
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA rapid synthesis of low-nanomolar divalent LecA inhibitors in four linear steps from D-galactose pentaacetate†
Eva
Zahorska
 abc,
Sakonwan
Kuhaudomlarp
d,
Saverio
Minervini
a,
Sultaan
Yousaf
a,
Martin
Lepsik
d,
Thorsten
Kinsinger
a,
Anna K. H.
Hirsch
bce,
Anne
Imberty
d and
Alexander
Titz
*abc
abc,
Sakonwan
Kuhaudomlarp
d,
Saverio
Minervini
a,
Sultaan
Yousaf
a,
Martin
Lepsik
d,
Thorsten
Kinsinger
a,
Anna K. H.
Hirsch
bce,
Anne
Imberty
d and
Alexander
Titz
*abc
aChemical Biology of Carbohydrates, Helmholtz Institute for Pharmaceutical Research Saarland, Helmholtz Centre for Infection Research, 66123 Saarbrücken, Germany. E-mail: alexander.titz@helmholtz-hzi.de
bDeutsches Zentrum für Infektionsforschung (DZIF), Standort Hannover-Braunschweig, 38124 Braunschweig, Germany
cDepartment of Pharmacy, Saarland University, 66123 Saarbrücken, Germany
dUniversité Grenoble Alpes, CNRS, CERMAV, 38000 Grenoble, France
eDrug Design and Optimization, Helmholtz Institute for Pharmaceutical Research Saarland, Helmholtz Centre for Infection Research, 66123 Saarbrücken, Germany
First published on 6th July 2020
Abstract
Chronic infections with Pseudomonas aeruginosa are associated with the formation of bacterial biofilms. The tetrameric P. aeruginosa lectin LecA is a virulence factor and an anti-biofilm drug target. Increasing the overall binding affinity by multivalent presentation of binding epitopes can enhance the weak carbohydrate–ligand interactions. Low-nanomolar divalent LecA ligands/inhibitors with up to 260-fold valency-normalized potency boost and excellent selectivity over human galectin-1 were synthesized from D-galactose pentaacetate and benzaldehyde-based linkers in four linear steps.
Pseudomonas aeruginosa has been classified as a priority-1 pathogen by the World Health Organization due to its high antimicrobial resistance and the lack of new drugs to treat multidrug-resistant strains.1 New strategies against these bacterial infections are being explored to overcome the current antimicrobial-resistance crisis.2 The so-called anti-virulence therapy aims to neutralize bacterial virulence factors instead of increasing the selection pressure imposed by targeting essential cellular functions with antibiotics and thereby circumvents the advent of new resistances whilst preserving commensal bacteria.3,4 This strategy is investigated for P. aeruginosa infections by targeting its tetravalent lectins LecA and LecB.2,5,6 Both proteins are virulence factors regulated by quorum sensing, mediate bacterial host-cell adhesion and are essential structural components of P. aeruginosa biofilms.7–9 Whereas the best L-fucose/D-mannose-based LecB antagonists bind in the nanomolar range, monovalent D-galactose-based LecA inhibitors only reach binding affinities in the mid to low micromolar range.10–15 In Nature, the rather weak lectin–carbohydrate binding is often overcome by increasing valency, and thus enhancing apparent affinity.16,17 Likewise, a boost in target-binding affinity was achieved with multivalent inhibitors of LecA and LecB.6,18 Since LecA is a tetramer and pairs of binding sites are geometrically favorably oriented, simultaneously binding divalent inhibitors can boost binding affinity through favorable binding entropy.19,20 Notably, Pieters and co-workers have developed divalent LecA inhibitors based on complex and rigid repeating units of carbohydrate-triazole spacers with potent binding affinities ranging from 12 to 220 nM,21–23 while a divalent inhibitor with a more flexible linker reaches an affinity of 80 nM.24 In another report, an oligoproline-spaced digalactoside bound to LecA with Kd of 71 nM.25
In this work, we aimed to develop divalent LecA ligands with a focus on drug-like properties, synthetic accessibility and linker simplicity enabling future lead optimization. Spacer length and flexibility are important factors contributing to the overall potency of multivalent inhibitors.6 An optimized linker connecting two neighboring binding sites within one LecA tetramer and avoiding unwanted cross-linking between different LecA tetramers is desired (Fig. 1a). β-Linked aryl aglycons increase the binding strength of galactosides to LecA by establishing CH–π interactions with His50.15 The co-crystal structure of LecA with phenyl β-D-galactoside (PDB code: 5d21) showed possible growth vectors in meta- and para-position at the phenyl aglycon (Fig. 1b).26
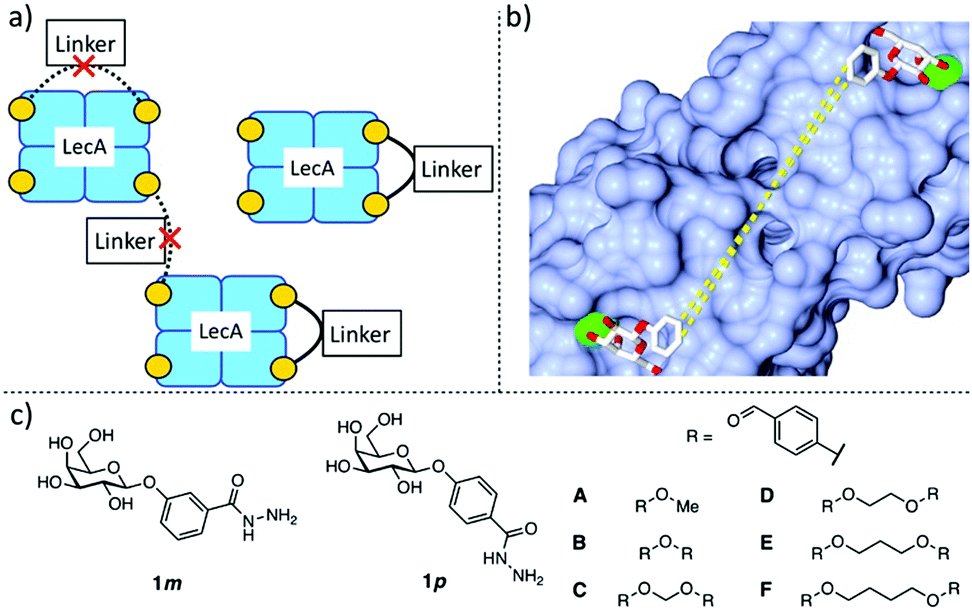 | ||
| Fig. 1 Design of divalent LecA inhibitors accessible through a short synthetic route with acylhydrazone coupling chemistry. (a) Possible binding modes of divalent LecA inhibitors with desired linkage for two adjacent binding sites. (b) The crystal structure of phenyl β-D-galactoside in complex with LecA (pdb code: 5d21) and distances between two ligands within one pair of binding sites in LecA (from meta to meta: 23 Å, from para to para: 25 Å). (c) Building blocks of LecA inhibitors: m/p-hydrazinecarbonylphenyl β-D-galactopyranoside (1m, 1p) and benzaldehydes A–F. Bis-benzaldehyde linkers B–F with systematic variation of length and number of rotatable bonds to optimize distance and flexibility. | ||
Protein-templated dynamic combinatorial chemistry (DCC) is an elegant method for the identification of potent ligands from a combinatorial library of building blocks with suitable linking chemistry in presence of a given protein.27,28 To apply this method to LecA, we introduced hydrazides at the para- or meta-position of phenyl β-D-galactoside in order to allow for acylhydrazone formation in DCC. For this purpose, we chose two galactoside building blocks with meta- or para-attached hydrazides, 1m and 1p, and varied linker length, rigidity and number of rotatable bonds by systematically increasing the number of methylene units in the corresponding benzaldehyde spacers B–F. The corresponding monovalent control A was included (Fig. 1c).
Benzaldehydes A and B were commercially available and bis-benzaldehydes C–F were obtained in one step using 4-hydroxybenzaldehyde in a double nucleophilic substitution reaction on aliphatic α,ω-di-halogenated C1–C4 hydrocarbons under microwave irradiation (Scheme 1). Lewis acid-promoted glycosylation of methyl meta- or para-hydroxybenzoate with β-D-galactose pentaacetate (2) yielded glycosides 3m and 3p in 69% and 47% yield, respectively. Removal of the acetates under Zemplén conditions gave galactosides 4m and 4p quantitatively. Subsequent ester hydrazinolysis resulted in hydrazides 1m and 1p in very good yields. These building blocks were then used in DCC reactions in presence of LecA: since the addition of LecA to the library caused precipitation, all divalent molecules were individually synthesized in absence of protein. Acylhydrazone formation of aldehydes A–F with excess hydrazide 1m or 1p under acidic conditions yielded the mono- and divalent LecA ligands A5m–F5m and A5p–F5p. The reduced solubility of the meta-series compared to the para-series and more difficult purifications could explain the lower yields despite nearly quantitative turnover during the individual reactions.
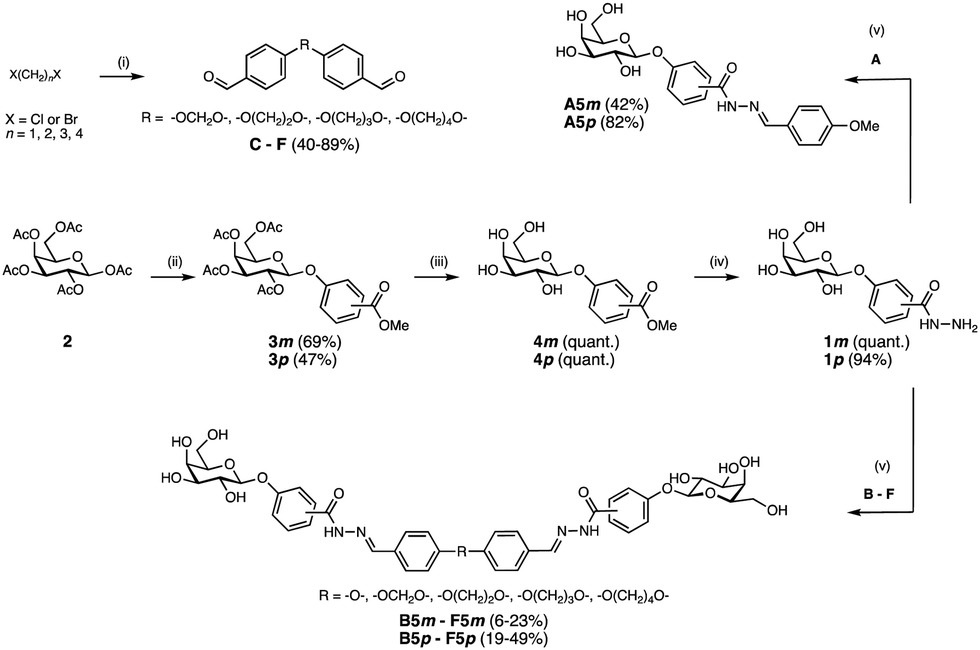 | ||
| Scheme 1 Synthesis of divalent LecA ligands and their monovalent analogs. Reagents and conditions: (i) 4-hydroxy benzaldehyde, K2CO3, DMF, 70 °C, microwave, 3–10 h; (ii) methyl m/p-hydroxybenzoate, BF3·Et2O, CH2Cl2, 0 °C – r.t., o.n.; (iii) NaOMe, MeOH, r.t., o.n.; (iv) NH2NH2·H2O, MeOH, 70 °C, o.n. (v) formic acid, DMSO, r.t., 4 h, for D5m: DMSO/MeCN, for F5m: H2O/MeCN. | ||
All synthesized galactosides, A5m–F5m and A5p–F5p, were then analyzed in the previously established competitive LecA binding assay based on fluorescence polarization (Fig. 2).11 Monovalent meta-ligand A5m (IC50 = 21.6 ± 4.5 μM) was twice as potent as its para-isomer A5p (IC50 = 55.5 ± 4.4 μM). The divalent ligands B5m–F5m and B5p–F5p showed a very similar profile in the competitive binding assay with very similar IC50 values in the single-digit micromolar range and a very steep Hill slope of the fit. These observations are likely a result of reaching the lower assay limit since the low affinity of the fluorescent primary ligand (Kd = 7.4 μM) required a relatively high LecA concentration of 20 μM. Therefore, ligand affinities with orders-of-magnitude higher potencies than the primary competitively displaced ligand cannot be reliably determined.
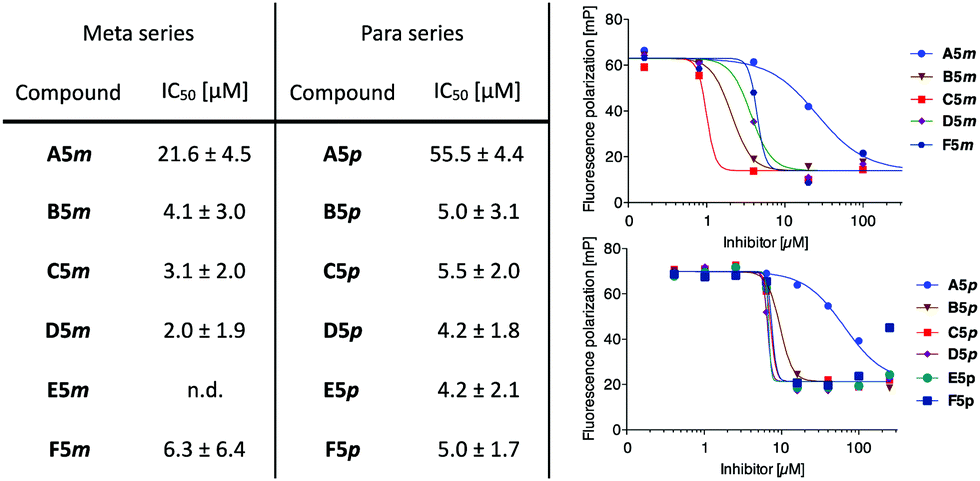 | ||
| Fig. 2 Evaluation in a competitive binding assay. One representative titration is shown for each series (right) – steep titration slopes for divalent inhibitors indicate the lower assay limit was reached. Aver. and std. dev. from at least 3 independent titrations of triplicates each. n.d. = not determined. | ||
To overcome the competitive binding assay's limitation, we analyzed all inhibitors in a direct LecA binding experiment using surface plasmon resonance (SPR). In case of the monovalent inhibitors A5m and A5p, rapid changes in the binding response during the association and dissociation phases were observed, indicating fast association/dissociation kinetics of the monovalent inhibitors to immobilized LecA (Fig. 3a and b). Due to this fast association/dissociation behavior, kon and koff for their interaction with LecA could not be accurately determined and affinity analysis was performed instead of determining Kd at steady-state binding. In SPR, the monovalent ligands A5m and A5p have Kd values of 4.9 ± 0.1 and 5.6 ± 0.3 μM, respectively (Table 1). The binding affinities for those monovalent compounds were validated using isothermal titration microcalorimetry (ITC) as an orthogonal method and similar values to SPR data were obtained (ITC: A5mKd = 2.7 ± 1.3 μM, A5pKd = 6.1 ± 0.5 μM, Table 1 and Fig. S1, ESI†). In both analyses, a meta-substitution of the phenyl aglycon in A5m resulted in higher affinities to LecA compared to the para-isomer A5p.
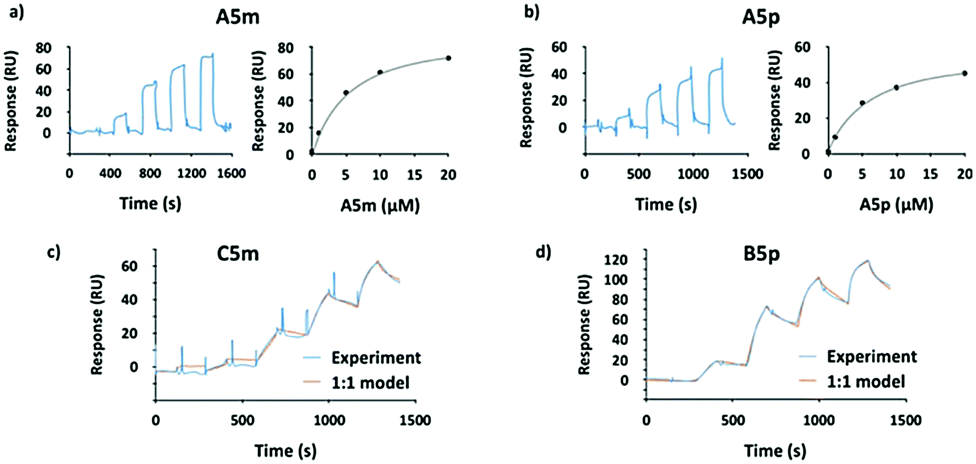 | ||
| Fig. 3 SPR analyses of the interaction between the monovalent inhibitors (a) A5m and (b) A5p. The sensorgrams are shown on the left panel and the affinity analyses on the right. (c) Sensorgrams of the most potent divalent inhibitors from meta-series (C5m) and (d) from para-series (B5p) at five different concentrations (0, 10, 50, 100, 200 nM). | ||
| meta series | para series | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| k on (×103 M−1 s−1) | k off (×10−3 s−1) | K d (nM) | r.p. | k on (×103 M−1 s−1) | k off (×10−3 s−1) | K d (nM) | r.p. | ||
| a Averages and std. dev. from three independent experiments. Relative potencies (r.p.) were calculated compared to monovalent compound in each series (A5m and A5p) and valency-normalized. b ITC determination. | |||||||||
| A5m | — | — | 4900 ± 100 | 1 | A5p | — | — | 5600 ± 300 | 1 |
| — | — | 2700 ± 1300b | — | — | — | 6100 ± 500b | — | ||
| B5m | 196 ± 6 | 5.34 ± 0.28 | 27.3 ± 1.6 | 90 | B5p | 152 ± 3 | 1.64 ± 0.13 | 10.8 ± 1.0 | 259 |
| C5m | 59 ± 3 | 1.11 ± 0.08 | 18.9 ± 1.6 | 130 | C5p | 114 ± 2 | 2.33 ± 0.02 | 20.5 ± 0.2 | 137 |
| D5m | 120 ± 41 | 2.67 ± 0.38 | 23.8 ± 7.1 | 103 | D5p | 121 ± 9 | 2.57 ± 0.20 | 21.4 ± 1.0 | 131 |
| E5m | 45 ± 4 | 3.64 ± 0.35 | 80.7 ± 11.4 | 30 | E5p | 103 ± 2 | 2.19 ± 0.45 | 22.5 ± 9.0 | 124 |
| F5m | 104 ± 53 | 3.39 ± 1.5 | 33.4 ± 2.3 | 73 | F5p | 79 ± 5 | 3.98 ± 0.16 | 50.1 ± 1.2 | 56 |
In contrast to the monovalent hydrazides, the SPR sensorgrams of the divalent inhibitors clearly indicated much slower association/dissociation of the compounds from LecA, demonstrating the benefit of divalent binding and enabling determination of kinetic parameters (kon and koff) as well as Kd (calculated from koff/kon, Fig. 3c, d and Fig. S2, ESI†). The divalent inhibitors B5m–F5m and B5p–F5p showed a strong increase in potency into the low nanomolar range (Table 1). In the para-series, compound B5p was the most potent ligand (Kd = 10.8 ± 1.0 nM), with a 520-fold increase (260-fold valency-corrected) compared to its monovalent congener A5p (Kd = 5600 ± 300 nM). The divalent para-ligands showed a slight decrease in potency with increasing spacer length, resulting from gradually decreasing kon and increasing koff (Table 1). In the meta-series, similar trends were absent. C5m with one central methylene unit had the optimal length and showed a Kd of 18.9 ± 1.6 nM. Shortening or increasing spacer length resulted in reduced affinities and surprisingly the second longest compound E5m was the least efficient inhibitor (Kd of 80.7 ± 11.4 nM). Comparing across the meta-series, the kon values were surprisingly 2–4 times smaller for compounds C5m and E5m. The koff values were gradually increasing going from C5m–F5m. The koff for B5m was 5-times higher than that of C5m.
We then studied compound selectivity in binding experiments towards human galectin-1, a homodimeric lectin that specifically recognizes β-galactoside containing glycans such as Me-β-lactoside (Kd = 187 μM).29 The most potent inhibitors from the para- and meta-series, B5p and C5m, together with their respective monovalent counterparts (A5p and A5m), were analyzed by SPR for their interaction with human galectin-1. Neither the monovalent (at 250 μM) nor the divalent compounds (at 25 μM) had a detectable interaction with the immobilized galectin-1 (Fig. S3, ESI†).
Since we did not succeed in obtaining crystal structures of LecA complexed with the divalent inhibitors, we carried out modeling on pairs of para- and meta-compounds, selecting the high-affinity binder C5m and its para-counterpart C5p; and the longer, less active E5m and E5p. The dynamics of these four compounds were simulated in the free state and in the modeled complex with LecA. In the free state, the ligands adopted a broad range of semi-extended to fully extended conformations characterized by Gal:C1⋯Gal:C1 distances ranging from 20 to 31 Å at a cutoff of 5% frequency (Fig. 4a, solid curves). A distance close to the crystallographic value of 29 Å (PDB: 5d21) is desired for optimal divalent binding of LecA. In the longer compounds, E5m and E5p, there was a small population (around 5%) of folded conformations (see the peaks at 5 Å in Fig. 4a). The unfolding of these conformations prior to LecA binding may be in part responsible for their slower on-rate (kon).
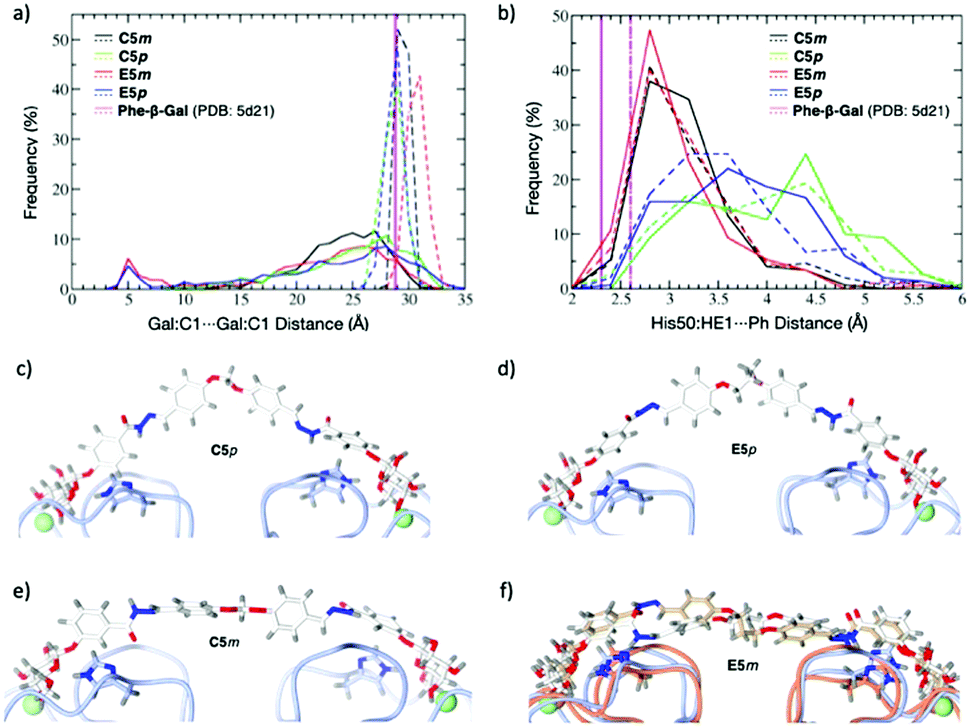 | ||
| Fig. 4 Molecular dynamics simulations of C5m, E5m; C5p; E5p: (a) distribution of Gal:C1⋯Gal:C1 distances of compounds in the free state (solid lines) and in complex with LecA (dashed lines). (b) Distribution of distances between His50 and the phenyl aglycon of ligands in complex with LecA (solid lines for LecA protein chain A, dashed lines for chain B). (c–f) Snapshot from trajectories of C5p (c), E5p (d), C5m (e), and two snapshots of E5m (f) indicating the conformational change of LecA. | ||
The four ligands modeled in complex with LecA showed a narrow distribution of the Gal:C1⋯Gal:C1 distances (Fig. 4a, dashed lines). C5m, C5p and E5p sampled distances of 28.9 ± 0.4 Å, 28.1 ± 0.9 Å, and 28.2 ± 0.7 Å, respectively, close to the 29 Å observed in crystal structures, thus indicating that the linker lengths are well-suited to bridge two LecA monomers. The narrow range for C5m (28–31 Å) may explain the lower affinity of shorter B5m. In contrast, C5p and E5p have larger range (26–31 Å) and shortening is beneficial such as in B5p. E5m with LecA displayed a higher mean distance of 30.2 ± 0.8 Å (range of 28 to 33 Å). Such longer meta linker pushes the two LecA monomers slightly apart, which is not favourable. The T-shaped CH–π interaction between His50 and the phenyl aglycon was observed with higher frequency for the meta compounds C5m and E5m compared to their para analogues C5p and E5p (Fig. 4b). On the opposite, the para ligands, C5p and E5p, preferentially adopted an inverted V-shape (Fig. 4c and d) in which their phenyl aglycons sampled a variety of arrangements with respect to His50 (parallel, diagonal, T-shape).
To conclude, we designed and synthesized highly potent divalent LecA inhibitors in four linear chemical steps from galactose pentaacetate. These simple and rapidly accessible divalent inhibitors B5m–F5m and B5p–F5p have comparable or superior activity to the previously reported and structurally complex di- and multivalent LecA ligands. Monovalent analogs A5m and A5p showed binding to LecA in SPR and ITC experiments in the low micromolar range (Kd = 2.7–6.1 μM). Divalent display of these epitopes in B5m–F5m and B5p–F5p boosted binding affinity with LecA to low nanomolar values. Molecular dynamics simulations gave insights into the interplay of linker geometry and length for an optimal divalent binding. To the best of our knowledge, compound B5p with a Kd of 10.8 nM is the most potent divalent LecA ligand reported to date with confirmed selectivity for LecA over galectin-1. Due to the simplicity of our synthetic design and readily accessible building blocks, further fine tuning and optimization of drug-like properties can be readily implemented. Future optimization of these compounds targeting LecA may provide a treatment of biofilm-associated P. aeruginosa infections.
The authors are grateful to Dirk Hauck (HIPS) for excellent technical assistance and Varsha R. Jumde (HIPS) for assisting with DCC. M. L. acknowledges a EU H2020 Marie Sklodowska-Curie grant (795605). Computations were run on GRICAD infrastructure, HPC-EUROPA3 project (H2020-INFRAIA-2016-1-730897), and EPCC at the University of Edinburgh, Scotland, and HPC resources from GENCI-IDRIS (Grant 2019-A0070711040). A. T. thanks the European Research Council (ERC Starting Grant, Sweetbullets) and Deutsche Forschungsgemeinschaft (Ti756/5-1). S. Y. acknowledges a RISE fellowship, German Academic Exchange Service DAAD. The work was supported by the ANR/DFG French-German GLYCOMIME project (ANR-AAPG-2017, DFG Ti756/5-1). A. I. and S. K. acknowledge support from Glyco@Alps (ANR-15-IDEX02), Labex Arcane/CBH-EUR-GS (ANR-17-EURE-0003). A. K. H. H. gratefully acknowledges the ERC Starting Grant (757913).
Conflicts of interest
There are no conflicts to declare.Notes and references
- WHO, https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed, accessed 9 April 2020.
- S. Wagner, R. Sommer, S. Hinsberger, C. Lu, R. W. Hartmann, M. Empting and A. Titz, J. Med. Chem., 2016, 59, 5929–5969 CrossRef CAS PubMed.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548 CrossRef CAS PubMed.
- M. B. Calvert, V. R. Jumde and A. Titz, Beilstein J. Org. Chem., 2018, 14, 2607–2617 CrossRef CAS PubMed.
- J. Meiers, E. Siebs, E. Zahorska and A. Titz, Curr. Opin. Chem. Biol., 2019, 53, 51–67 CrossRef CAS PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- K. Winzer, C. Falconer, N. C. Garber, S. P. Diggle, M. Camara and P. Williams, J. Bacteriol., 2000, 182, 6401–6411 CrossRef CAS PubMed.
- S. P. Diggle, R. E. Stacey, C. Dodd, M. Cámara, P. Williams and K. Winzer, Environ. Microbiol., 2006, 8, 1095–1104 CrossRef CAS PubMed.
- D. Tielker, S. Hacker, R. Loris, M. Strathmann, J. Wingender, S. Wilhelm, F. Rosenau and K. E. Jaeger, Microbiology, 2005, 151, 1313–1323 CrossRef CAS PubMed.
- J. Rodrigue, G. Ganne, B. Blanchard, C. Saucier, D. Giguère, T. C. Shiao, A. Varrot, A. Imberty and R. Roy, Org. Biomol. Chem., 2013, 11, 6906–6918 RSC.
- I. Joachim, S. Rikker, D. Hauck, D. Ponader, S. Boden, R. Sommer, L. Hartmann and A. Titz, Org. Biomol. Chem., 2016, 14, 7933–7948 RSC.
- R. Sommer, S. Wagner, K. Rox, A. Varrot, D. Hauck, E. C. Wamhoff, J. Schreiber, T. Ryckmans, T. Brunner, C. Rademacher, R. W. Hartmann, M. Brönstrup, A. Imberty and A. Titz, J. Am. Chem. Soc., 2018, 140, 2537–2545 CrossRef CAS PubMed.
- R. Sommer, K. Rox, S. Wagner, D. Hauck, S. S. Henrikus, S. Newsad, T. Arnold, T. Ryckmans, M. Brönstrup, A. Imberty, A. Varrot, R. W. Hartmann and A. Titz, J. Med. Chem., 2019, 62, 9201–9216 CrossRef CAS PubMed.
- S. Wagner, D. Hauck, M. Hoffmann, R. Sommer, I. Joachim, R. Müller, A. Imberty, A. Varrot and A. Titz, Angew. Chem., Int. Ed., 2017, 56, 16559–16564 CrossRef CAS PubMed.
- R. U. Kadam, D. Garg, J. Schwartz, R. Visini, M. Sattler, A. Stocker, T. Darbre and J.-L. Reymond, ACS Chem. Biol., 2013, 8, 1925–1930 CrossRef CAS PubMed.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef PubMed.
- K. Drickamer and M. E. Taylor, Curr. Opin. Struct. Biol., 2015, 34, 26–34 CrossRef CAS PubMed.
- A. Bernardi, J. Jiménez-Barbero and A. Casnati, et al. , Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- G. Cioci, E. P. Mitchell, C. Gautier, M. Wimmerová, D. Sudakevitz, S. Pérez, N. Gilboa-Garber and A. Imberty, FEBS Lett., 2003, 555, 297–301 CrossRef CAS PubMed.
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef CAS PubMed.
- F. Pertici and R. J. Pieters, Chem. Commun., 2012, 48, 4008–4010 RSC.
- G. Yu, A. C. Vicini and R. J. Pieters, J. Org. Chem., 2019, 84, 2470–2488 CrossRef CAS PubMed.
- R. Visini, X. Jin, M. Bergmann, G. Michaud, F. Pertici, O. Fu, A. Pukin, T. R. Branson, D. M. E. Thies-Weesie, J. Kemmink, E. Gillon, A. Imberty, A. Stocker, T. Darbre, R. J. Pieters and J. L. Reymond, ACS Chem. Biol., 2015, 10, 2455–2462 CrossRef CAS PubMed.
- A. Novoa, T. Eierhoff, J. Topin, A. Varrot, S. Barluenga, A. Imberty, W. Römer and N. Winssinger, Angew. Chem., Int. Ed., 2014, 53, 8885–8889 CrossRef CAS PubMed.
- S.-F. Huang, C.-H. Lin, Y.-T. Lai, C.-L. Tsai, T.-J. R. Cheng and S.-K. Wang, Chem. – Asian J., 2018, 13, 686–700 CrossRef CAS PubMed.
- M. Bergmann, G. Michaud, R. Visini, X. Jin, E. Gillon, A. Stocker, A. Imberty, T. Darbre and J. L. Reymond, Org. Biomol. Chem., 2016, 14, 138–148 RSC.
- M. Mondal and A. K. H. Hirsch, Chem. Soc. Rev., 2015, 44, 2455–2488 RSC.
- A. M. Hartman, R. M. Gierse and A. K. H. Hirsch, Eur. J. Org. Chem., 2019, 3581–3590 CrossRef CAS PubMed.
- I. Cumpstey, E. Salomonsson, A. Sundin, H. Leffler and U. J. Nilsson, ChemBioChem, 2007, 8, 1389–1398 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc03490h |
| This journal is © The Royal Society of Chemistry 2020 |