
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conformational selection vs. induced fit: insights into the binding mechanisms of p38α MAP Kinase inhibitors†
Patrick
Roser‡
 a,
Jörn
Weisner‡
b,
Juliane
Stehle
a,
Daniel
Rauh
*b and
Malte
Drescher
*a
a,
Jörn
Weisner‡
b,
Juliane
Stehle
a,
Daniel
Rauh
*b and
Malte
Drescher
*a
aDepartment of Chemistry and Konstanz Research School Chemical Biology (KoRS-CB), University of Konstanz, Universitätsstraße 10, 78457 Konstanz, Germany. E-mail: malte.drescher@uni-konstanz.de
bFaculty of Chemistry and Chemical Biology, TU Dortmund University, Drug Discovery Hub Dortmund (DDHD) am Zentrum für Integrierte Wirkstoffforschung (ZIW), Otto-Hahn-Strasse 4a, 44227 Dortmund, Germany. E-mail: daniel.rauh@tu-dortmund.de; Web: www.twitter.com/DDHDortmund
First published on 6th July 2020
Abstract
The conformational dynamics of a kinase's activation loop have been challenging to assess due to the activation loop's intrinsic flexibility. To directly probe the conformational equilibrium of the activation loop of mitogen-activated protein kinase p38α, we present an approach based on site-directed spin labeling, electron paramagnetic resonance (EPR) distance restraints, and multilateration. We demonstrate that the activation loop of apo p38α resides in a highly flexible equilibrium state and we reveal that binding of small molecules significantly alters this equilibrium and the populated sub-states.
Mitogen-activated protein kinases (MAPKs) are involved in intracellular signaling cascade mediating, e.g., stress-induced cellular responses.1 Dysfunctions of these mechanisms are linked to multiple human diseases including inflammatory diseases2 and cancer.3 Thus, the development of small molecule modulators of protein kinase activity and function is essential.4 The catalytic activity of protein kinases is often triggered by structural rearrangement of the activation loop (A-loop) that can be induced upon substrate binding or post-translational modifications such as phosphorylation. The most commonly described conformational sub-states based on crystallographic studies are termed according to the orientation of the consensus sequence Asp-Phe-Gly within the A-loop, i.e., DFG-in and DFG-out.5 In the DFG-in conformation, the phenylalanine side chain points towards the αC helix thereby occupying the hydrophobic back pocket while the aspartate side chain is oriented towards the ATP-binding pocket enabling the Mg2+ coordination of the phosphate groups of the cofactor ATP. In contrast, the DFG-out conformation is represented by a flipped phenylalanine side chain occupying the active site and thus preventing the binding of ATP.6 The MAPK p38α was reported to exist in a structural equilibrium of several activation loop states that is shifted upon binding of small molecule inhibitors.7–9 These states are classified into types I–VI based on their impact on the A-loop structure and conformation of the DFG-motif.6,10,11 Inhibitor specificity and potency are defined by the binding mechanism and A-loop structuring, highlighting the tremendous interest in understanding the underlying kinase's structural features and conformational dynamics. As the highly dynamic A-loop is rarely found to be completely resolved in X-ray crystallographic studies, complementary techniques such as NMR, IR, EPR or fluorescence spectroscopy are required to investigate its conformational equilibria and transitions.12–17 While the analysis of ligand binding mechanisms on an atomistic level remains challenging, such investigations could substantially support the structure-based design and optimization of conformation-specific small molecule ligands.18,19
Here, we report an electron paramagnetic resonance (EPR) spectroscopy based approach utilizing unphosphorylated p38α MAP kinase to directly probe the conformational flexibility within the A-loop region in the apo protein as well as the effects of small molecule type I and type II inhibitors on A-loop dynamics and conformations (SLiK, Spin Labels in Kinases). Site-specific introduction of two spin labels enabled the precise analysis and evaluation of distance distributions for the apo and the ligand-bound states, thereby pinpointing conformational states and changes of the labeled residues relative to each other. This approach not only facilitated the structural assessment of the disordered A-loop region, but also allowed the subsequent three-dimensional mapping by means of multilateration, illustrating the distinct conformational selection- and induced fit-like binding mechanisms of type I and type II modulators, respectively.
First, a panel of five different p38α MAPK constructs bearing two cysteine point mutations each were generated and subjected to site-directed spin labeling with MTSSL ((1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl)methanethiosulfonate). These constructs were labeled at residue 172 (DFG + 2) located in the flexible A-loop in combination with a second labeling site at alternating positions in the protein sequence; the respective constructs are termed p38α 119, 251, 277, 309, and 347, depending on the position of the second label (Fig. 1). The rigidity and thus the positional stability of the structural element containing the second labeling site were ensured by aligning all crystal structures of p38α MAPK available in the protein data bank (Fig. S1, ESI†). The functional integrity of the kinase after mutagenesis and subsequent spin labeling at the A-loop has been previously demonstrated.9 Although activation of p38α by phosphorylation at Thr180 and Tyr182 results in altered A-loop dynamics compared to unphosphorylated p38α, phosphorylation does not significantly affect the binding affinities of diverse type I and type II ligands.20–22 We therefore utilized unphosphorylated p38α for the EPR-based analyses.
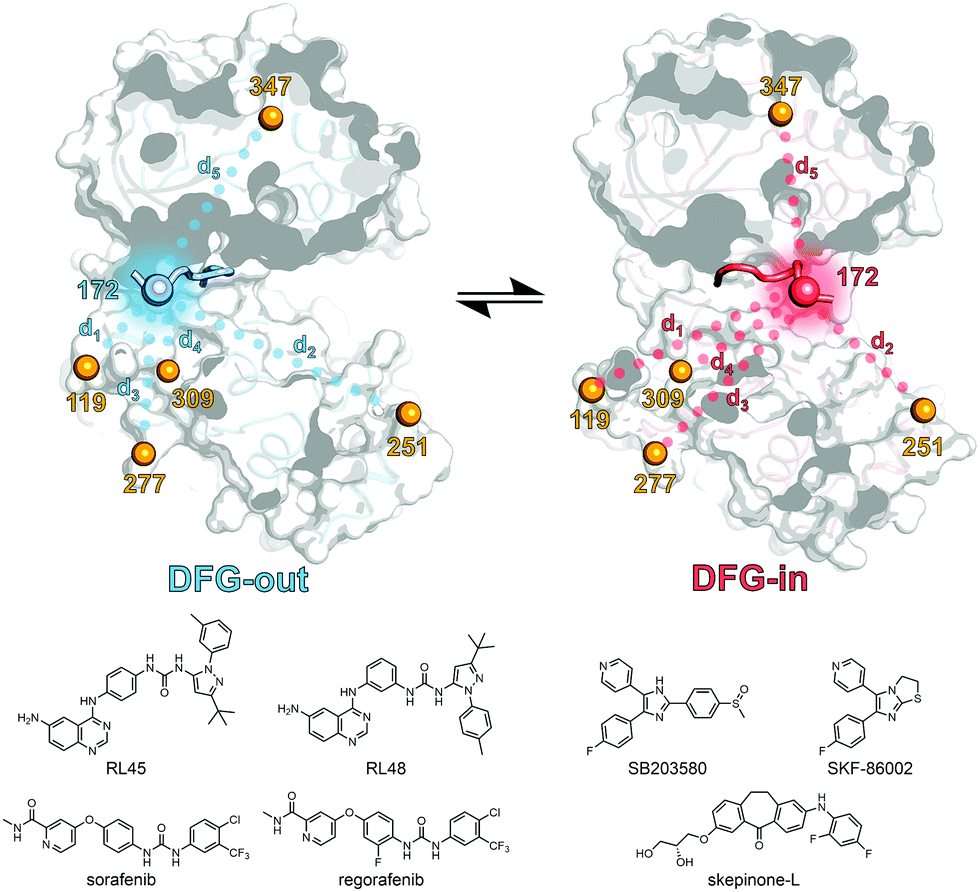 | ||
| Fig. 1 Schematic representation of the EPR based approach. The kinase inhibitors are shown on the left and right, the spin labeling positions (depicted as orange, red and blue spheres) used for the structural analysis of the A-loop region are shown in blue and red for the DFG-out and DFG-in structures, respectively (based on PDB 4KIN and 3BV2). Dotted lines depict the spin label distances (d1–d5) determined by EPR. Parts of the activation loop that are resolved in both crystal structures including the DFG-motif as well as the primary labeling site at position 172 are shown as opaque loops and spheres, respectively. | ||
After isolation and labeling of the kinase following standard procedures,9 we conducted pulsed EPR distance measurements (double electron electron resonance, DEER, also known as PELDOR)23 upon shock-freezing to trap the conformational ensemble for all five constructs of p38α in its apo state (Fig. 2 and Fig. S2–S6, ESI†). Additionally, we investigated the effects of three type I (SKF-86002, SB203580, skepinone-L; Fig. S7–S11, ESI†) and four type II small molecule kinase inhibitors (RL45, RL48, sorafenib, and regorafenib; Fig. S12–S16, ESI†) on the distance distributions.24–29 Analysis of the DEER data resulted in broad distance distributions observed for apo p38α, which was in line with previous studies suggesting a structural equilibrium between the DFG-in and DFG-out state in the absence of exogenous ligands.7,8,21 Bi- or multimodal restraints were observed for all five double label combinations, supporting the assumption of a broad conformational ensemble of the A-loop of apo p38α. Of note, the presence of bimodal distance restraints is not unequivocal proof for a two-state structural model as the inherent flexibility of the spin label tether must be taken into account. Here, modality caused by spin label dynamics was excluded by rotamer simulations and subsequent prediction of distance distributions for all labeling combinations based on a hyptothetical rigid protein structure (Fig. S17, ESI†). These simulated distance distributions showed no bimodal behavior comparable to the experimental data, suggesting that the observed bimodality was caused by the flexibility in the A-loop, not by rotamers of the spin label.
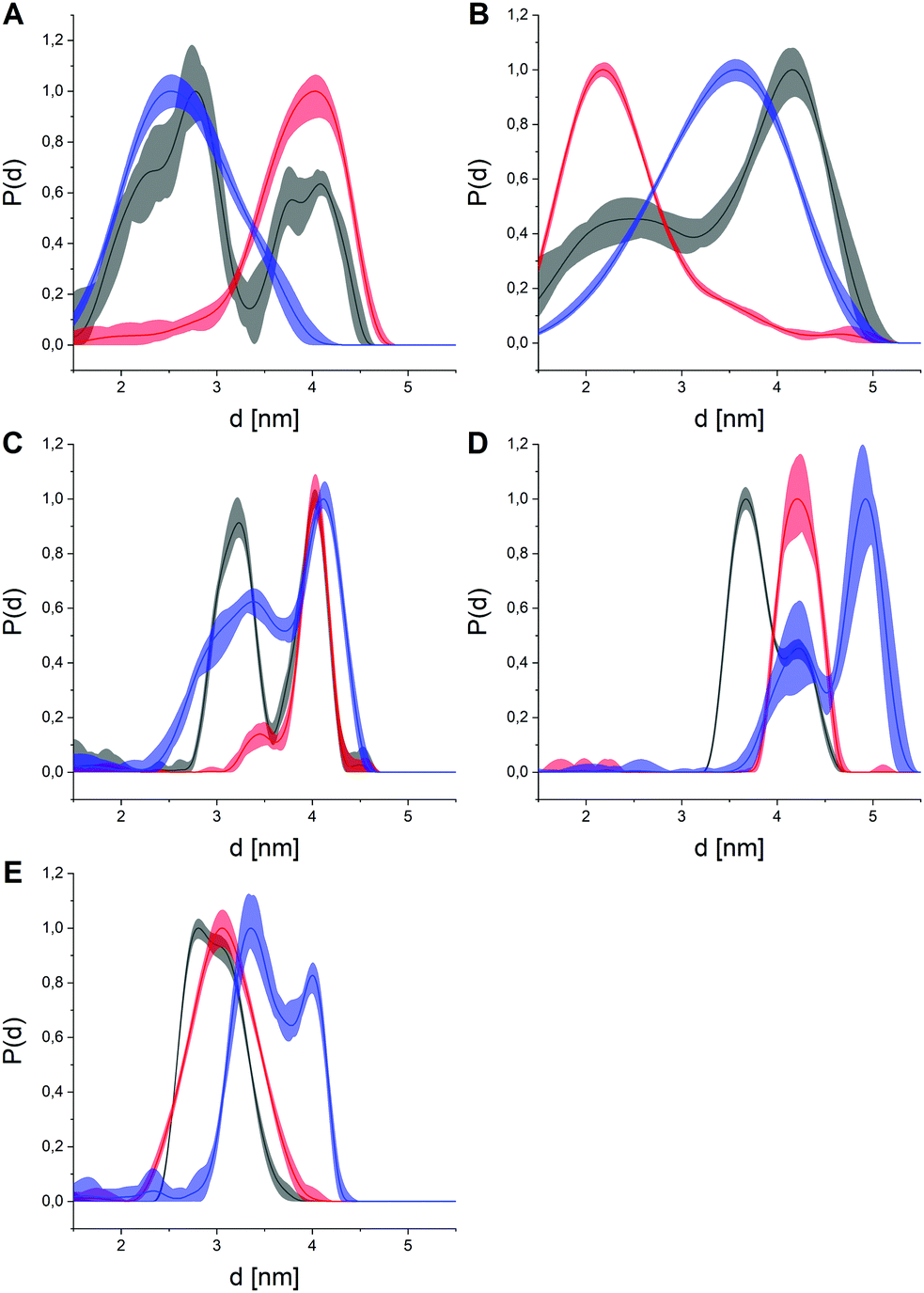 | ||
| Fig. 2 Distance distributions obtained for all five double labeled p38α constructs (p38α- A: 119, B: 251, C: 277, D: 309, E: 347) in the apo state (black), with representative type I (SB203580, red) or type II inhibitors (sorafenib, blue). The distances between the spin moieties report on conformational changes between the respective residues. Distributions are maximum normalized to facilitate interpretation. The light colored areas represent the uncertainty of the distance distributions. For raw data, validation and analysis details, see ESI† and Fig. S2–S16. | ||
The presence of small molecule inhibitors induced profound effects on the distance distributions for all five double mutant constructs (Fig. 2). The addition of saturating concentrations of type I inhibitors led to narrow, mostly unimodal distributions that were remarkably well-conserved for all three compounds, i.e., SKF-86002, SB203580, skepinone-L (Fig. S18, ESI†). The high conformity of distance distributions obtained for these type I inhibitors hints at a distinct and uniform ligand binding mechanism. These narrow distance distributions obtained for all five label combinations report on an increased structural rigidity of the A-loop in the presence of these inhibitors. Comparison of the experimental data with simulated distance distributions based on a crystal structure of p38α bound to the same type I inhibitor resulted in a very high agreement (Fig. S17, ESI†). Strikingly, the restraints obtained with type I inhibitors not only overlapped with distance regimes already occupied in the apo p38α, but in many cases even the shapes of the corresponding parts of the distance distributions were conserved. This outcome implies that a fraction of the conformational ensemble of the apo protein is selectively stabilized by type I modulators. Due to this pronounced enrichment of a specific sub-state, we attributed the binding mechanism of type I ligands with unphosphorylated p38α to a conformational selection-like process. To further assess this hypothesis, we combined the full set of distance distributions to conduct multilaterations/localizations.30–32 Therefore, we used the MMM toolbox (Multiscale Modeling of Macromolecules).33 Multilateration enabled the localization of the A-loop-tethered spin label based on experimental distance restraints with respect to the reference labels in the rigid regions of the protein while accounting for uncertainties introduced by spin label tether flexibility. Based on our experimental data, we were able to localize the A-loop-attached spin label as a marker to monitor the loop restructuring both in the presence and absence of small molecule ligands (Fig. 3). For p38α in the apo state, the broad multimodal distributions led to a large probability volume for the spin position, i.e., a broad distribution of loop sub-states.
 | ||
| Fig. 3 Multilateration results for p38α in the absence and presence of inhibitors from two different perspectives, based on PDB structure 3GCU. The multilateration for the spin label attached to the A-loop at position 172 of p38α in its apo state (dark grey volume), in the presence of type I inhibitor skepinone-L (red) and type II inhibitor sorafenib (blue) is shown as a probability volume, representing 50% of the total probability. The small spheres represent the locations of maximum probability. | ||
The probability volumes shown in Fig. 3 are artificially amplified due to the approximation of the distance constraints using Gaussians in the data processing, thus reducing the achievable precision. Still, the localization translated into a broad distribution without a pronounced population of a distinct sub-state in the structural ensemble, supporting the hypothesis, based on the distance distributions, that apo p38α resides in a multi-state equilibrium. A similar approach, used by Peter et al., resulted in a comparable probability volume of a spin label in the A-loop of the YopO kinase.16 The estimated volume for type I inhibitors indicated a narrow localization of the A-loop label, restricting possible loop conformations to a spatially confined volume (Fig. 3). The majority of the probability volumes resides within the localization volume obtained for apo p38α, corroborating a conformational selection mechanism upon ligand binding.
In contrast, type II inhibitors led to distance restraints that were not exclusively unimodal and retained significant distribution width (Fig. 2), hinting at a higher remaining conformational freedom in the A-loop compared to type I ligands. This observation is consistent with the fact that type II modulators are known to stabilize the DFG-out conformation which is structurally more flexible than the DFG-in state.34 All four tested type II modulators clearly diverged from the apo datasets and the obtained distance distributions were mostly similar within this inhibitor group (Fig. S19, ESI†). Variations within this group illustrated that the structural flexibility of the A-loop is retained upon binding of type II compounds, highlighting the relatively disordered A-loop in the DFG-out state. This flexibility of the DFG-out state might also explain the deviations from the crystal structure of a type II inhibitor bound p38α (Fig. S17, ESI†), which represents only a single structure of the broader DFG-out structural ensemble. In the presence of type II inhibitors, the obtained distance restraints (Fig. 2) clearly differ from the apo dataset with sub-states that are vacant in the apo protein, but which become significantly populated solely upon type II inhibitor binding.
Multilateration of datasets obtained for type II inhibitors revealed that the spin label populates a significantly smaller volume than observed for apo p38α (Fig. 3). The deviations between distance datasets of the four type II compounds translated into only minor changes in the localization volumes (Fig. S20, ESI†). Notably, the majority of the probability volumes for type II inhibitors was separated from the label localization in apo p38α. This separation of sub-states has already been observed by the lack of overlap of populated areas in the distance constraints, providing evidence that binding of type II inhibitors could induce the adaptation of sub-states that are not populated in the structural equilibrium for the apo protein, indicating at least a partial induced-fit binding mechanism. Type II inhibitors are expected to bind to the inactive DFG-out conformation, which has been proposed to be already populated in the structural equilibrium of apo p38α.7–9 This coexistence of loop states is clearly reflected by our results obtained via DEER, however, the transition between DFG-in and DFG-out appears to follow a non-binary equilibrium with multiple available sub-states present in the apo state as well as in the type II ligand-bound state. Therefore, our DEER-based approach yielded novel insights into the structural dynamics and conformational plasticity of p38α MAPK that were not resolved with other techniques such as X-ray crystallography. In general, our SLiK methodology is suited to assess and map flexible and dynamic (sub-)structures in kinases and, potentially, in other proteins.
In summary, using pulsed EPR distance measurements in combination with multilateration we were able to discriminate the structural ensembles adopted by the A-loop in the apo state and upon inhibitor binding. We have shown direct evidence of a highly flexible and dynamic A-loop in apo p38α MAPK hinting at multiple occupied sub-states. For type I ligands we observed a narrowing of distance restraints, while retaining the majority of the distribution shape correlating with the stabilization of a specific sub-state represented by a small localization volume which is also present in the apo equilibrium. In contrast, type II modulators, induced a narrow localization volume of the spin label that only partially overlapped with the probability volume observed for the apo protein. Furthermore, the calculated probability volumes of the spin label were mutually exclusive for type I and type II modulators. These observations hint at structurally distinct binding modes for type I and type II ligands that we assigned to conformational selection- and induced fit-like mechanisms, respectively. Our results highlight the potential of this method for the assessment of conformational ensembles and the identification of novel binding pockets. This study additionally underscores the potential challenges associated with structure-based drug design.35
This work was supported by the Konstanz Research School Chemical Biology. D. R. is grateful to funds by the German Federal Ministry for Education and Research (NGFNPlus and e:Med) (Grant No. BMBF 01GS08104, 01ZX1303C) and thanks the German federal state North Rhine Westphalia (NRW) and the European Union (European Regional Development Fund: Investing In Your Future) (EFRE-800400). We thank Frederike Kisters for experimental contributions and Andreas Arndt for the support with protein expression and purification.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. F. Chang and M. Karin, Nature, 2001, 410, 37–40 CrossRef CAS PubMed.
- S. Kumar, J. Boehm and J. C. Lee, Nat. Rev. Drug Discovery, 2003, 2, 717–726 CrossRef CAS PubMed.
- E. F. Wagner and A. R. Nebreda, Nat. Rev. Cancer, 2009, 9, 537–549 CrossRef CAS PubMed.
- M. Huse and J. Kuriyan, Cell, 2002, 109, 275–282 CrossRef CAS PubMed.
- D. K. Treiber and N. P. Shah, Chem. Biol., 2013, 20, 745–746 CrossRef CAS PubMed.
- R. Roskoski, Pharmacol. Res., 2016, 103, 26–48 CrossRef CAS PubMed.
- Y. M. M. Huang, W. Chen, M. J. Potter and C. E. A. Chang, Biophys. J., 2012, 103, 342–351 CrossRef CAS PubMed.
- M. Vogtherr, K. Saxena, S. Hoelder, S. Grimme, M. Betz, U. Schieborr, B. Pescatore, M. Robin, L. Delarbre, T. Langer, K. U. Wendt and H. Schwalbe, Angew. Chem., Int. Ed., 2006, 45, 993–997 CrossRef CAS PubMed.
- P. Roser, J. Weisner, J. R. Simard, D. Rauh and M. Drescher, Chem. Commun., 2018, 54, 12057–12060 RSC.
- Z. Fang, C. Grütter and D. Rauh, ACS Chem. Biol., 2012, 8, 58–70 CrossRef PubMed.
- S. Müller, A. Chaikuad, N. S. Gray and S. Knapp, Nat. Chem. Biol., 2015, 11, 818–821 CrossRef PubMed.
- J. A. H. Gilburt, H. Sarkar, P. Sheldrake, J. Blagg, L. Ying and C. A. Dodson, Angew. Chem., Int. Ed., 2017, 56, 11409–11414 CrossRef CAS PubMed.
- E. F. Ruff, J. M. Muretta, A. R. Thompson, E. W. Lake, S. Cyphers, S. K. Albanese, S. M. Hanson, J. M. Behr, D. D. Thomas, J. D. Chodera and N. M. Levinson, eLife, 2018, 7, e32766 CrossRef PubMed.
- E. W. Lake, J. M. Muretta, A. R. Thompson, D. M. Rasmussen, A. Majumdar, E. B. Faber, E. F. Ruff, D. D. Thomas and N. M. Levinson, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E11894 CrossRef CAS PubMed.
- L. M. Pegram, J. C. Liddle, Y. Xiao, M. Hoh, J. Rudolph, D. B. Iverson, G. P. Vigers, D. Smith, H. Zhang, W. Wang, J. G. Moffat and N. G. Ahn, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15463–15468 CrossRef CAS PubMed.
- M. F. Peter, A. T. Tuukkanen, C. A. Heubach, A. Selsam, F. G. Duthie, D. I. Svergun, O. Schiemann and G. Hagelueken, Structure, 2019, 27, 1416–1426.e3 CrossRef CAS PubMed.
- D. M. Yin, J. S. Hannam, A. Schmitz, O. Schiemann, G. Hagelueken and M. Famulok, Angew. Chem., Int. Ed., 2017, 56, 8417–8421 CrossRef CAS PubMed.
- S. Gianni, J. Dogan and P. Jemth, Biophys. Chem., 2014, 189, 33–39 CrossRef CAS PubMed.
- M. A. Morando, G. Saladino, N. D’Amelio, E. Pucheta-Martinez, S. Lovera, M. Lelli, B. López-Méndez, M. Marenchino, R. Campos-Olivas and F. L. Gervasio, Sci. Rep., 2016, 6, 24439 CrossRef CAS PubMed.
- K. M. Sours, S. C. Kwok, T. Rachidi, T. Lee, A. Ring, A. N. Hoofnagle, K. A. Resing and N. G. Ahn, J. Mol. Biol., 2008, 379, 1075–1093 CrossRef CAS PubMed.
- G. S. Kumar, M. W. Clarkson, M. B. A. Kunze, D. Granata, A. J. Wand, K. Lindorff-Larsen, R. Page and W. Peti, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4655–4660 CrossRef CAS PubMed.
- J. R. Simard, C. Grütter, V. Pawar, B. Aust, A. Wolf, M. Rabiller, S. Wulfert, A. Robubi, S. Klüter, C. Ottmann and D. Rauh, J. Am. Chem. Soc., 2009, 131, 18478–18488 CrossRef CAS PubMed.
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed.
- A. Cuenda, J. Rouse, Y. N. Doza, R. Meier, P. Cohen, T. F. Gallagher, P. R. Young and J. C. Lee, FEBS Lett., 1995, 364, 229–233 CrossRef CAS PubMed.
- C. Pargellis, L. Tong, L. Churchill, P. F. Cirillo, T. Gilmore, A. G. Graham, P. M. Grob, E. R. Hickey, N. Moss, S. Pav and J. Regan, Nat. Struct. Biol., 2002, 9, 268–272 CrossRef CAS PubMed.
- L. Tong, S. Pav, D. M. White, S. Rogers, K. M. Crane, C. L. Cywin, M. L. Brown and C. A. Pargellis, Nat. Struct. Biol., 1997, 4, 311–316 CrossRef CAS PubMed.
- S. C. Koeberle, J. Romir, S. Fischer, A. Koeberle, V. Schattel, W. Albrecht, C. Grütter, O. Werz, D. Rauh and T. Stehle, Nat. Chem. Biol., 2012, 8, 141–143 CrossRef CAS PubMed.
- J. R. Simard, M. Getlik, C. Grütter, V. Pawar, S. Wulfert, M. Rabiller and D. Rauh, J. Am. Chem. Soc., 2009, 131, 13286–13296 CrossRef CAS PubMed.
- D. Strumberg and B. Schultheis, Expert Opin. Invest. Drugs, 2012, 21, 879–889 CrossRef CAS PubMed.
- P. P. Borbat, H. S. McHaourab and J. H. Freed, J. Am. Chem. Soc., 2002, 124, 5304–5314 CrossRef CAS PubMed.
- S. Bleicken, G. Jeschke, C. Stegmueller, R. Salvador-Gallego, A. J. Garcia-Saez and E. Bordignon, Mol. Cell, 2014, 56, 496–505 CrossRef CAS PubMed.
- D. Abdullin, N. Florin, G. Hagelueken and O. Schiemann, Angew. Chem., Int. Ed., 2015, 54, 1827–1831 CrossRef CAS PubMed.
- G. Jeschke, Protein Sci., 2018, 27, 76–85 CrossRef CAS PubMed.
- Z. Zhao, H. Wu, L. Wang, Y. Liu, S. Knapp, Q. S. Liu and N. S. Gray, ACS Chem. Biol., 2014, 9, 1230–1241 CrossRef CAS PubMed.
- W. Pitsawong, V. Buosi, R. Otten, R. V. Agafonov, A. Zorba, N. Kern, S. Kutter, G. Kern, R. A. Padua, X. Meniche and D. Kern, eLife, 2018, 7, e36656 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc02539a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |