
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A transition metal–gallium cluster formed via insertion of “GaI”†
Toby J.
Blundell
 a,
Laurence J.
Taylor
a,
Andrew J.
Valentine
a,
William
Lewis
b,
Alexander J.
Blake
a,
Jonathan
McMaster
a and
Deborah L.
Kays
*a
a,
Laurence J.
Taylor
a,
Andrew J.
Valentine
a,
William
Lewis
b,
Alexander J.
Blake
a,
Jonathan
McMaster
a and
Deborah L.
Kays
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Deborah.Kays@nottingham.ac.uk
bThe University of Sydney, F11 Eastern Ave, Sydney, NSW 2006, Australia
First published on 12th June 2020
Abstract
The reaction between a two-coordinate Co(II) diaryl complex and “GaI” affords 2,6-Pmp2C6H3CoGa3I5, in a new geometry for a heavier group 13-transition metal cluster. Experimental and computational investigations show that this compound is best described as a nido metalla-group 13 cluster.
Cluster compounds featuring metal–metal bonds are of significant interest as they serve as intermediate species between discrete molecules and bulk metals.1 Such compounds provide potentially valuable models for bulk-phase reactions, such as that seen in the reaction of Al13− with dioxygen.2 In addition to homometallic compounds, the development of mixed-metal cluster species offers potential insight into alloys and intermetallic phases.3
In previous work, the reaction between a 1,2-diaminoethane (en) solution of the Zintl phase precursor K4Ge9 with the coordinatively unsaturated m-terphenyl complex (2,6-Mes2C6H3)2Fe (Mes = 2,4,6-Me3C6H2) and 2,2,2-crypt (4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]-hexacosane) afforded [K(2,2,2-crypt)]3[Fe@Ge10]·2(en).4a The endohedral Zintl ion [Fe@Ge10]3− exhibits a pentagonal prismatic 10-atom germanium cage with an interstitial Fe atom in the central cavity. An analogous Co species, [Co@Ge10]3−, was published at approximately the same time by a different route.4b This synthesis of [Fe@Ge10]3− serves as one example of how low-coordinate transition metal diaryl complexes can facilitate a wide range of stoichiometric and catalytic reactvity.5
Further to this, the stoichiometric reactions of Fe(II) and Co(II) terphenyl complexes with carbon monoxide has furnished several new metal complexes6 and ketones7 through M–C bond breaking (M = Fe, Co) and C–C bond forming reactions. These have generated new stereogenic centres and, more recently, squaraines featuring a C4 unit entirely derived from CO through C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O scission and homologation.8 Given this reactivity, and the parallels in bonding that have been drawn between CO and the group 13 diyls such as the gallium monohalides GaX (X = Cl, Br, I),9 we were interested in the chemistry between m-terphenyl transition metal complexes and “GaI”.
O scission and homologation.8 Given this reactivity, and the parallels in bonding that have been drawn between CO and the group 13 diyls such as the gallium monohalides GaX (X = Cl, Br, I),9 we were interested in the chemistry between m-terphenyl transition metal complexes and “GaI”.
A freshly prepared suspension of excess “GaI” in toluene10 was reacted with 1 [(2,6-Pmp2C6H3)2Co, Pmp = C6Me5] in toluene at room temperature [Scheme 1(i)] to afford a deep red solution over a black suspension after 5 days. The removal of volatiles in vacuo and the extraction of the dark green/black solid into hexane yields 2,6-Pmp2C6H3CoGa3I5 (2, 8% yield) and into diethyl ether affords (2,6-Pmp2C6H3)2GaI (3, 21% yield). Performing the analogous reaction in a mixture of toluene and THF [Scheme 1(ii)] again yields 2 [now co-crystallised with 1 eq. GaI3(THF), denoted as 2·GaI3(THF), in 6% yield] but affords a different side product, [(2,6-Pmp2C6H3)2Ga]+[GaI4]− (4). The extractions left an insoluble black powder, which is likely to be elemental gallium, presumably originating from “GaI”.11 Linti has described the difficulties in rationalising the reactivity of the subvalent species “GaI”,12 which has been demonstrated to be a mixture of Ga(0), Ga(I) and Ga(III) species.13 Although we have been unable to determine an exact mechanism for the formation of the cluster 2 and its by-products from 1 and “GaI”, given the previous isolation of Ga3I5·(PEt3)3 from “GaI”,14 one could postulate a Ga3I5 moiety reacting with 1 to afford 2 and the organic radical 2,6-Pmp2C6H3˙. Given their structures, it is plausible that side products 3 and 4 may result from transmetallation and/or halide abstraction reactions involving Ga(III) species in the “GaI” mixture.15 In our hands, complexes 2–4 are consistently isolated, and these are the only compounds detected on repeated attempts.
 | ||
| Scheme 1 Reactions between 1 and “GaI”. Reaction conditions: (i) excess “GaI”, toluene, room temperature, 5 days; (ii) excess “GaI”, toluene/THF. Pmp = C6Me5. | ||
Single crystals of 3 were obtained from a saturated solution of 3 in diethyl ether (Fig. 1). 3 features a Ga(III) centre coordinated to two m-terphenyl ligands and an iodide donor. 3 is highly sterically encumbered, as demonstrated by the twisting of the flanking C6Me5 substituents. Thus, the C(7) ring is twisted from planarity; ortho-methyl C(17) sits 0.382 Å and meta-methyl C(14) sits 0.343 Å out of the best mean plane for the phenyl ring defined by C(7), presumably to avoid a steric clash with I(1) [C(17)⋯I(1) = 3.506(3) Å].
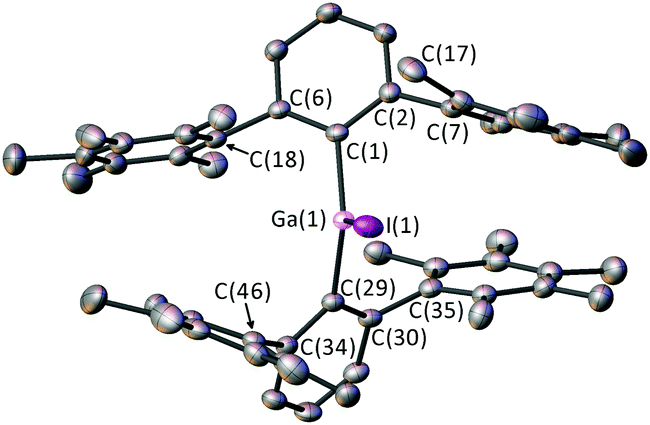 | ||
| Fig. 1 Crystal structure of 3 with displacement ellipsoids set at 50% probability. Hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Ga(1)–I(1) 2.5357(5), Ga(1)–C(1) 1.990(3), Ga(1)–C(29) 1.989(3), C(1)–Ga(1)–C(29) 131.1(1), C(1)–Ga(1)–I(1) 114.02(8), C(29)–Ga(1)–I(1) 114.89(9). | ||
4 (Fig. 2) is a rare example of an unsolvated two-coordinate gallium cation and is an analogue of the highly reactive alumenium cations (R2Al+).16 The mesityl-substituted complex [(2,6-Mes2C6H3)2Ga]+ has been stabilised by the highly weakly-coordinating Li[Al{OCH(CF3)2}4]−,17 but it appears from the isolation of 4 that such weakly coordinating anions18 are unnecessary if the ligands are sufficiently bulky. The [GaI4]− counterion is non-coordinating in 4, with a very large separation between Ga(1) and the closest I atom (6.4541(7) Å). The Ga–C bonds [1.920(4) Å] in 4 are ca. 3.5% shorter than for 3, and the C(1)–Ga(1)–C(29) angle [173.9(2)°] is deviated from linearity.
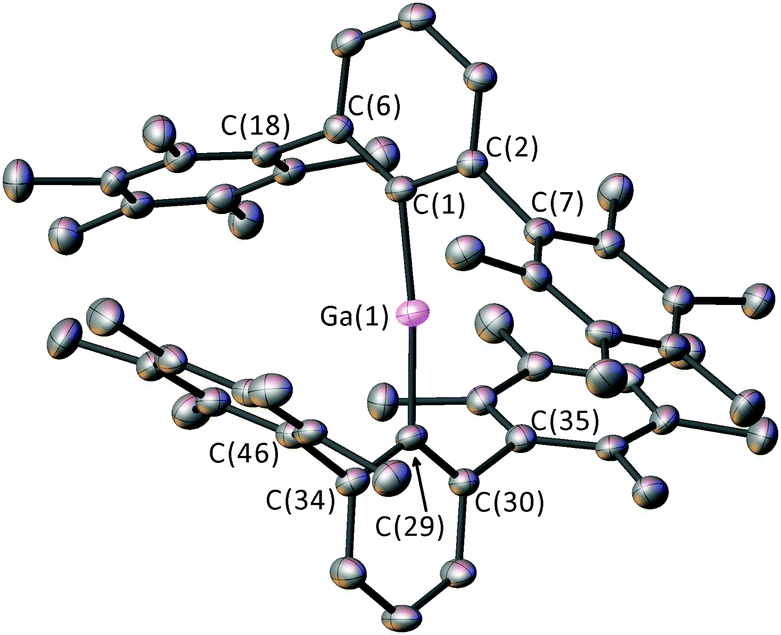 | ||
| Fig. 2 Crystal structure of 4 with displacement ellipsoids set at 50% probability. Hydrogen atoms, solvent of crystallisation and [GaI4]− omitted for clarity. Selected bond lengths (Å) and angles (°): Ga(1)–C(1) 1.920(4), Ga(1)–C(29) 1.920(4), C(1)–Ga(1)–C(29) 173.9(2). | ||
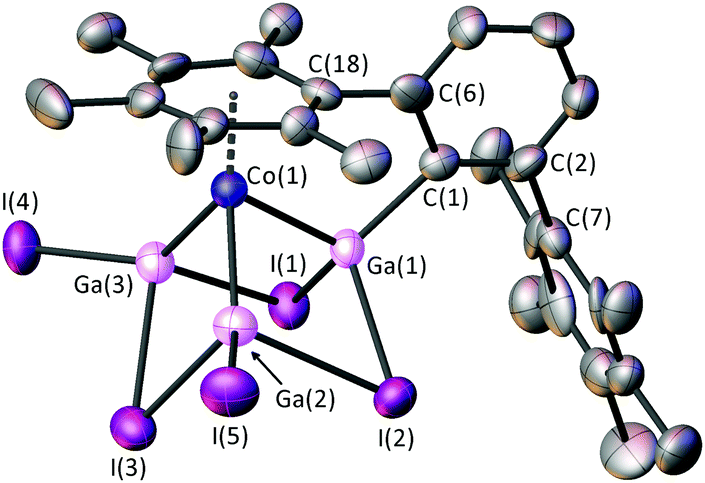 | ||
| Fig. 3 Crystal structure of 2·GaI3(THF) with displacement ellipsoids set at 50% probability. Hydrogen atoms and lattice GaI3·(THF) omitted for clarity. Selected bond lengths (Å): Co(1)–Ga(1) 2.374(3), Co(1)–Ga(2) 2.296(3), Co(1)–Ga(3) 2.277(3), Ga(1)–C(1) 1.96(2), Ga(1)–I(1) 2.661(2), Ga(1)–I(2) 2.698(2), Ga(2)–I(2) 2.862(3), Ga(3)–I(1) 2.917(3), Ga(2)–I(5) 2.532(3), Ga(3)–I(4) 2.540(2), Ga(1)⋯Ga(2) 2.966(3), Ga(2)⋯Ga(3) 2.948(3), Ga(1)⋯Ga(3) 3.017(3), Co(1)–C(arene) 2.07(2)–2.16(2). | ||
Single crystals of 2 [or 2·GaI3(THF)] can be obtained from the work-up of both of the reactions as detailed above (Scheme 1). 2 yielded poor quality crystals, but crystals of 2·GaI3(THF) obtained from hexane afforded higher quality diffraction data. The cobalt-containing moieties in the crystal structures of 2 and 2·GaI3(THF) are isostructural, therefore discussions will focus on the 2·GaI3(THF) data. The cluster adopts a cubane-type structure that contains four metal atoms and three I−, with one corner of the cube remaining unoccupied, in a similar manner as that found for the anionic moiety in [Li(THF)]+[R4Ga4I3]− (R = Si(SiMe3)3).12 There are close contacts between the Co centre and the three Ga atoms, with two of these exhibiting terminal Ga–I bonds, the other being bound terminally to the m-terphenyl ligand. The coordination sphere of the Co centre is completed by an interaction with one of the flanking aryl substituents of the terphenyl. We note that 2 is a 12e− cluster (2e− per GaI, 3e− from the Co-arene and 3e− from the bridging iodides) and adopts a nido geometry as predicted by Wade–Mingos rules.19
Within the cluster there are significant differences between the three Co–Ga distances, the longest being to Ga(1) [Co(1)–Ga(1) = 2.374(3) Å], with shorter bonds to Ga(2) and Ga(3) [Co(1)–Ga(2) = 2.296(3) Å, Co(1)–Ga(3) = 2.277(3) Å]. Concomitant with this, the bridging iodine distances to Ga(1) are shorter than those to Ga(2) or Ga(3) [2.661(2) and 2.698(2) Å vs. 2.862(3) and 2.917(3) Å]. These differences in Co–Ga and Ga–I distances could be due to the influence of the strong σ-donor C(1), or possibly the rigid steric restraints of the m-terphenyl ligand. The Co(1)–Ga(2) and Co(1)–Ga(3) distances are in the range exhibited by [Cp*Co(GaCp*)3][BArf]2 [Cp* = η5-C5Me5; Arf = 3,5-(CF3)2C6H3] [2.2798(13)–2.3168(16) Å].20
The Ga(1)–C(1) distance [1.96(2) Å] is similar to those for the paramagnetic cluster Ga11(C6H3-2,6-Mes2)4 [1.991(6), 1.996(6) Å], which features four-coordinate Ga–C(terphenyl) moieties.21 There is some variation within the lengths of the three Ga⋯Ga edges [2.948(3)–3.017(3) Å], the shortest of these being opposite to the terphenyl moiety [Ga(2)⋯Ga(3)]. We also note that the Ga–I bridging distances [2.661(2)–2.917(2) Å] are significantly longer than the terminal Ga–I distances [2.532(3) and 2.540(2) Å].
The average Co–C arene ring distances (2.12 Å) is shorter than those observed in metallaboranes 2-(η6-C6H5Me)-1-[(Me3Si)2CH]-2,1-CoCB10H10 (2.14 Å) and Co(η6-C6H5Me)(η5-7-NHtBu-7-CB10H10) (2.15 Å).22 There is a difference of ca. 0.09 Å between the shortest and longest Co–C distances in 2 due to the asymmetry of the η6-arene and the rigidity of the m-terphenyl ligand. There is also a pronounced tilting between the planes of the η6-arene and the Ga3 rings of 11.6(4)°. Whilst relatively common for Co,23 the incorporation of (η6-arene)metal units is unknown for heavier group 13-transition metal clusters.
X-ray photoelectron spectroscopy (XPS) measurements were performed on 2 to probe its electronic structure (see ESI,† Section S4). Complex 2 shows binding energies of Ga 2p3/2 (1119.1 eV) and Ga 3d5/2 (20.7 eV), with the kinetic energy for the Ga LMMa transition of 1061.7 eV and the Auger parameter = 1082.4 eV. Oxidation states for gallium iodides are difficult to assign from XPS parameters, potentially due to the electron-donating ability of iodide ligands or the polarisability of iodine,24b but 2 has broadly similar binding and kinetic energy values to other gallium iodides such as [Ga][GaI4] and GaI3.24
To support the formulation of 2, a series of geometry optimisations were carried out on models of 2 in the S = 0, 1, 2 spin-states using the ADF software package.25 Geometry optimisation of the model of 2 in the S = 0 state afforded a geometry that most closely matched the experimental crystal structure (ESI,† Section S6.1), with optimisation in higher spin states resulting in significant distortion (Fig. S8 and Table S4, ESI†). This supports a diamagnetic, S = 0 formulation of 2 the ground state, in line with our NMR spectroscopic measurements.
The bonding in 2 was analysed using the Quantum Theory of Atoms in Molecules (QTAIM)26 on a DFT optimised structure (see ESI,† Section S6.2).27 A plot of located bond paths, bond critical points (BCPs) and ring critical points (RCPs) for the central [CoGa3I5C] core of compound 2 is shown in Fig. 4. Bond paths and BCPs are found linking the 3 Ga atoms to the central Co, and between the Ga and I atoms, indicating attractive interactions (which are not, necessarily, classical 2c,2e− bonds). However, there is no evidence of Ga–Ga bonds, with an RCP located at the centre of each [CoGa2I] face of the molecule. We also note the substantial HOMO–LUMO gap calculated for this compound (1.9501 eV), consistent with the view that 2 is a cluster compound in which all bonding MOs are filled (see ESI,† Fig. S9).
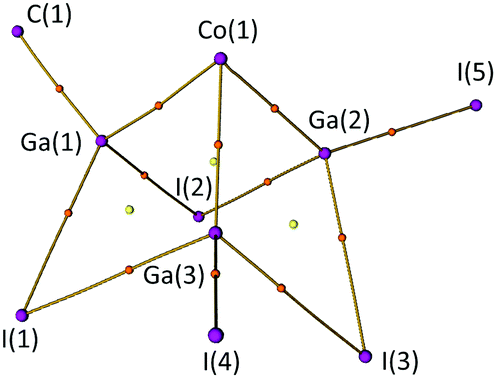 | ||
| Fig. 4 Plot of computed bond paths, nuclear attractors (NAs, purple), bond critical points (BCPs, orange) and ring critical points (RCPs, yellow) for the [CoGa3I5C] core of compound 2. Plot generated by the MultiWFN software package.35 | ||
A selection of QTAIM parameters calculated for the atomic interactions in the [CoGa3I5C] core of 2 is presented in Table S5 of the ESI.† Of note are the bond degree parameter (BD = HBCP/ρBCP) and the ratio |VBCP|/GBCP.28,29 In 2, all bonds within the cluster have 1 < |VBCP|/GBCP < 2 and negative BD values, indicating that they are transit shared shell interactions with a significant degree of covalency (which is typical for heavy-element interactions).28,29 Delocalisation indices (DIs) have also been computed (ESI,† Table S5). These are a measure of the degree of electron sharing between two atoms30 which has been proposed as a quantum mechanical measure of the classical concept of “bond order”.31 Here, we observe the terminal Ga–C (0.717) and Ga–I (0.805, 0.813) bonds have substantially higher DIs than the bridging Ga–I bonds (0.443–0.592). We also note that Co(1)–Ga(2) and Co(1)–Ga(3) have higher DIs than Co(1)–Ga(1). This suggests that Ga(2) and Ga(3) form stronger bonds to Co(1) than Ga(1), which correlates with the crystallographically observed difference in bond lengths. Conversely, Ga(1) appears to form stronger bonds with I(1) and I(2) than either Ga(2) or Ga(3) (see Table S5, ESI†).
From the experimental bond lengths (Fig. 3) and computed delocalisation indices (Table S5, ESI†), we can determine approximate bond orders for the cluster, which allows us to assign formal oxidation states using the methods outlined by Karen.32 If we consider each Co–Ga, Ga–C, and terminal Ga–I to have a bond order of 1, while the bridging Ga–I interactions have a bond order of 0.5, we can assign charges using Allen electronegativities33 and arrive at formal oxidation states of (−I) for C(1) and all I atoms, (+III) for the Ga atoms, and (−III) for the Co. This is consistent with the observed neutrality of 2, as well as its diamagnetism, as it assigns 12 valence electrons to Co to fill both the 4s and 3d orbitals. This view of the oxidation states is supported by the DDEC6 computed atomic charges,34 which show large partial positive charges on the Ga atoms (+0.321 to +0.394) and a substantial negative charge on the Co (−0.434), consistent with the assigned (+III)/(−III) oxidation states. Full data on the atomic charges is given in the ESI,† Table S6.
In conclusion, a metal-gallium cluster complex (2,6-Pmp2C6H3CoGa3I5, 2) featuring a previously unseen coordination geometry has been synthesized via the reaction between (2,6-Pmp2C6H3)2Co and “GaI”. It is formed alongside Ga-containing by-products, which are all formed consistently under the reaction conditions employed. Based on X-ray diffraction measurements, QTAIM analysis, and atomic charge calculations we conclude that the bonding in 2 is best described as a nido metalla-group 13 cluster with formal oxidation states of Ga(III) and Co(−III).
This work was supported by the Engineering and Physical Sciences Research Council [grant number EP/R004064/1 to D. L. K. and EP/K005138/1 for the Kratos LiPPS XPS instrument] and the University of Nottingham. We thank Dr Emily Smith at the Nanoscale and Microscale Research Centre (nmRC), University of Nottingham for acquiring the XPS spectra and data interpretation, Dr Mick Cooper at the University of Nottingham and EPSRC NMSF at Swansea University for mass spectrometry, Mr Stephen Boyer (London Metropolitan University) for elemental microanalysis and MEDAC Ltd for ICP analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. Linti, H. Schöckel, W. Uhl and N. Wiberg, in Molecular Clusters of the Main Group Elements, ed. M. Driess and H. Nöth, Wiley-VCH, Weinheim, 2004, ch. 2.3, p. 126 Search PubMed; (b) S. Schulz, Chem. – Eur. J., 2010, 16, 6416 CrossRef CAS PubMed; (c) H. Schnöckel and A. Schnepf, in The Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities, ed. S. Aldridge and A. J. Downs, John Wiley & Sons Ltd, Chichester, 2011, ch. 7, p. 402 Search PubMed.
- R. Burgert, H. Schnöckel, A. Grubisic, X. Li, S. T. Stokes, K. H. Bowen, G. F. Ganteför, B. Kiran and P. Jena, Science, 2008, 319, 438 CrossRef CAS PubMed.
- See, for example: B. J. L. Witzel, W. Klein, J. V. Dums, M. Boyko and T. F. Fässler, Angew. Chem., Int. Ed., 2019, 58, 12908 CrossRef CAS PubMed; C. Ganesamoorthy, J. Weßing, C. Kroll, R. W. Seidel, C. Gemel and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7943 CrossRef PubMed; T. Bollermann, M. Molon, C. Gemel, K. Freitag, R. W. Seidel, M. von Hopffgarten, P. Jerabek, G. Frenking and R. A. Fischer, Chem. – Eur. J., 2012, 18, 4909 CrossRef PubMed; T. Cadenbach, C. Gemel and R. A. Fischer, Angew. Chem., Int. Ed., 2008, 47, 9146 CrossRef PubMed.
- (a) B. Zhou, M. S. Denning, D. L. Kays and J. M. Goicoechea, J. Am. Chem. Soc., 2009, 131, 2802 CrossRef CAS PubMed; (b) J.-Q. Wang, S. Stegmaier and T. F. Fässler, Angew. Chem., Int. Ed., 2009, 48, 1998 CrossRef CAS PubMed.
- (a) D. L. Kays, Dalton Trans., 2011, 40, 769 RSC; (b) P. P. Power, Chem. Rev., 2012, 112, 3482 CrossRef CAS; (c) L. J. Taylor and D. L. Kays, Dalton Trans., 2019, 48, 12365 RSC.
- C. Ni and P. P. Power, Chem. Commun., 2009, 5543 RSC.
- B. M. Gridley, A. J. Blake, A. L. Davis, W. Lewis, G. J. Moxey and D. L. Kays, Chem. Commun., 2012, 48, 8910 RSC.
- H. R. Sharpe, A. M. Geer, L. J. Taylor, B. M. Gridley, T. J. Blundell, A. J. Blake, E. S. Davies, W. Lewis, J. McMaster, D. Robinson and D. L. Kays, Nat. Commun., 2018, 9, 3757 CrossRef PubMed.
- See, for example: K. K. Pandey and S. Aldridge, Inorg. Chem., 2011, 50, 1798 CrossRef CAS PubMed; K. K. Pandey, P. Patidar and S. Aldridge, J. Phys. Chem. A, 2010, 114, 12099 CrossRef PubMed; J. A. Gámez, R. Tonner and G. Frenking, Organometallics, 2010, 29, 5676 CrossRef; K. K. Pandey, P. Patidar and H. Braunschweig, Inorg. Chem., 2010, 49, 6994 CrossRef; D. Vidovic and S. Aldridge, Angew. Chem., Int. Ed., 2009, 48, 3669 CrossRef; N. D. Coombs, D. Vidovic, J. K. Day, A. L. Thompson, D. D. Le Pevelen, A. Stasch, W. Clegg, L. Russo, L. Male, M. B. Hursthouse, D. J. Willock and S. Aldridge, J. Am. Chem. Soc., 2008, 130, 16111 CrossRef PubMed; N. R. Bunn, S. Aldridge, D. L. Kays, N. D. Coombs, A. Rossin, D. J. Willock, J. K. Day, C. Jones and L.-L. Ooi, Organometallics, 2005, 24, 5891 CrossRef; S. Aldridge, A. Rossin, D. L. Coombs and D. J. Willock, Dalton Trans., 2004, 2649 RSC; N. R. Bunn, S. Aldridge, D. L. Coombs, A. Rossin, D. J. Willock, C. Jones and L.-L. Ooi, Chem. Commun., 2004, 1732 RSC; H. Braunschweig, M. Colling, C. Hu and K. Radacki, Angew. Chem., Int. Ed., 2002, 41, 1359 CrossRef.
- M. L. H. Green, P. Mountford, G. J. Smout and S. R. Speel, Polyhedron, 1990, 9, 2763 CrossRef CAS.
- M. R. Lichtenthaler, F. Stahl, D. Kratzert, L. Heidinger, E. Schleicher, J. Hamann, D. Himmel, S. Weber and I. Krossing, Nat. Commun., 2015, 6, 8288 CrossRef CAS.
- W. Köstler and G. Linti, Angew. Chem., Int. Ed. Engl., 1997, 36, 2644 CrossRef.
- B. J. Malbrecht, J. W. Dube, M. J. Willans and P. J. Ragogna, Inorg. Chem., 2014, 53, 9644 CrossRef CAS PubMed.
- A. Schnepf, C. Doriat, E. Möllhausen and H. Schnöckel, Chem. Commun., 1997, 2111 RSC.
- See, for example: (a) A. M. Feliz, D. A. Dickie, I. S. Horne, G. Page and R. A. Kemp, Inorg. Chem., 2012, 51, 4650 CrossRef; (b) P. A. Gray, J. W. Saville, K. D. Krause, N. Burford, R. McDonald and M. J. Ferguson, Can. J. Chem., 2017, 95, 346 CrossRef CAS; (c) F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Inorg. Chem., 2007, 46, 7215 CrossRef CAS PubMed.
- See, for example: K.-C. Kim, C. A. Reed, G. S. Long and A. Sen, J. Am. Chem. Soc., 2002, 124, 7662 CrossRef CAS PubMed; J. D. Young, M. A. Khan and R. J. Wehmschulte, Organometallics, 2004, 23, 1965 CrossRef; M. Kessler, C. Knapp and A. Zogaj, Organometallics, 2011, 30, 3786 CrossRef.
- R. J. Wehmschulte, J. M. Steele, J. D. Young and M. A. Khan, J. Am. Chem. Soc., 2003, 125, 1470 CrossRef CAS PubMed.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982 CrossRef CAS PubMed.
- (a) K. Wade, J. Chem. Soc. D, 1971, 792 RSC; (b) D. M. P. Mingos, Nat. Phys. Sci., 1972, 236, 99 CrossRef CAS.
- T. Bollerman, A. Puls, C. Gemel, T. Cadenbach and R. A. Fischer, Dalton Trans., 2009, 1372 RSC.
- N. J. Hardman, R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 2667 CrossRef CAS PubMed.
- (a) W. Quintana, R. L. Ernest, P. J. Carroll and L. G. Sneddon, Organometallics, 1988, 7, 166 CrossRef; (b) J. C. Jeffery, V. N. Lebedev and F. G. A. Stone, Inorg. Chem., 1996, 35, 2967 CrossRef CAS.
- (a) T. Nguyen, W. A. Merrill, C. Ni, H. Lei, J. C. Fettinger, B. D. Ellis, G. J. Long, M. Brynda and P. P. Power, Angew. Chem., Int. Ed., 2008, 47, 9115 CrossRef CAS PubMed; (b) C. Jones, C. Schulten, R. P. Rose, A. Stasch, S. Aldridge, W. D. Woodul, K. S. Murray, B. Moubaraki, M. Brynda, G. La Macchia and L. Gagliardi, Angew. Chem., Int. Ed., 2009, 48, 7406 CrossRef CAS PubMed; (c) T. R. Dugan, X. Sun, E. V. Rybak-Akimova, O. Olatunji-Ojo, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2011, 133, 12418 CrossRef CAS PubMed.
- (a) J. L. Bourque, P. D. Boyle and K. M. Baines, Chem. – Eur. J., 2015, 21, 9790 CrossRef CAS PubMed; (b) J. L. Bourque, M. C. Biesinger and K. M. Baines, Dalton Trans., 2016, 45, 7678 RSC.
- (a) C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391 Search PubMed; (b) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- (a) R. F. W. Bader, Chem. Rev., 1991, 91(5), 893 CrossRef CAS; (b) R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, USA, 1994 Search PubMed; (c) P. L. A. Popelier, Atoms in Molecules: An Introduction, Prentice Hall, UK, 2000 Search PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed; (c) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- E. Espinosa, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- C. Lepetit, P. Fau, K. Fajerwerg, M. L. Kahn and B. Silvi, Coord. Chem. Rev., 2017, 345, 150 CrossRef CAS.
- X. Fradera, M. A. Austen and R. F. W. Bader, J. Phys. Chem. A, 1999, 103, 304 CrossRef CAS.
- C. Outeiral, M. A. Vincent, A. M. Pendás and P. L. A. Popelier, Chem. Sci., 2018, 9, 5517 RSC.
- (a) P. Karen, P. McArdle and J. Takats, Pure Appl. Chem., 2014, 86, 1017 CAS; (b) P. Karen, Angew. Chem., Int. Ed., 2015, 54, 4716 CrossRef CAS PubMed.
- L. C. Allen, J. Am. Chem. Soc., 1989, 111, 9003 CrossRef CAS.
- (a) T. A. Manz and N. G. Limas, RSC Adv., 2016, 6, 45727 RSC; (b) T. A. Manz and N. G. Limas, RSC Adv., 2016, 6, 47771 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details for the synthesis, XPS, DFT calculations, crystallographic data and CIF files. CCDC 2004404–2004408. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03559a |
| This journal is © The Royal Society of Chemistry 2020 |