
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the antimicrobial activity of low molecular weight hydrogels using dopamine autoxidation†
Emily R.
Cross
 a,
Sophie M.
Coulter
b,
Ana M.
Fuentes-Caparrós
a,
Kate
McAulay
a,
Ralf
Schweins
c,
Garry
Laverty
*b and
Dave J.
Adams
*a
a,
Sophie M.
Coulter
b,
Ana M.
Fuentes-Caparrós
a,
Kate
McAulay
a,
Ralf
Schweins
c,
Garry
Laverty
*b and
Dave J.
Adams
*a
aSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
bSchool of Pharmacy, Queen's University Belfast, Medical Biology Centre, 97 Lisburn Road, Belfast, BT9 7BL, Northern Ireland, UK. E-mail: garry.laverty@qub.ac.uk
cLarge Scale Structures Group, Institut Laue – Langevin, 71 Avenue des Martyrs, CS 20156, 38042 Grenoble Cedex 9, France
First published on 10th June 2020
Abstract
We present a method to trigger the formation of dipeptide-based hydrogels by the simple addition of dopamine. Dopamine undergoes oxidation in air, reducing the pH to induce gelation. The production of polydopamine and release of reactive oxygen species such as hydrogen peroxide confers antimicrobial activity. Gel stiffness can be controlled by modulating the initial starting pH of the gelator solution. We can use this method to tune the antimicrobial activity of the gels, with gels that are less stiff demonstrating increased bactericidal efficacy against Gram-positive bacteria.
The use of medical implants such as intravascular and urinary catheters, heart valve prostheses, artificial hip joints, dental implants and intraocular lenses have increased in recent years due to both an ageing population and an improvement in medical healthcare.1,2 Following the increase in medical implant usage, rates of infection due to bacterial adherence followed by biofilm formation on the implant surface has also increased.2–4 Gels with antimicrobial properties can be used to coat medical implants to reduce the formation of microbial biofilms which lead to infectious disease.5–7
The biocompatible nature of low molecular weight gelators (LMWG) makes them useful building blocks for antimicrobial gels.8 Antimicrobial gels can either be formed by encapsulation of a known antimicrobial agent into the gel or by developing the hydrogel network to possess antimicrobial activity. Encapsulation of silver and gold nanoparticles can lead to antimicrobial activity.3,9–11 However, the mechanism which leads to these antimicrobial properties is unclear.8 It is possible that bacterial membrane damage is caused by the generation of reactive oxygen species binding to bacterial cell membranes.8 Furthermore, antibiotics such as amoxicillin,12 vancomycin,13 and gentamicin,14 can be also encapsulated into the gel network. Controlled release of these antibiotics into the area surrounding the biomaterial causes bacteria death.12,14,15
However, antimicrobial gels formed by the physical encapsulation of antimicrobial agents can be problematic.16 Accumulation of nanoparticles can be toxic and can cause various health problems and such formulations are prone to a high proportion of burst release.16–18 In order to prevent antimicrobial resistance development in the use of antibiotic encapsulated gels, it is critical that a sufficient concentration of antibiotic is released, that is above clinically effective concentrations for a sufficient period of time.19 At least 1% of bacteria in the stationary phase of biofilm development are tolerant to antibiotics; over time, the number of resistant microorganisms within the stationary phase can increase due to exposure to sub-therapeutic concentrations of antibiotics and therefore greater resistance can develop.20
Recently, there has been growing interest in the use of dopamine for surface modification to prevent microbial fouling.21 The production of polydopamine has been reported to display antimicrobial activity owing to the auto-oxidation of catechol in the presence of molecular oxygen to form semiquinone and quinone. During this oxidation process reactive oxygen species such as superoxide anions and hydrogen peroxide (H2O2) are generated as by-products.22 Despite widespread interest in the use of polydopamine as an antimicrobial agent, there is still dispute as to the exact mechanism by which it develops, however, it is known that the self-polymerization process requires alkaline conditions and the presence of oxygen.23 H2O2 is well known for its antimicrobial activity,3,24 and the sustained release of H2O2 generated as a result of the dopamine self-polymerization process has demonstrated broad spectrum antimicrobial activity against Gram-positive and Gram-negative organisms.22
One group of gels that has been used to form antimicrobial gels are LMWGs. Specifically here, we work with LMWG with a hydrophobic group attached to a dipeptide; this class can be tailored to specific requirements.25 Hydrogels are formed when these gelators self-assemble into one-dimensional aggregates, most commonly fibres, which entangle and immobilise water.26 The properties of the gels are determined by the nature of the fibres as well as the cross-links and microstructure.
The self-assembly of the gelators to form LMWG is controlled by a decrease in the solubility of the gelator in solution. There are many different methods to reduce the solubility of the gelators.26,27 Here, we use a gelator where the solubility is controlled by pH. At high pH, the gelator disperses in water. When the pH is lowered, gelation occurs. Conventionally, this would be carried out using a mineral acid or via in situ hydrolysis of a lactone. Here, we show that the auto-oxidation of dopamine can be used to trigger a reduction in the pH and hence the solubility of gelator 1 in solution (Fig. 1). We also show how we can control the antimicrobial properties of a gel by controlling the rate of gelation and gel stiffness.
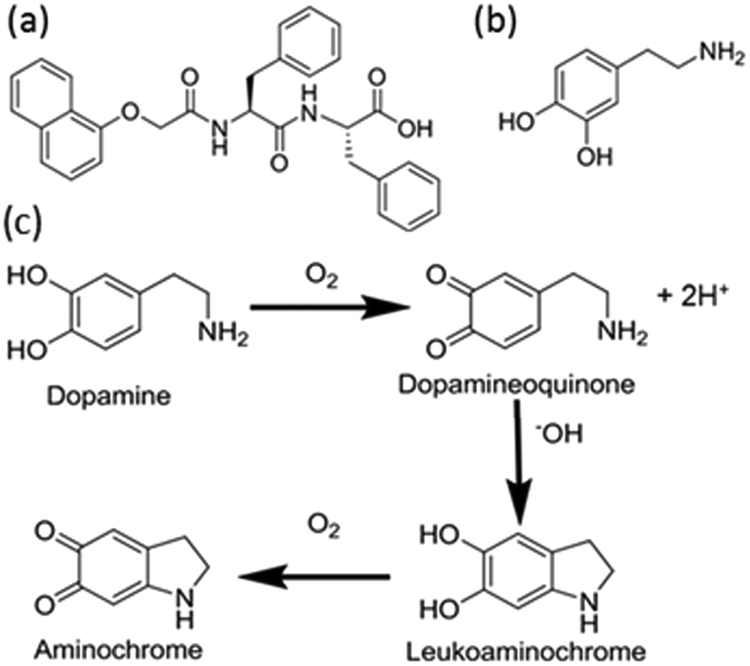 | ||
| Fig. 1 Chemical structures of (a) gelator 1 and (b) dopamine. (c) Oxidation pathway of dopamine, initial oxidation step produces protons which are used here to lower the pH of the bulk gelator solution, triggering gelation. A Michael addition and further oxidation forms the brown pigment leukoaminochrome and aminochrome. | ||
Dopamine oxidises in air to produce dopaminoquinone and protons (Fig. 1c).28,29 The quinone undergoes intramolecular Michael addition forming aminochrome.29 This aminochrome can further polymerise into neuromelanin which is dark brown in colour. Several studies on the kinetic behaviour of dopamine autoxidation have been carried out.30–32 The summary of these kinetic studies suggest that the autoxidation of dopamine is strongly pH-dependent with the rate of autoxidation faster at higher pH.32 The increase in rate is due to the abundance of hydroxide ions which are required in the intramolecular Michael addition step. Babbit, Lloyd and more recently Salomäki et al. report the rate constant for the formation of aminochrome is first order.30,31,33 At low pH, the Michael addition is the rate limiting step.30 If we are able to control the rate of dopamine oxidation, we should be able to control the gelation kinetics of self-assembly.
Solutions of 1 were prepared at different initial pH values of 7, 8, 9 and 10 at a concentration of 8 mg mL−1. The apparent pKa of 1 is 6.8 and hence 1 is expected to be deprotonated in all these solutions (we refer to an apparent pKa since this is the pKa of the aggregate, see discussion in Fig. S3, ESI†). All solutions were viscous, as expected as 1 assembles into worm-like micelles at these pH values.34 In all cases, no gelation occurs with time for these solutions. However, a gel was formed when dopamine (3 mg mL−1) was added to a solution of gelator 1 (16 mg in 2 mL) at pH 8. To determine whether the effect of gelation was due to the oxidation products of dopamine or the electrostatic interactions between dopamine and the gelator in solution, two gels were prepared, one under a nitrogen atmosphere and the other under air. After 16 hours, the solution of 1 and dopamine which was under nitrogen did not form a gel whereas a gel was formed from the mixture in air. Furthermore, a brown colour gradient could be observed with a dark brown colour at the gel and air interface which faded into the bulk solution as shown in Fig. 2. This led us to conclude that the oxidation of dopamine in air triggers gelation.
 | ||
| Fig. 2 Image of gelator solutions of 1 (8 mg mL−1, 2 mL sample) with dopamine (6 mg) under an atmosphere of (a) N2 and (b) air. The left-hand sample in both (a) and (b) show the solution before gelation and the right-hand sample shows the solution after 16 hours. Oxidation products of dopamine cause brown colour when the solution is left in air, which is absent under N2. | ||
To investigate the effect of pH on the rate of dopamine oxidation and gelation, dopamine (3 mg mL−1) was added to stock solutions of 1 (8 mg mL−1) at a pH of 7, 8, 9 or 10 (for full experimental see ESI†). After 16 hours, in all cases, the sample could be inverted without flow. Again, the dark brown colour gradient starting from the gel/air interface was observed. For the samples at the higher initial pH, the brown colour extended further into the bulk solution; for the sample starting at pH 7 the brown colour was only observed near the gel–air interface. We assume that this brown colour corresponds to the presence of dopamine oxidation products and polydopamine which are highly coloured.35 In all cases, after 16 hours, the pH of the solution was between 7.0 and 7.3.
Rheological analysis was carried out to further investigate the effect of the initial pH of the gelator solution pH on the final gel properties. Frequency and strain sweeps were used to determine the stiffness and breaking points of the gels (Fig. 3a). All gels show frequency independence at the measured 0.1–100 rad s−1 angular frequency. Remarkably, a linear relationship between the gel stiffness and the pH of the starting solution was observed (Fig. 3b). We describe this linear relationship as an effect of the first order rate kinetics of dopamine oxidation.30 As the pH decreases, the rate of dopamine oxidation is slower. The resulting gels are stiffer as the oxidation rate is slower (Fig. 3b). This phenomenon of gel stiffness being controlled with the rate of gelation has been previously seen in other low molecular weight hydrogels.36 There are small changes to the rheology and pH of the gels over time (Fig. S3, ESI†).
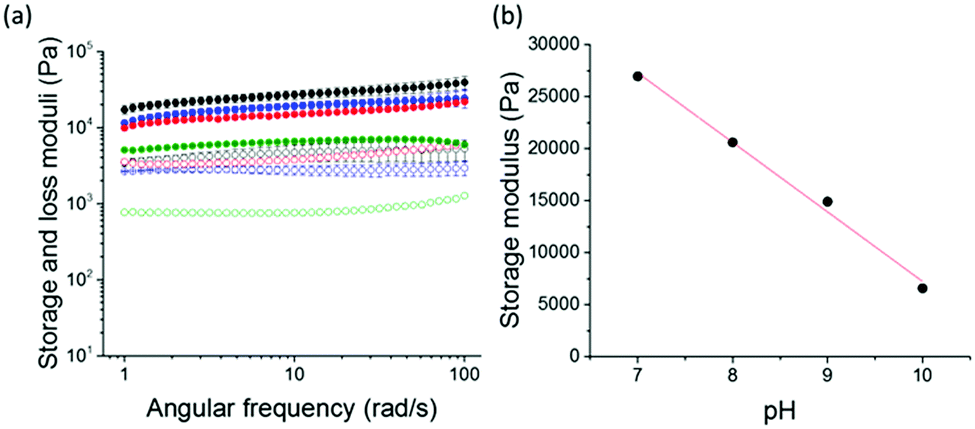 | ||
| Fig. 3 (a) Frequency sweep showing the storage and loss moduli of gels formed from 1 on adding dopamine to at an initial pH of 7 (black), 8 (blue), 9 (red) and 10 (green), storage moduli have closed circles, loss moduli have open circles. (b) Storage moduli at a frequency of 10 rad s−1versus pH. The linear regression (r2) is 0.98988. | ||
Drying such gels can lead to morphological changes;37 as such, we use small angle neutron scattering (SANS) to access information as to the morphology of the structures that are underpinning the gel network. This can be used directly on hydrated gels. For the gels formed from initial pH values of 7, 8, and 9, the data can be fitted to a core–shell model combined with a power law to take into account the scattering at low Q. The full scattering data and fits are shown in the ESI.† The fits imply that the core is larger for the gels formed from the initial pH 7 solutions, whilst those formed from the solutions at pH 8 and 9 are very similar. There is a hydrated shell around the fibres, which increases in size at lower initial pH. The scattering length density (SLD) is higher than would be expected from the structure of polydopamine; we suspect that this implies that the coating is highly hydrated. This suggests that the slow oxidation at the initially lower pH results in a polydopamine ‘shell’ forming around the fibres. The data for the sample that was initially pH 10 fits best to a cylinder combined with a power law implying that at the initially high pH, the fast rate means no coating forms. Hence, it is likely that the gel stiffness is affected by the presence of a polydopamine coating in some cases, which may result in cross-links between fibres.
Bacterial susceptibility assays were performed to assess antimicrobial activity of the gels and to examine whether there was any correlation between final gel stiffness, initial pH, polydopamine/H2O2 production and antimicrobial activity. In order to do this, bacterial susceptibility assays were performed against clinically relevant Gram-positive and Gram-negative organisms with the ability to reduce bacterial viability measured using a colony counting method providing bacterial numbers as log10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) colony forming units per mL (log10CFU per mL). A (hydroxypropyl)methyl cellulose (HPMC) control was employed as an inert hydrogel to study the effect of the presence of a gelator on bacterial viability.38,39 All data were collected on gels formed after 24 hours, removing any potential ageing issues.
colony forming units per mL (log10CFU per mL). A (hydroxypropyl)methyl cellulose (HPMC) control was employed as an inert hydrogel to study the effect of the presence of a gelator on bacterial viability.38,39 All data were collected on gels formed after 24 hours, removing any potential ageing issues.
We observed antimicrobial activity against the Gram-positive bacteria Staphylococcus aureus when pH 10 was used to initiate gelation and Staphylococcus epidermidis when both pH 9 and 10 were used to initiate gelation (Fig. 4). We highlight again that all gels once formed were at a pH of between 7.0 and 7.3. In each case, at least a three log reduction in bacterial counts was observed and this was used to denote clinical significance.40 Remarkably, the antimicrobial properties increased as the gel stiffness decreased. This contrasts with other studies relating to the antimicrobial activity of peptide gelators. For example, work by Jiang et al. demonstrated a correlation between an increase in storage modulus and more effective bacterial inhibition and suggested that increased storage modulus provides the required mechanical support for individual nanofibres and fibrous networks to direct their desirable chemical and biological functionalities against bacteria.38 This leads us to believe that the differences observed in antimicrobial activity seen here are likely due to the production of polydopamine and reactive oxygen species rather than the gelator itself. If the gelator alone were responsible for the antimicrobial activity, then it would be expected that an increase in stiffness should result in an increase in antimicrobial activity in line with other studies.
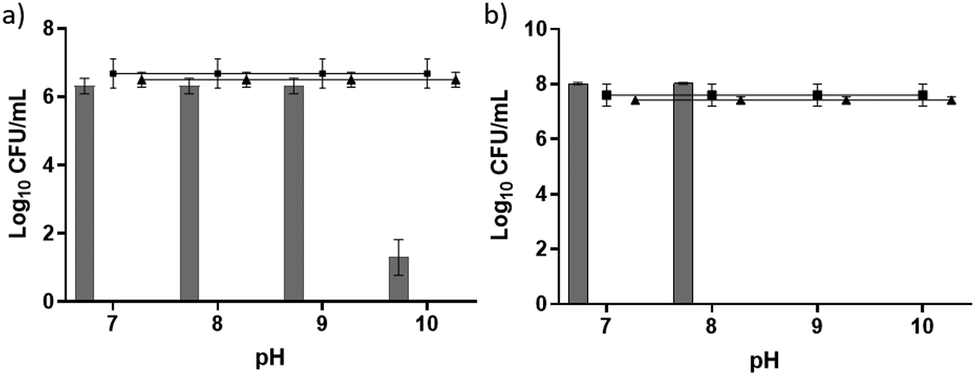 | ||
| Fig. 4 Bacterial susceptibility assays for (a) Staphylococcus aureus NCTC 10788 and (b) Staphylococcus epidermidis ATCC 12228. Results displayed as bacterial counts in log10 colonyforming units per mL (log10CFU per mL). In both parts, gels of 1 formed at different initial pH values with dopamine are shown as grey bars, negative PBS control as black triangles, HPMC controls as black square. | ||
We suggest that with increasing pH used for initiating gelation, there is an increase in the rate of polydopamine production and therefore a subsequent increase in H2O2 release. Ball et al. investigated the kinetics of the formation of polydopamine films under various pH conditions and found that the thickness of the film formed increased from pH 5.5 to pH 8 indicating that more polydopamine was produced under increasing alkaline conditions.41 The decrease in gel stiffness and fibre entanglement observed for gels produced under a higher pH may promote diffusion of H2O2 through the network pores to enable increased interaction with bacterial cell membranes and intracellular targets (e.g. DNA) thereby enabling a bactericidal effect.42 The lack of activity observed for Gram-negative organisms (Fig. S5, ESI†) may be due to differences in the membrane architecture or detoxification of reactive oxygen species. The additional outer lipopolysaccharide membrane in Gram-negative organisms is well documented for its ability to limit the influx and uptake of antibiotic molecules, including reactive oxygen species.43 Bacteria also demonstrate an ability to reduce the damaging effects of reactive oxygen species through the production of neutralising molecules such as the exopolysaccharide Psl in biofilm forming isolates of P. aeruginosa.44 Interestingly, work by Forooshani et al. found the sustained release of low doses of H2O2, generated during polydopamine production, was sufficient to achieve broad spectrum activity against both Gram-positive and Gram-negative bacteria.22 Antibacterial efficacy may therefore be a concentration dependent effect reliant on the rate at which H2O2 or other reactive oxygen species are produced within the system. Gram-negative microorganisms demonstrate increased resistance to reactive oxygen species, requiring increased exposure time or concentration to achieve significant kill.22 Unfortunately, it is not possible to determine due to the colour of our gels as well as the assays only being suitable for fully solubilised systems.
In conclusion, we present a new gelation trigger method for low molecular weight hydrogels. Using this method the gel stiffness can be controlled by the initial starting pH of the gelator solution. SANS data show how the composition of the gel fibres are similar at low Q therefore, the differences in gel stiffness are due to the entanglement of the fibres. We demonstrate that less stiff gels show greater antimicrobial properties towards Gram-positive bacteria and attribute this to the production of ROS as a result of the auto-oxidation of dopamine to produce polydopamine. This suggests potential to control the antimicrobial properties of a gel by controlling the mechanical properties of gelation.
ERC and AMFC thanks the University of Glasgow for studentships. DJA thanks the EPSRC for a Fellowship (EP/L021978/1), which also funded KM. GL also thanks the EPSRC (EP/S031561/1) for a research grant which also funds SMC. The experiment at the Institut Laue-Langevin was allocated beam time under experiment number 9-11-1905 (DOI: 10.5291/ILL-DATA.9-11-1905). This work benefitted from SasView software, originally developed by the DANSE project under NSF award DMR-0520547.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. P. McCloskey, B. F. Gilmore and G. Laverty, Pathogens, 2014, 791–821 CrossRef CAS PubMed.
- C. Potera, Science, 1999, 283, 1837–1839 CrossRef CAS PubMed.
- E. M. Hetrick and M. H. Schoenfisch, Chem. Soc. Rev., 2006, 35, 780–789 RSC.
- E. M. Kojic and R. O. Darouiche, Clin. Microbiol. Rev., 2004, 17, 255–267 CrossRef PubMed.
- A. Rai, S. Pinto, M. B. Evangelista, H. Gil, S. Kallip, M. G. S. Ferreira and L. Ferreira, Acta Biomater., 2016, 33, 64–77 CrossRef CAS PubMed.
- I. C. Saldarriaga Fernández, H. C. van der Mei, M. J. Lochhead, D. W. Grainger and H. J. Busscher, Biomaterials, 2007, 28, 4105–4112 CrossRef PubMed.
- G. P. Giammona Gaetano, P. Fabio Salvatore, M. Susanna, S. Sara and R. Carlo Luca, Hydrogels, 2018, 9, 180–200 Search PubMed.
- A. Salomé Veiga and J. P. Schneider, Pept. Sci., 2013, 100, 637–644 CrossRef PubMed.
- S. Li, S. Dong, W. Xu, S. Tu, L. Yan, C. Zhao, J. Ding and X. Chen, Adv. Sci., 2018, 5, 1700527 CrossRef PubMed.
- A. Panáček, L. Kvítek, R. Prucek, M. Kolář, R. Večeřová, N. Pizúrová, V. K. Sharma, T. J. Nevěčná and R. Zbořil, J. Phys. Chem. B, 2006, 110, 16248–16253 CrossRef PubMed.
- J. F. Hernández-Sierra, F. Ruiz, D. C. Cruz Pena, F. Martínez-Gutiérrez, A. E. Martínez, A. de Jesús Pozos Guillén, H. Tapia-Pérez and G. Martínez Castañón, Nanomedicine, 2008, 4, 237–240 CrossRef PubMed.
- C.-H. Chang, Y.-H. Lin, C.-L. Yeh, Y.-C. Chen, S.-F. Chiou, Y.-M. Hsu, Y.-S. Chen and C.-C. Wang, Biomacromolecules, 2010, 11, 133–142 CrossRef CAS PubMed.
- C.-H. Liao, C. S. Chen, Y.-C. Chen, N.-E. Jiang, C. J. Farn, Y.-S. Shen, M.-L. Hsu and C.-H. Chang, J. Microbiol., Immunol. Infect. DOI:10.1016/j.jmii.2019.08.008.
- H. Li, J. Yang, X. Hu, J. Liang, Y. Fan and X. Zhang, J. Biomed. Mater. Res., Part A, 2011, 98, 31–39 CrossRef PubMed.
- S. Marchesan, Y. Qu, L. J. Waddington, C. D. Easton, V. Glattauer, T. J. Lithgow, K. M. McLean, J. S. Forsythe and P. G. Hartley, Biomaterials, 2013, 34, 3678–3687 CrossRef CAS PubMed.
- J. L. Fox, Nat. Biotechnol., 2013, 31, 379–382 CrossRef CAS PubMed.
- S. Prabhu and E. K. Poulose, Int. Nano Lett., 2012, 2, 32 CrossRef.
- D. M. Wen, H. Yang, Y. Lyu, J. Shao, A. Cheng, X. Chen and L. Xu, PLoS One, 2017, 19, e0185554 CrossRef PubMed.
- C. Pan, Z. Zhou and X. Yu, J. Orthop. Surg. Res., 2018, 13, 220 CrossRef PubMed.
- E. Maisonneuve and K. Gerdes, Cell, 2014, 157, 539–548 CrossRef CAS PubMed.
- L. Su, Y. Yu, Y. Zhao, F. Liang and X. Zhang, Sci. Rep., 2016, 6, 24420 CrossRef CAS PubMed.
- P. Kord Forooshani, E. Polega, K. Thomson, M. S. A. Bhuiyan, R. Pinnaratip, M. Trought, C. Kendrick, Y. Gao, K. A. Perrine, L. Pan and B. P. Lee, Front. Chem., 2019, 7, 631 CrossRef PubMed.
- W. Zheng, H. Fan, L. Wang and Z. Jin, Langmuir, 2015, 31, 11671–11677 CrossRef CAS PubMed.
- B. J. Juven and M. D. Pierson, J. Food Prot., 1996, 59, 1233–1241 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- S. Senoh, C. R. Creveling, S. Udenfriend and B. Witkop, J. Am. Chem. Soc., 1959, 81, 6236–6240 CrossRef CAS.
- E. Herlinger, R. F. Jameson and W. Linert, J. Chem. Soc., Perkin Trans. 2, 1995, 259–263 RSC.
- M. Salomäki, L. Marttila, H. Kivelä, T. Ouvinen and J. Lukkari, J. Phys. Chem. B, 2018, 122, 6314–6327 CrossRef PubMed.
- R. V. Lloyd, Chem. Res. Toxicol., 1995, 8, 111–116 Search PubMed.
- D. C. Tse, R. L. McCreery and R. N. Adams, J. Med. Chem., 1976, 19, 37–40 CrossRef CAS PubMed.
- T. E. Young and B. W. Babbitt, J. Org. Chem., 1983, 48, 562–566 CrossRef CAS.
- E. R. Draper, O. O. Mykhaylyk and D. J. Adams, Chem. Commun., 2016, 52, 6934–6937 RSC.
- T.-F. Wu and J.-D. Hong, Biomacromolecules, 2015, 16, 660–666 CrossRef CAS PubMed.
- E. R. Cross, S. Sproules, R. Schweins, E. R. Draper and D. J. Adams, J. Am. Chem. Soc., 2018, 140, 8667–8670 CrossRef CAS PubMed.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS PubMed.
- L. Jiang, D. Xu, T. J. Sellati and H. Dong, Nanoscale, 2015, 7, 19160–19169 RSC.
- A. P. McCloskey, S. M. Gilmore, J. Zhou, E. R. Draper, S. Porter, B. F. Gilmore, B. Xu and G. Laverty, RSC Adv., 2016, 6, 114738 RSC.
- G. A. Pankey and L. D. Sabath, Clin. Infect. Dis., 2004, 38, 864–870 CrossRef CAS PubMed.
- V. Ball, D. D. Frari, V. Toniazzo and D. Ruch, J. Colloid Interface Sci., 2012, 386, 366–372 CrossRef CAS PubMed.
- H. I. Zgurskaya, C. A. López and S. Gnanakaran, ACS Infect. Dis., 2015, 1, 512–522 CrossRef CAS PubMed.
- J. van der Heijden, L. A. Reynolds, W. Deng, A. Mills, R. Scholz, K. Imami, L. J. Foster, F. Duong and B. B. Finlay, mBio, 2016, 7, e01238 CrossRef CAS PubMed.
- S. L. Chua, Y. Ding, Y. Liu, Z. Cai, J. Zhou, S. Swarup, D. I. Drautz-Moses, S. C. Schuster, S. Kjelleberg, M. Givskov and L. Yang, Open Biol., 2016, 6, 160162 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc02569k |
| This journal is © The Royal Society of Chemistry 2020 |