitut Lavoisier, UMR CNRS 8180, Université de Versailles St-Quentin-en-Yvelines, Université Paris-Saclay, 78035 Versailles, France
First published on 3rd August 2020
Abstract
Zirconium tetramandelate (2-hydroxy-2-phenylacetate) has been used for selective gravimetric analysis of zirconium for over 70 years. Herein its crystal structure is reported from synchrotron powder X-ray diffraction and 13C solid-state NMR. The complex is a rare example of isolated zirconium cations, rather than the clusters prevalent in aqueous solutions.
In 1947, Kumins reported the precipitation of zirconium by mandelic acid (2-hydroxy-2-phenylacetic acid),1 and most importantly, this was in the presence of a number of other metal cations, meaning it could be used as a selective and quantitative gravimetric analysis reagent for zirconium. Alternative methods of separation or determination of zirconium content include spectrophotometrically, using alizarin,2,3 or with other gravimetric reagents such as 8-hydroxyquinoline,4 or the first to be reported, cupferron.5 Kumins’ method arguably has become the most commonly used, subject to various improvements over time.6–9 The affinity of mandelic acid for zirconium is such that it can selectively operate in the presence of a range of other metal cations in solution including titanium, iron, vanadium, aluminium, chromium, thorium, cerium, tin, barium, calcium, copper, bismuth, antimony and cadmium.1 Kumins proposed that zirconium mandelate was most likely a tetramandelate, a simple salt containing four anions per zirconium atom: Zr(C6H5CH(OH)CO2H)4, but its crystal structure has never been determined.
A number of other authors expanded on Kumins’ work; Hahn found that the method was still applicable in strongly acidic solution, and that hafnium was also precipitated by mandelic acid.10 Feigl proposed a chelate-type structure whereby coordination of mandelate to zirconium took place through the α-hydroxy oxygen and the acidic carboxylate oxygen,11 an idea later confirmed by Hahn and Weber.12 It was subsequently found that the use of strongly acidic conditions is key in ensuring the formation of the tetramandelate; otherwise other compositions such as Zr(OH)(C6H5CH(OH)CO2H)3 result.13,14 In contrast to the idea of a simple ML4 complex, Larsen and Homeier formed the hypothesis that zirconium tetramandelate is in fact polymeric, despite the use of concentrated hydrochloric acid during synthesis that would solubilise Zr4+.15 This is consistent with the oxophilic nature of zirconium and its propensity for hydrolysis in aqueous solution.16 Indeed, the most studied solution species are based upon the so-called zirconium tetramer, a slightly distorted square array of zirconium atoms linked together by double hydroxy bridges. Such an arrangement was first found to be the basis of the structure of solid zirconium oxychloride,17 and then later found to also exist in aqueous solution.18 It is common in the solid state to find zirconium intricately linked with oxygen in chains or clusters, for example, the [Zr6O4(OH)4] octahedral clusters that form part of the structure of the archetypal metal–organic framework (MOF) UiO-66,19 and in other materials may also be present in individual clusters capped by carboxylate ligands.20,21 Chains featuring oxygen and anion-linked zirconium atoms can be found in the layered structures of the hydrated zirconium sulfates and nitrates.22–24 Chains of edge-sharing ZrO7 polyhedra are found in the structure of the MOF MIL-140,25 and chains of edge-shared ZrO8 units commonly feature in MOFs with phenolate linkers.26 Herein we present the synthesis and structural characterisation of zirconium tetramandelate, which presents the uncommon situation of isolated zirconium centres.
The reaction between zirconium salts and mandelic acid in acidic aqueous solution is rapid and produces a white crystalline solid. We tested a number of approaches to producing zirconium mandelate, including varying reaction temperature, the stereochemical form of the mandelic acid used and the choice of zirconium salt precursor. In all cases, the mandelic acid was present in slight excess, a 4.5× molar ratio relative to zirconium, and the reaction medium was a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of deionised water and 35% hydrochloric acid, respectively, in an attempt to prohibit the formation of condensed zirconium species by hydrolysis. Solid reagents were added to a Teflon™-lined autoclave, followed by the hydrochloric acid solution and stirred magnetically for 20 minutes. The autoclave was then heated in an oven at 90 °C for 18 hours and allowed to cool slowly (1 °C min−1) to room temperature. The most crystalline product was made from zirconium sulfate tetrahydrate and (R)-mandelic acid (Fig. S1 and S2, ESI†). It was observed that the initial precipitation of solid when using the sulfate was delayed compared to with other zirconium precursors such as zirconium oxychloride and zirconium acetylacetonate, suggesting a kinetic effect in the solution formation of the salt, possibly a reason for the increased quality of the product. Higher reaction temperatures resulted in decomposition of the mandelic acid and no crystalline product. The resulting solid was collected by vacuum filtration and washed with deionised water and acetone, and then dried at 75 °C in air before further study.
:1 mix of deionised water and 35% hydrochloric acid, respectively, in an attempt to prohibit the formation of condensed zirconium species by hydrolysis. Solid reagents were added to a Teflon™-lined autoclave, followed by the hydrochloric acid solution and stirred magnetically for 20 minutes. The autoclave was then heated in an oven at 90 °C for 18 hours and allowed to cool slowly (1 °C min−1) to room temperature. The most crystalline product was made from zirconium sulfate tetrahydrate and (R)-mandelic acid (Fig. S1 and S2, ESI†). It was observed that the initial precipitation of solid when using the sulfate was delayed compared to with other zirconium precursors such as zirconium oxychloride and zirconium acetylacetonate, suggesting a kinetic effect in the solution formation of the salt, possibly a reason for the increased quality of the product. Higher reaction temperatures resulted in decomposition of the mandelic acid and no crystalline product. The resulting solid was collected by vacuum filtration and washed with deionised water and acetone, and then dried at 75 °C in air before further study.
Initial characterisation by thermogravimetric analysis (Fig. S3, ESI†) confirmed the number of mandelate ligands per each zirconium cation to be 4. Fourier-transform infrared spectroscopy (Fig. S4, ESI†) showed that bands relating to the α-O–H stretch27 at 3438 cm−1, and that for the free carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1712 cm−1 as seen in the spectrum for mandelic acid, were absent for the zirconium mandelate product. A very broad band possibly from the coordinated α-OH was seen in the product spectrum centred at 3392 cm−1, and the presence of a pair of CO stretches at ∼1579 cm−1 and ∼1381 cm−1, separated by approx. 202 cm−1, was consistent with unidentate coordination of the carboxylate group.28 Such a bonding mode is also known for magnesium and barium mandelates, although in these cases the α-OH group does not participate in ligand-metal coordination.29 Zirconium K-edge EXAFS spectroscopy (Beamline B18, Diamond Light Source, Fig. S6, ESI†), detected only Zr–O correlations and no Zr–Zr correlations, as seen for the zirconium tetramer, in ZrOCl2·8H2O, for example, implying the presence of isolated zirconium centres in the material.
O stretch at 1712 cm−1 as seen in the spectrum for mandelic acid, were absent for the zirconium mandelate product. A very broad band possibly from the coordinated α-OH was seen in the product spectrum centred at 3392 cm−1, and the presence of a pair of CO stretches at ∼1579 cm−1 and ∼1381 cm−1, separated by approx. 202 cm−1, was consistent with unidentate coordination of the carboxylate group.28 Such a bonding mode is also known for magnesium and barium mandelates, although in these cases the α-OH group does not participate in ligand-metal coordination.29 Zirconium K-edge EXAFS spectroscopy (Beamline B18, Diamond Light Source, Fig. S6, ESI†), detected only Zr–O correlations and no Zr–Zr correlations, as seen for the zirconium tetramer, in ZrOCl2·8H2O, for example, implying the presence of isolated zirconium centres in the material.
Reaction at room temperature with stirring resulted in a product that was of inferior crystallinity compared to that made hydrothermally; this has been observed also in the crystallisation of other metal mandelates, such as those of divalent transition-metal cations.30 Even for the most crystalline sample, scanning electron microscopy showed the material to be made up of submicron-sized crystallites (Fig. S5, ESI†), which were not suitable for single crystal diffraction studies. High-resolution X-ray powder diffraction data were measured from a capillary-mounted sample of zirconium mandelate (Beamline I11, Diamond Light Source) and a model for the crystal structure of the material was determined (see ESI†). Successive improvements to structural models were aided by NMR crystallography,31,32 whereby measured spectra were compared to those calculated from the model structure. Adjustment of torsional angles within the ligands and Zr–C distances was made to give the best possible match between experimental and gauge-including projector augmented wave (GIPAW)33 calculated NMR spectra, and consistency with powder diffraction data (see ESI†). Fig. 1 shows the Rietveld fit to the powder XRD data, Table 1 the crystal structure parameters, and Fig. 2 the 1H & 13C cross polarisation (CP) magic-angle spinning (MAS) NMR spectra with the GIPAW calculated peak positions from the final crystal structure model. Not seen by PXRD directly was the location of the α-OH hydrogen atom, inferred from IR spectroscopy, the presence of which was confirmed by 1H NMR (Fig. 2). It was located by considering the possibility of hydrogen bond formation between α-OH groups and CO groups on neighbouring complexes in the unit cell.
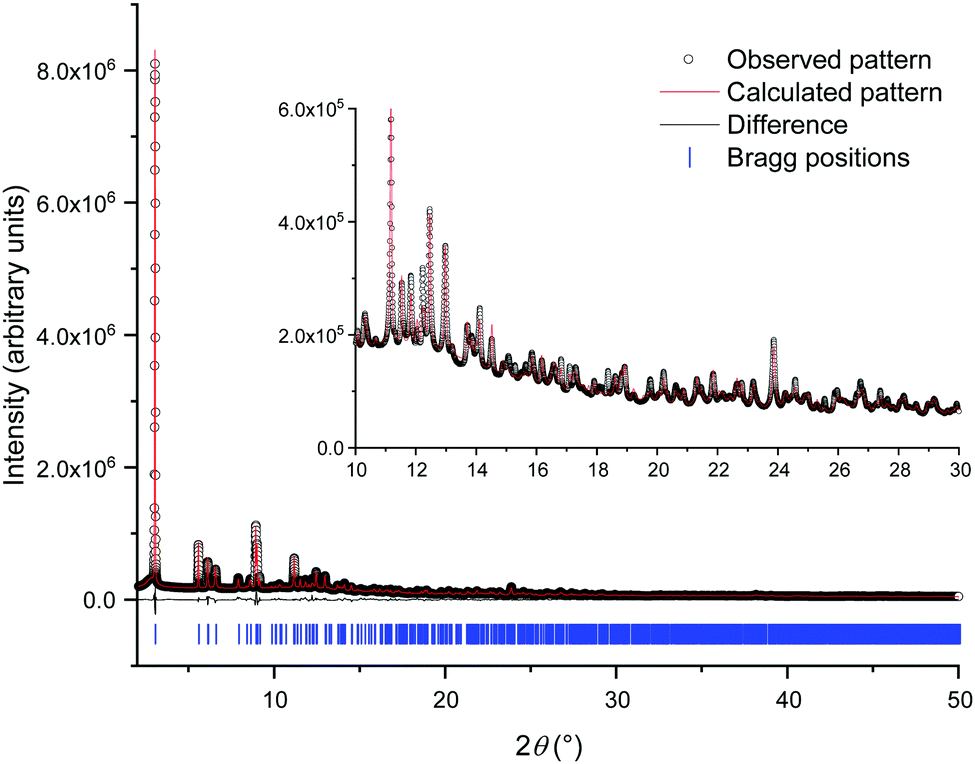 | ||
| Fig. 1 Final Rietveld fit to the PXRD data. The open circles are the measured data, the red line the fit and the black line the difference curve. The blue ticks show the position of allowed Bragg peaks. | ||
| Crystal structure determined from | Powder data |
| CCDC | 2004875 |
| M w/g mol−1 | 695.78 |
| Crystal system | Monoclinic |
| Space group (no.) | Pc (7) |
| a/Å | 17.0775(7) |
| b/Å | 5.63423(10) |
| c/Å | 16.9860(4) |
| β (°) | 114.885(3) |
| V/Å3 | 1482.62(8) |
| Z | 2 |
| ρ calc/mg m−3 | 1.558 |
| T/K | 298 |
| Radiation type | Synchrotron |
| λ/Å | 0.82445 |
| 2θ range/° | 2–50 |
| R p/% | 3.5 |
| R wp/% | 4.9 |
| R B/% | 1.7 |
| GoF | 17 |
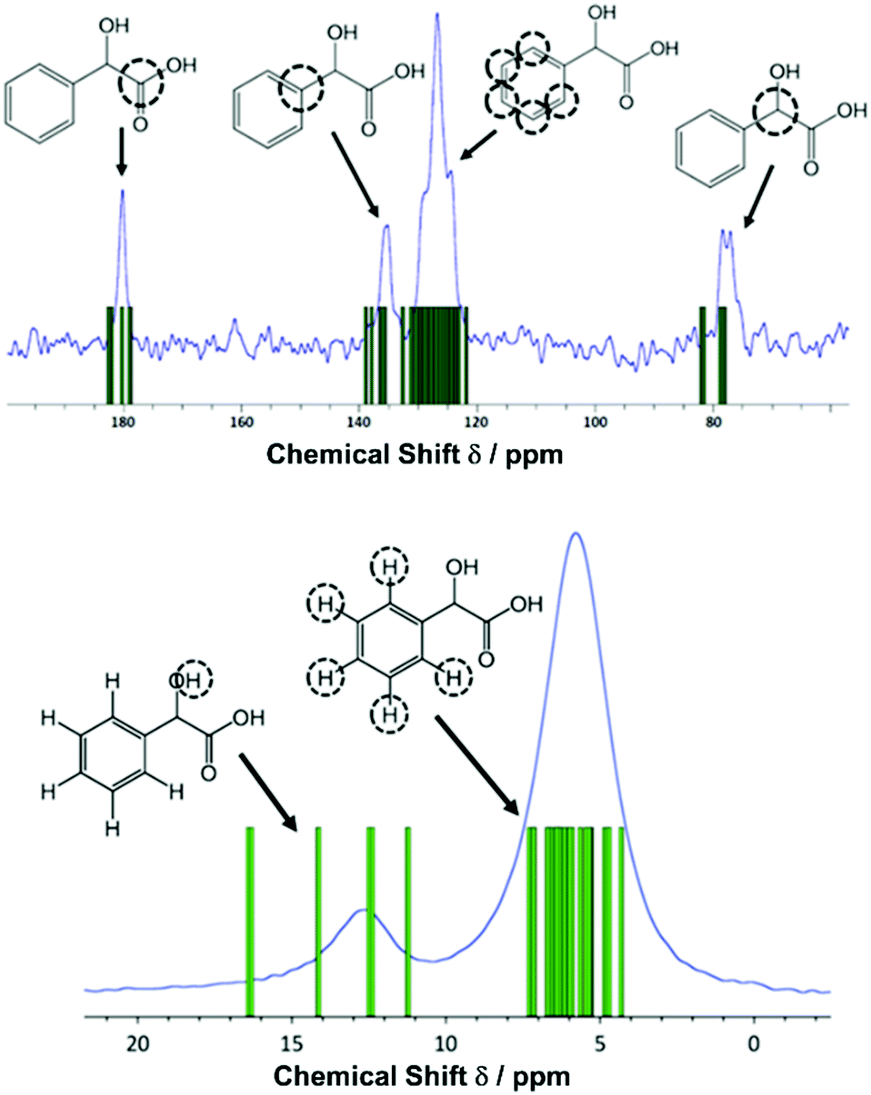 | ||
| Fig. 2 Solid-state NMR spectra (ν0(1H) = 600 MHz) of zirconium mandelate with assignments: top: 1H–13C CP MAS (νR = 12.5 kHz, contact time = 1.5 ms, 16 co-added transients for a recycle delay of 60 s). Bottom: 1H one-pulse (νR = 60 kHz, 4 co-added transients for a recycle delay of 103 s). The experimental data (blue plots) are overlaid with GIPAW calculated chemical shifts (green bars). The noticeable discrepancies for the α-OH protons is attributed to the close proximity of these to the zirconium cation. | ||
The crystal structure shows the presence of isolated zirconium centres coordinated by two oxygen donor atoms from each of the four mandelates (Fig. 3a). Each of the mandelate ligands is bonded via the oxygen of the α-hydroxy group and by one of the carboxylate oxygens. The second carboxylate oxygen is terminal. Upon inspection of the unit cell, it can be seen that one of the two zirconium centres is coordinated by only (R)-mandelate, and the other only (S)-mandelate ligands (Fig. 3b). Thus, racemisation of mandelic acid must take place in solution during the course of the reaction, as only (R)-mandelic acid (or (S)-) was used as a precursor, yet both stereochemical forms are present in the final product. Powder X-ray diffraction patterns of the obtained solids showed that the same material was produced regardless of whether the R or S form of mandelic acid was used in the synthesis (Fig. S1, ESI†).
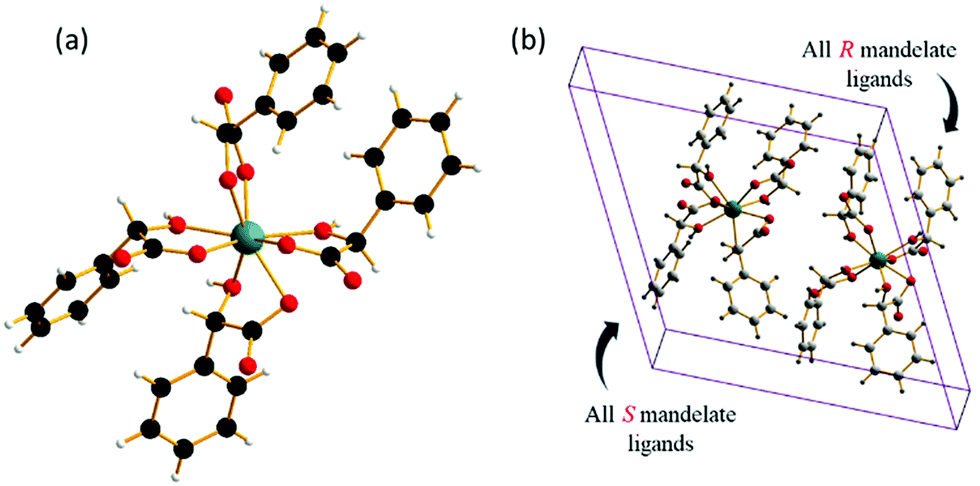 | ||
| Fig. 3 (a) View of the four mandelate ligands around each zirconium atom. The zirconium centre is coloured turquoise, with oxygen, hydrogen, and carbon atoms in red, white, and black, respectively. (b) A single unit cell of zirconium mandelate, with the different stereochemical forms of the mandelate ligands on each of the zirconium centres indicated. | ||
Mandelic acid is known to racemise readily in both acidic and basic solution,34 and the occurrence of this during synthesis of mandelate-containing materials to generate both stereochemical forms in the final product has been seen previously in mandelates of manganese, iron, cobalt, nickel and copper.30 In these compounds, which have layered structures, each metal(II) cation is surrounded by six oxygen atoms, four of which arise from two mandelates that both coordinate via the α-OH oxygen and a carboxylate oxygen. The remaining two oxygen atoms arise from the coordination of a further two mandelate ligands (each via a carboxylate oxygen), both of which bridge to other metal centres (Fig. 4a). A number of other metal mandelates are also known, including zinc and beryllium mandelate,35 neodymium mandelate,36 gadolinium, terbium, dysprosium and erbium mandelate,37 europium mandelate38 and rhodium mandelates.39 Coordination through both the carboxylate and α-hydroxy groups is common amongst these, and the mandelate may also bridge two metal centres, see for example Fig. 4b. A few structures are known for materials featuring a combination of mandelate and other ligands, and these contain similar bonding motifs for mandelate, for example in complexes of Cu2+ (Fig. 4c and d).40,41
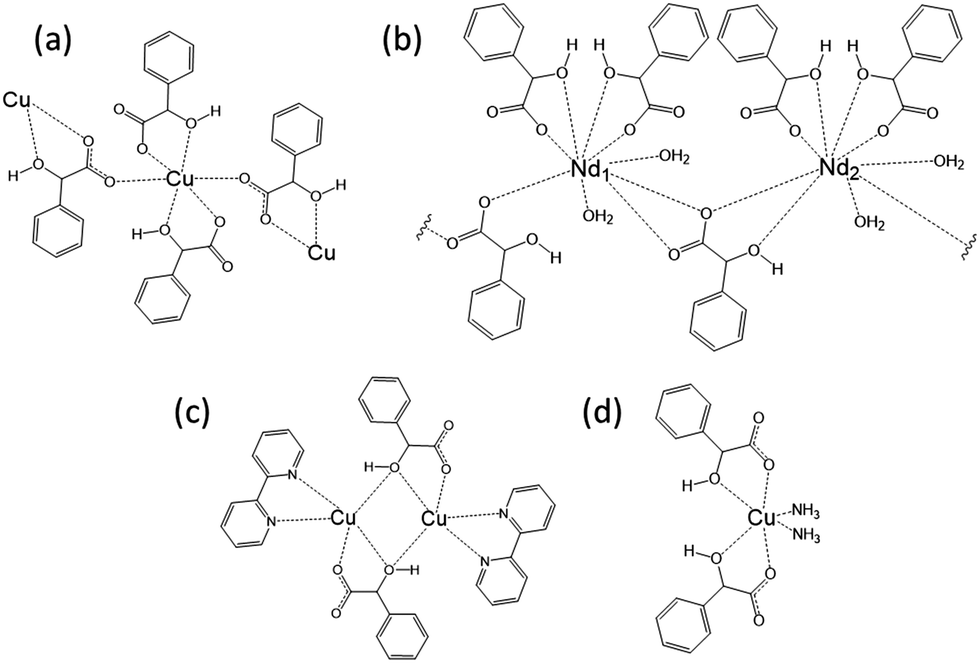 | ||
| Fig. 4 Examples of the coordination of mandelate in the solid state. (a) The arrangement of mandelate ligands around Cu(II) as described by Beghidja et al.30 (b) A schematic of the repeat unit of the layered structure of neodymium mandelate.36 (c) The dinuclear Cu complex [Cu(bpy)(mandelate)]2·8H2O.41 (d) The mononuclear Cu mandelate complex Cu(mandelate)2(NH3)2.40 | ||
In summary, we have demonstrated that with a strongly chelating ligand it is possible to avoid the hydrolysis of zirconium in aqueous solution, which would otherwise occur so readily leading to zirconium oxyclusters in the solid state. To our knowledge, complexes that contain isolated Zr centres have rarely been reported. In solution, EXAFS has been used to detect the existence of monomeric carbonate complexes under specific concentrations,42,43 and potassium salts of zirconium malonate and of zirconium 2,6-dicarboxypicolinate (the latter also containing a nitrogen donor) have been reported, where the monomeric zirconium complexes are separated by the additional cation.44 Our results suggest there is more to learn about the aqueous chemistry of zirconium and that with ligand design, novel coordination complexes and networks may be possible.
JMC thanks MEL Luxfer Technologies for award of a PhD studentship. We thank Diamond Light Source for access to diffraction and EXAFS beamlines, and the University of Warwick's Research Technology Platforms. The I11 beamtime was obtained through the Diamond Light Source Block Allocation Group award “Oxford/Warwick Solid State Chemistry BAG to probe composition–structure–property relationships in solids” (EE18786). We also thank Lu Jia for collection of HRPXRD data and Ehsan Ghadim for SEM imaging. EKC thanks EPSRC, AstraZeneca and Syngenta for a PhD studentship through the EPSRC Centre for Doctoral Training in Molecular Analytical Science, grant number EP/L015307/1. The research data underpinning this article can be accessed at: http://wrap.warwick.ac.uk/138907.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. A. Kumins, Anal. Chem., 1947, 19, 376–377 CrossRef CAS
.
- D. E. Green, Anal. Chem., 1948, 20, 370–372 CrossRef CAS
- J. F. Flagg, H. A. Liebhafsky and E. H. Winslow, J. Am. Chem. Soc., 1949, 71, 3630–3632 CrossRef CAS
- W. W. Wendlandt, Anal. Chim. Acta, 1957, 16, 129–134 CrossRef CAS
- W.M. Thornton and E. M. Hayden, Z. Anorg. Chem., 1914, 89, 377–382 CrossRef
- R. Belcher, A. Sykes and J. C. Tatlow, Anal. Chim. Acta, 1954, 10, 34–47 CrossRef CAS
- R. S. Barbieri, V. R. Terra, A. M. Netto and J. C. Rocha, Quim. Nova, 1995, 18, 40–43 CAS
- A. A. Astanina and E. A. Ostroumov, Chem. Abstr., 1951, 45, 6121 Search PubMed
- A. A. Astanina and E. A. Ostroumov, Zh. Anal. Khim., 1951, 6, 27–33 CAS
- R. B. Hahn, Anal. Chem., 1949, 21, 1579–1580 CrossRef CAS
-
F. Feigl, Chemistry of Specific, Sensitive, and Selective Reactions, Academic Press Inc., New York, 1949 Search PubMed
- R. B. Hahn and L. Weber, J. Am. Chem. Soc., 1955, 77, 4777–4779 CrossRef CAS
- R. B. Hahn and E. S. Baginski, Anal. Chim. Acta, 1956, 14, 45–47 CrossRef CAS
- R. N. Kapoor and R. C. Mehrotra, J. Am. Chem. Soc., 1958, 80, 3569–3573 CrossRef CAS
- E. M. Larsen and E. H. Homeier, Inorg. Chem., 1972, 11, 2687–2692 CrossRef CAS
- A. S. Solovkin and S. V. Tsvetkova, Russ. Chem. Rev., 1962, 31, 655–669 CrossRef
- A. Clearfield and P. A. Vaughan, Acta Crystallogr., 1956, 9, 555–558 CrossRef CAS
- G. M. Muha and P. A. Vaughan, J. Chem. Phys., 1960, 33, 194–199 CrossRef CAS
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- C. Hennig, S. Weiss, W. Kraus, J. Kretzschmar and A. C. Sheinhost, Inorg. Chem., 2017, 56, 2473–2480 CrossRef CAS PubMed
- C. Artner, M. Czakler and U. Schubert, Inorg. Chim. Acta, 2015, 432, 208–212 CrossRef CAS
- J. Singer and D. T. Cromer, Acta Crystallogr., 1959, 12, 719–723 CrossRef CAS
- D. B. McWhan and G. Lundgren, Inorg. Chem., 1966, 5, 284–289 CrossRef CAS
- P. Benard, M. Louer and D. Louer, J. Solid State Chem., 1991, 94, 27–35 CrossRef CAS
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef CAS PubMed
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC
- H. M. Badawi and W. Forner, Spectrochim. Acta, Part A, 2011, 78, 1162–1167 CrossRef PubMed
-
N. Nakamoto, Infrared and Raman
Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, New York, 4th edn, 1986 Search PubMed
- R. C. Silva, F. J. Caires, D. J. C. Gomes, T. A. D. Colman and M. Ionashiro, J. Therm. Anal. Calorim., 2014, 117, 217–221 CrossRef CAS
- A. Beghidja, S. Hallynck, R. Welter and P. Rabu, Eur. J. Inorg. Chem., 2005, 662–669 CrossRef CAS
- P. Hodgkinson, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118-119, 10–53 CrossRef CAS
- S. E. Ashbrook and D. McKay, Chem. Commun., 2016, 52, 7186–7204 RSC
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef
- A. N. Campbell and A. J. R. Campbell, J. Am. Chem. Soc., 1932, 54, 4581–4585 CrossRef CAS
- P. V. Khadikar, R. L. Ameria, M. G. Kekre and S. D. Chauhan, J. Inorg. Nucl. Chem., 1973, 35, 4301–4304 CrossRef CAS
- M. Babij, P. Starynowicz and A. Mondry, J. Mol. Struct., 2011, 1006, 672–677 CrossRef CAS
- M. Babij and A. Mondry, Opt. Mater., 2012, 34, 2061–2065 CrossRef CAS
- M. Babij and A. Mondry, J. Rare Earths, 2011, 29, 1188–1191 CrossRef CAS
- F. Pruchnik, B. R. James and P. Kvintovics, Can. J. Chem., 1986, 64, 936–939 CrossRef CAS
- H.-Q. Hao, W.-T. Liu, W. Tan, Z.-J. Lin and M.-L. Tong, CrystEngComm, 2009, 11, 967–971 RSC
- S. Balboa, R. Carballo, A. Castineiras, J. M. Gonzalez-Perez and J. Niclos-Gutierrez, Polyhedron, 2008, 27, 2921–2930 CrossRef CAS
- F. Takasaki, K. Fujiwara, Y. Nakajima, T. Nishikawa, H. Masu, M. Imanari, Y. Hidaka and N. Ogawa, Dalton Trans., 2015, 44, 645–652 RSC
- F. Takasaki, K. Fujiwara, T. Kikuchi, T. Tanno, Y. Nakajima, Y. Toyoda and N. Ogawa, Anal. Sci., 2017, 33, 1007–1012 CrossRef CAS PubMed
- G. R. Willey, T. J. Woodman, M. Fisher and M. G. B. Drew, Transition Met. Chem., 1998, 23, 467–471 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and further characterisation data. CCDC 2004875. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03647a |
| This journal is © The Royal Society of Chemistry 2020 |