
Conversion of racemic alcohols to optically pure amine precursors enabled by catalyst dynamic kinetic resolution: experiment and computation†
Luis Miguel
Azofra
 *a,
Mai Anh
Tran
b,
Viktoriia
Zubar
b,
Luigi
Cavallo
c,
Magnus
Rueping
bc and
Osama
El-Sepelgy
*b
*a,
Mai Anh
Tran
b,
Viktoriia
Zubar
b,
Luigi
Cavallo
c,
Magnus
Rueping
bc and
Osama
El-Sepelgy
*b
aInstituto de Estudios Ambientales y Recursos Naturales (i-UNAT), Universidad de Las Palmas de Gran Canaria (ULPGC), Campus de Tafira, 35017, Las Palmas de Gran Canaria, Spain. E-mail: luismiguel.azofra@ulpgc.es
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: Osama.Elsepelgy@rwth-aachen.de
cKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
First published on 29th June 2020
Abstract
An unprecedented base metal catalysed asymmetric synthesis of α-chiral amine precursors from racemic alcohols is reported. This redox-neutral reaction utilises a bench-stable manganese complex and Ellman's sulfinamide as a versatile ammonia surrogate. DFT calculations explain the unusual finding of the highly stereoselective transformation enabled by a catalyst that undergoes an unusual dynamic kinetic resolution.
 Luis Miguel Azofra | Luis Miguel Azofra loves Quantum Chemistry as much as the sea. The latter was a gift he received for being born in the Canary Islands, the first is a passion he learned during his PhD in Prof. Alkorta's lab (CSIC, Spain). In 2015, Dr Sun and Prof. MacFarlane introduced him to the field of catalysis (Monash University, Australia), and in 2016 he joined Prof. Cavallo's group as an in silico designer (KAUST, Saudi Arabia). Currently, Dr Azofra is an early-career research leader at Universidad de Las Palmas de Gran Canaria, where he combines his efforts as a researcher and lecturer. |
The hydrogen autotransfer (HA) strategy has captured much attention during the last few years, mainly due to its synthetic importance as a powerful environmentally benign approach for the construction of C–C and C–N bonds. In this regard, progress has been made in transition-metal catalysed C-alkylations and N-alkylations with non-activated alcohols. The reactions mainly rely on the production of achiral or racemic products.2
The direct asymmetric amination of alcohols to produce chiral aniline,3 amino alcohols,4 oxazolidinones,5 and hydrazones6 has been demonstrated using ruthenium and iridium catalysis. In contrast, the direct production of chiral primary amines from racemic alcohols is significantly more challenging. In this regard, a ruthenium catalysed protocol was disclosed. However, this noble metal method is limited to the methyl substituted chiral amines.7 Apart from metal catalysis, the use of a dual enzymatic system for the direct amination of secondary alcohols has been reported.8 Therefore, the development of a new catalytic system, ideally based on a non-precious metal catalyst,9 which can transform a broad range of racemic alcohols to enantiopure primary amines, is an elusive goal.10
Ellman's sulfinamide represents as an industrially relevant reagent, frequently used as an ammonia surrogate in the synthesis of enantiopure primary amines.11 This transformation typically requires three chemical steps, i.e., stoichiometric oxidation, condensation using stoichiometric titanium reagent and stoichiometric reduction, producing large quantities of waste.
Encouraged by recent studies on metal catalysed hydrogen autotransfer,2,12,13 we envisioned that an effective base-metal catalyst might potentially catalyse the oxidative dehydrogenation of the sec-alcohols (rac-1), to form the corresponding ketone and shuttle the abstracted hydrogen required for the stereoselective hydrogenation. The in situ formed ketone undergoes condensation with an ammonia equivalent to produce an imine and only water as a by-product. Finally, the imine will be reduced in a stereoselective fashion to produce the desired product. The chemoselectivity of the reaction depends on the potential of the metal catalyst to return the stored hydrogen to the final product.
Herein, we present a new manganese catalysed synthesis of optically active amines from racemic alcohols. Interestingly, our DFT mechanistic study on this stereoselective reaction explains how a racemic catalyst could lead to optically pure product. Notably, we found that the high stereocontrol is enabled by unusual catalyst dynamic kinetic resolution (Scheme 1a). The chiral imine intermediate kinetically discriminates the catalyst racemic mixture (Scheme 1b). While only one enantiomer of the catalyst can be involved in the stereoselective hydrogenation reaction, the ketone intermediate plays the crucial role in the racemisation of the second catalyst enantiomer (Scheme 1a). We are not aware of a literature report with such an unusual observation.
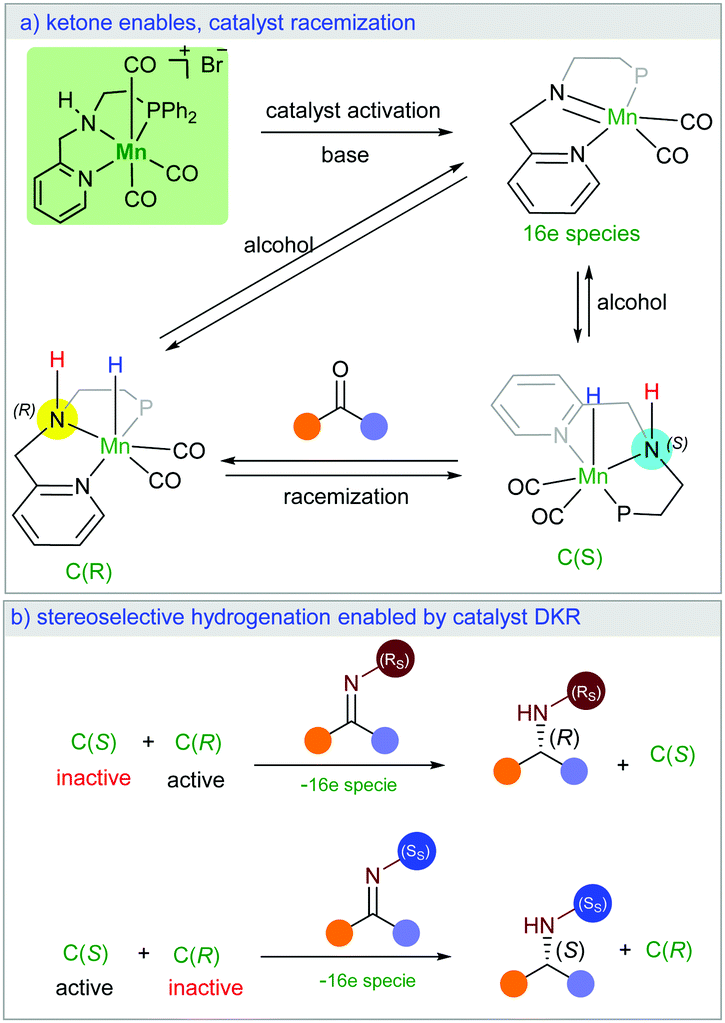 | ||
| Scheme 1 Manganese-catalysed diasteroselective hydrogen autotransfer. | ||
We started our study with the selection of the appropriate base metal catalyst and the optimisation of the suitable reaction parameters. After careful investigations, we found that the use of the air stable PNN-Mn complex (5 mol%) in combination with Cs2CO3 (10 mol%) and t-amyl alcohol (0.5 M) is the optimal combination for this reaction (see the ESI† for details).
Next, the variability and the applicability of the asymmetric hydrogen autotransfer reaction were investigated (Table 1). We initially explored different racemic benzylic alcohols. The alcohols 1a–1i bearing different electron donating and electron withdrawing groups were applicable without significant effect on the reactivity or the stereochemical outcome and all desired products 3a–3i were isolated in very good yields and optical purity. Similarly, the naphthyl substituted sulfinamide (R,Rs)-3j was isolated in very good yield and selectivity. Importantly, some of these amines are key intermediates in the synthesis of pharmaceuticals and bioactive molecules. For example, carpropamid,14 an agriculture fungicide, can be prepared from (R,Rs)-3b, whereas the Alzheimer's and Parkinson's drug rivastigmine can be produced using the sulfinylamine (S,Ss)-3g.15 It is noteworthy that the classical synthesis of these α-chiral amines involves the addition of MeLi to N-tert-butylsulfinyl aldimes. However, this reaction often suffers from unfavourable diastereocontrol, even at lower temperature. To our delight, our catalytic system was found not to be limited to the sec-phenethyl alcohol derivatives. Thus, 1-tetralol (1k) was converted to the desired product in 84% yield. This amine is of relevance as it is used in the synthesis of diverse bio-related compounds.16 The ethyl substituted alcohol 1l and the more challenging alcohol 1m were found to be reactive using the presented catalytic system. With this success, we turned our attention to the more demanding non-benzylic alcohols 1n–1q. The chiral amines bearing a cyclohexyl substituent (R,Rs)-3n and cyclopropyl substituent (R,Rs)-3p were produced in a very good diastereomeric ratio as well. Interestingly, the amphetamine, which is used in the treatment of attention deficit hyperactivity disorder, can be obtained from the rac-1q in 78% yield. Heterocycle-containing alcohols were also tolerated and underwent the reaction to afford the chiral amine precursors (R,Rs)-3r to (R,Rs)-3u in very good yields and with high diastereoselectivity. Interestingly, despite the importance of the optically pure 3s and 3r in the synthesis of HIV protease inhibitors,17 the asymmetric synthesis of the corresponding primary amines is not yet reported. It is noteworthy that even pyridine containing substrates and products, such as 3u are tolerated in this manganese catalysed reaction.
| a Reaction conditions: 1 (0.75 mmol), (R)-2a (0.5 mmol), [Mn] (0.025 mmol) and Cs2CO3 (0.05 mmol) in t-amyl alcohol (1 mL) were stirred at 140 °C (aluminum block), for 16 h in a glass tube under argon. Yields after column chromatography are given. b [Mn] (0.05 mmol), Cs2CO3 (0.1 mmol). c 48 h. d 1 (1 mmol). |
|---|
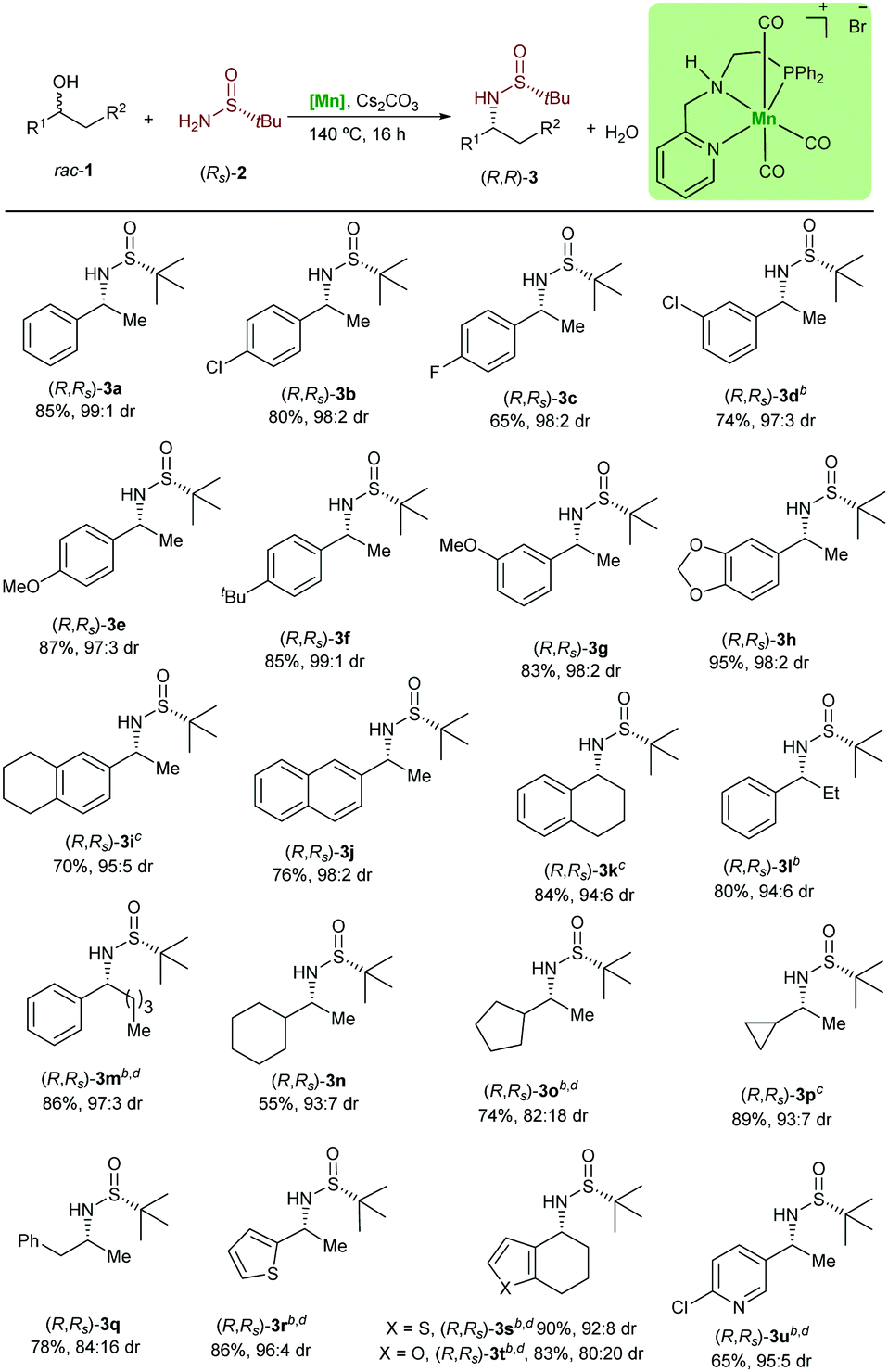
|
In order to understand the origin of the stereoselective hydrogenation, we have carried out DFT modelling for the asymmetric amination of 1-phenylethanol (Scheme 2). The 16e species A, which can be generated by treating the manganese pre-catalyst [Mn] with appropriate bases, dehydrogenates the alcohol substrate rac-1a to produce acetophenone and manganese hydride complex C. Based on our DFT results, the alcohol dehydrogenation step takes place in a stepwise fashion via the formation of four different diastereoisomers of manganese alkoxide intermediates B, bearing chiral nitrogen and carbon atoms. However, the proton transfer A–B is a barrier-less process. The calculated free activation energy for the hydride transfer B–C steps are between 19.9 and 20.1 kcal mol−1 at the M06/TZVP level of theory. The β-hydride elimination will lead to the formation of a racemic mixture of the hydrogenated catalyst C(S) and C(R).
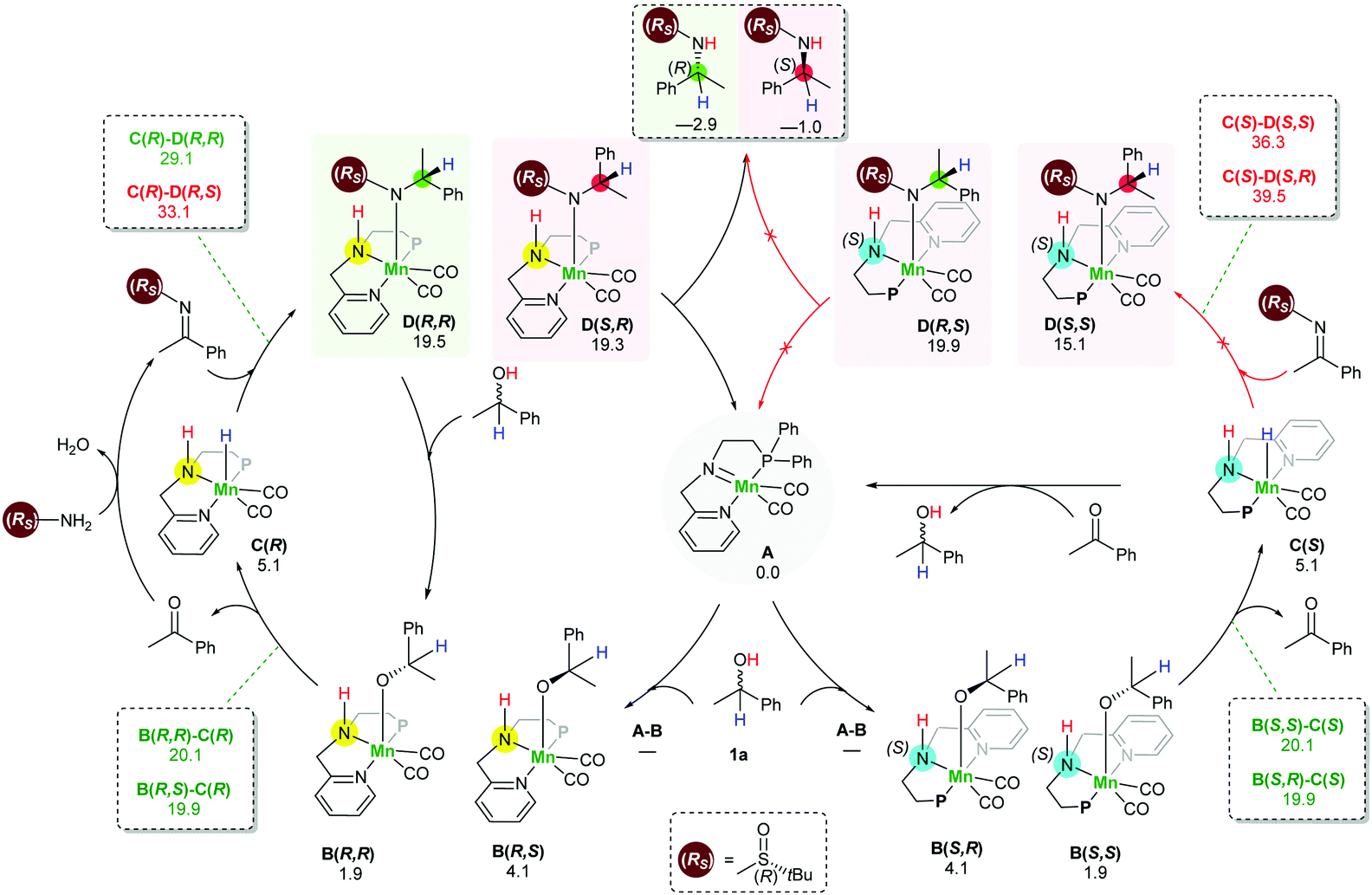 | ||
| Scheme 2 Proposed reaction mechanism for the stereoselective manganese catalysed hydrogen autotransfer amination of rac-alcohols. Free reaction and activation energies (at 140 °C) are shown in kcal mol−1 at the M06/TZVP level using toluene as a solvent. | ||
The condensation reaction between the in situ generated ketone and (Rs)-2 results in the generation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond which can be potentially hydrogenated by the action of racemic manganese catalyst C(S) and C(R). Similarly, to the alcohol substrate dehydrogenation, we found that the hydrogenation of the imine intermediate takes place via a stepwise mechanism. Since the hydrogenated catalyst exists as a racemic mixture, it establishes the possibility of the hydride transfer (C–D) to form four different intermediates D. Importantly, we found that the hydride transfer step (C–D) is the rate determining step as well as the stereodetermining step. When the manganese catalyst C(R) was used for the hydrogenation of an imine bearing (R)-sulfinamide group, the computed barrier for the hydride transfer is 29.1 kcal mol−1 for the creation of a new (R) stereogenic centre, while the barrier for the generation of the (S) stereogenic centre is 33.1 kcal mol−1. On the other hand, the barriers for the hydride transfer using the catalyst enantiomer C(S) are 39.5 and 36.3 kcal mol−1. Interestingly, when an imine bearing (S)-sulfinamide group is used as a substrate, only the (S) enatiomer of the catalyst is involved in the imine hydrogenation step and creates a new stereogenic centre with the (S) configuration. With this process being controlled by kinetics, it supposes a diasteoisomeric ratio equal to 99
N bond which can be potentially hydrogenated by the action of racemic manganese catalyst C(S) and C(R). Similarly, to the alcohol substrate dehydrogenation, we found that the hydrogenation of the imine intermediate takes place via a stepwise mechanism. Since the hydrogenated catalyst exists as a racemic mixture, it establishes the possibility of the hydride transfer (C–D) to form four different intermediates D. Importantly, we found that the hydride transfer step (C–D) is the rate determining step as well as the stereodetermining step. When the manganese catalyst C(R) was used for the hydrogenation of an imine bearing (R)-sulfinamide group, the computed barrier for the hydride transfer is 29.1 kcal mol−1 for the creation of a new (R) stereogenic centre, while the barrier for the generation of the (S) stereogenic centre is 33.1 kcal mol−1. On the other hand, the barriers for the hydride transfer using the catalyst enantiomer C(S) are 39.5 and 36.3 kcal mol−1. Interestingly, when an imine bearing (S)-sulfinamide group is used as a substrate, only the (S) enatiomer of the catalyst is involved in the imine hydrogenation step and creates a new stereogenic centre with the (S) configuration. With this process being controlled by kinetics, it supposes a diasteoisomeric ratio equal to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This is fully in agreement with our experimental results.
:1. This is fully in agreement with our experimental results.
The last step involves the proton transfer to the product nitrogen atom. The proton could be internally transferred from the catalyst NH group to generate the product and the 16e species A. Alternatively, in the presence of excess rac-1a, direct proton transfer from the alcohol substrate could take place to produce the desired product and the intermediates B without the regeneration of the 16e species A.18 Notably, calculations show that the undesired catalyst enantiomer C(S) can be easily racemised with the assistance of the acetophenone intermediate. In other words, the steps C(S)–B(S,R), C(S)–B(S,S), B(S,S)–A and B(S,R)–A are reversible. We are not aware of a previously reported mechanism with a similar observation of the catalyst racemisation assisted by the in situ generated intermediate.
In conclusion, we have developed an unprecedented base-metal catalysed stereoselective amination of racemic alcohols using the hydrogen autotransfer strategy. The produced enatiomerically pure sulfinamides could be easily converted to the corresponding α-chiral amine upon stirring in methanolic HCl at room temperature.19 Notably, the protocol uses an inexpensive earth-abundant manganese complex and readily available substrates. Our DFT calculations demonstrate the origin of high diastereoselectivity which is enabled by an unusual catalyst dynamic kinetic resolution process. We found that the alcohol dehydrogenation and imine hydrogenation reactions occur in a stepwise fashion, while the hydride transfer to the imine intermediate represents the rate and stereodetermining step of the whole reaction. Given the operational simplicity, the presented catalytic system will serve as a basis for further application in the synthesis of relevant optically pure α-chiral amines and heterocycles.
L. M. A. thanks Universidad de Las Palmas de Gran Canaria (ULPGC) for support. Gratitude is also due to the KAUST for use of the supercomputer Shaheen II for providing the computational resources.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. Aranda, G. Oksdath-Mansilla, F. R. Bisogno and G. de Gonzalo, Adv. Synth. Catal., 2020, 362, 1233–1257 CrossRef CAS; (b) D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284–300 CrossRef CAS; (c) C. E. Humphrey, M. Ahmed, A. Ghanem and N. J. Turner, in Separation of Enantiomers: Synthetic Methods, ed. M. Todd, Wiley-VCH, Weinheim, 2014, pp. 123–160 Search PubMed; (d) M. D. Truppo, N. J. Turner and J. D. Rozzell, Chem. Commun., 2009, 2127–2129 RSC; (e) T. C. Nugent, Chiral Amine Synthesis, Wiley-VCH, Weinheim, 2010 CrossRef; (f) M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781–3783 CrossRef CAS PubMed.
- (a) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed ; for the production of chiral products using biocatalysis: ; (b) E. Tassano and M. Hall, Chem. Soc. Rev., 2019, 48, 5596–5615 RSC.
- (a) Y. Zhang, C.-S. Lim, D. S. B. Sim, H.-J. Pan and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 1399–1403 CrossRef CAS PubMed; (b) Z.-Q. Rong, Y. Zhang, R. H. B. Chua, H.-J. Pan and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 4944–4947 CrossRef CAS PubMed; (c) C. S. Lim, T. T. Quach and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 7176–7180 CrossRef CAS PubMed.
- (a) A. E. Putra, Y. Oe and T. Ohta, Eur. J. Org. Chem., 2013, 6146–6151 CrossRef; (b) L.-C. Yang, Y.-N. Wang, Y. Zhang and Y. Zhao, ACS Catal., 2017, 7, 93–97 CrossRef CAS.
- M. Peña-López, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 7826–7830 CrossRef PubMed.
- P. Yang, C. Zhang, Y. Ma, C. Zhang, A. Li, B. Tang and J. S. Zhou, Angew. Chem., Int. Ed., 2017, 56, 14702–14706 CrossRef CAS PubMed.
- N. J. Oldenhuis, V. M. Dong and Z. Guan, J. Am. Chem. Soc., 2014, 136, 12548–12551 CrossRef CAS PubMed.
- (a) F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed; (b) S. L. Montgomery, J. Mangas-Sanchez, M. P. Thompson, G. A. Aleku, B. Dominguez and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 10491–10494 CrossRef CAS PubMed; (c) M. P. Thompson and N. J. Turner, ChemCatChem, 2017, 9, 3833–3836 CrossRef CAS; (d) W. Böhmer, T. Knaus and F. G. Mutti, ChemCatChem, 2018, 10, 731–735 CrossRef PubMed; (e) M. L. Corrado, T. Knaus and F. G. Mutti, Green Chem., 2019, 21, 6246–6251 RSC.
- (a) R. M. Bullock, Catalysis without Precious Metals, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 CrossRef; (b) B. Plietker, Iron Catalysis in Organic Chemistry: Reactions and applications, Wiley-VCH, Weinheim, 2008 CrossRef.
- M. Xiao, X. Yue, R. Xu, W. Tang, D. Xue, C. Li, M. Lei, J. Xiao and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 10528–10536 CrossRef CAS PubMed.
- (a) G. C. Liu, D. A. Cogan and J. A. Ellman, J. Am. Chem. Soc., 1997, 119, 9913–9914 CrossRef CAS; (b) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS PubMed; (c) M. Wakayama and J. A. Ellman, J. Org. Chem., 2009, 74, 2646–2650 CrossRef CAS PubMed.
- Recent reviews: (a) T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed; (b) A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS; (c) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed; (d) N. Gorgas and K. Kirchner, Acc. Chem. Res., 2018, 51, 1558–1569 CrossRef CAS PubMed; (e) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (f) M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 4344–4362 CrossRef CAS.
- Mn-Catalysed achiral N-alkylation with primary alcohols to produce achiral products: (a) S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed; (b) J. Neumann, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, Chem. – Eur. J., 2017, 23, 5410–5413 CrossRef CAS PubMed; (c) A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, J. Catal., 2017, 347, 57–62 CrossRef CAS; (d) M. Mastalir, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2017, 139, 8812–8815 CrossRef CAS PubMed; (e) R. Fertig, T. Irrgang, F. Freitag, J. Zander and R. Kempe, ACS Catal., 2018, 8, 8525–8530 CrossRef CAS; (f) K. Das, A. Mondal, D. Pal, H. K. Srivastava and D. Srimani, Organometallics, 2019, 38, 1815–1825 CrossRef CAS; (g) B. G. Reed-Berendt and L. C. Morrill, J. Org. Chem., 2019, 84, 3715–3724 CrossRef CAS PubMed.
- G. Tsuji, T. Takeda, I. Furusawa, O. Horino and Y. Kubo, Pestic. Biochem. Physiol., 1997, 57, 211–219 CrossRef CAS.
- C. M. Spencer and S. Noble, Drugs Aging, 1998, 13, 391–411 CrossRef CAS PubMed.
- R. Webster, A. Boyer, M. J. Fleming and M. Lautens, Org. Lett., 2010, 12, 5418–5421 CrossRef CAS PubMed.
- D. J. Kucera and R. W. Scott, US Pat. Appl. Publ., 20040204591, 2004 Search PubMed.
- (a) P. A. Dub, N. J. Henson, R. L. Martin and J. C. Gordon, J. Am. Chem. Soc., 2014, 136, 3505–3521 CrossRef CAS PubMed; (b) H.-J. Pan, Y. Zhang, C. Shan, Z. Yu, Y. Lan and Y. Zhao, Angew. Chem., Int. Ed., 2016, 55, 9615–9619 CrossRef CAS PubMed.
- A. Adamkiewicz and J. Mlynarski, Eur. J. Org. Chem., 2016, 1060–1065 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and computational details and optimised Cartesian coordinates. See DOI: 10.1039/d0cc02881a |
| This journal is © The Royal Society of Chemistry 2020 |