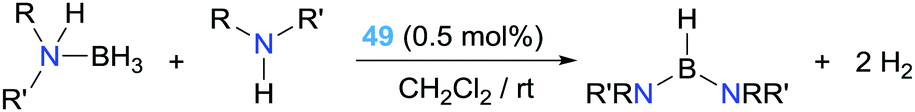DOI:
10.1039/D0CC01438A
(Feature Article)
Chem. Commun., 2020,
56, 5333-5349
Enhancing the catalytic properties of well-defined electrophilic platinum complexes
Received
24th February 2020
, Accepted 28th April 2020
First published on 28th April 2020
Abstract
Platinum complexes have been often considered as the least reactive of the group 10 triad metals. Slow kinetics are behind this lack of reactivity but, still, some industrially relevant catalytic process are dominated by platinum compounds and sometimes different selectivities can be found in comparison to Ni or Pd. Nevertheless, during the last years, it has been reported that the catalytic behaviour of well-defined platinum derivatives can be improved through a judicious choice of their electronic and steric properties, leading to highly electrophilic or low-electron count platinum systems. In this feature article, we highlight some catalytic processes in which well-defined electrophilic platinum complexes or coordinatively unsaturated systems play an important role in their catalytic activity.

Pablo Ríos
| Pablo Ríos received his BSc degree in Chemistry from the University of Seville (Spain) in 2013. Then, he moved to Bristol (United Kingdom) to carry out his MSc(R) studies in the group of Prof. Anthony P. Davis, where he worked on supramolecular chemistry developing pyrene-based receptors for neutral monosaccharides. In 2015, he went back to Seville to do his PhD on organometallic chemistry and homogeneous catalysis, under the supervision of Dr Salvador Conejero and Dr Amor Rodríguez. As part of his PhD studies, he also worked in the labs of Prof. Agustí Lledós at UAB (Spain) and Prof. Christopher C. Cummins at MIT (USA). Currently, he is a postdoctoral fellow at UC Berkeley (USA) in the group of Prof. T. Don Tilley thanks to a Marie Curie Global Fellowship. |

Amor Rodríguez
| Amor Rodríguez received her PhD in 2002 at the University of Oviedo under the supervision of Professors Víctor Riera and Julio A. Pérez. Shortly after, she started a three years postdoctoral stay at the University of California-Riverside with Professor Guy Bertrand. In January 2005, she joined the Group of Professor Ernesto Carmona at the Institute of Chemical Research of Seville (CSIC-University of Seville) and in September 2009 she became Tenured Scientist at the Spanish Research Council. Her research interests are mainly focused on the rational design of organometallic group 10 metal systems and the mechanism analysis of small molecules activation processes. |

Salvador Conejero
| Salvador Conejero received his PhD from the University of Oviedo, working with Prof. José Gimeno and Ma Pilar Gamasa. From 2002 to 2005 he was a post-doctoral fellow at the University of California-Riverside under the supervision of Prof. Guy Bertrand and in 2005 he was awarded with a Ramón y Cajal contract at the University of Seville-Instituto de Investigaciones Químicas (IIQ) in the group of Prof. Ernesto Carmona. Since 2007 he is Tenured Scientist at the IIQ. He has been awarded with the Young Research Award by the Spanish Royal Society of Chemistry (RSEQ) (2007) and with Excellence Research Award by the Orgamometallic Group of the RSEQ (2016). |
Introduction
Reports on homogeneous catalysis mediated by group 10 metals are widespread in the literature. Indeed, this triad is able to carry out a great number of chemical transformations, yet the catalytic properties of these elements dramatically vary from each other. In fact, there exists the notion that Pt is much less reactive than its lighter counterparts, and this is supported by different physicochemical data such as stronger Pt–ligand bond dissociation energies1 or higher kinetic barriers for oxidative addition and reductive elimination phenomena,2 apart from its low tendency to undergo one electron redox events.3 For these reasons, Pt is often employed as a mechanistic model so as to understand the successive elementary steps of a catalytic cycle by analysing the outcome of its corresponding stoichiometric reactions, for example.4 However, the role of Pt in catalysis is of paramount importance, given that it is the metal of choice in industrially relevant processes such as olefin hydrosilation5 and because of its ability to activate inert bonds such as those in methane (Shilov's6 and Periana's7 systems are representative cases of this type of transformation).
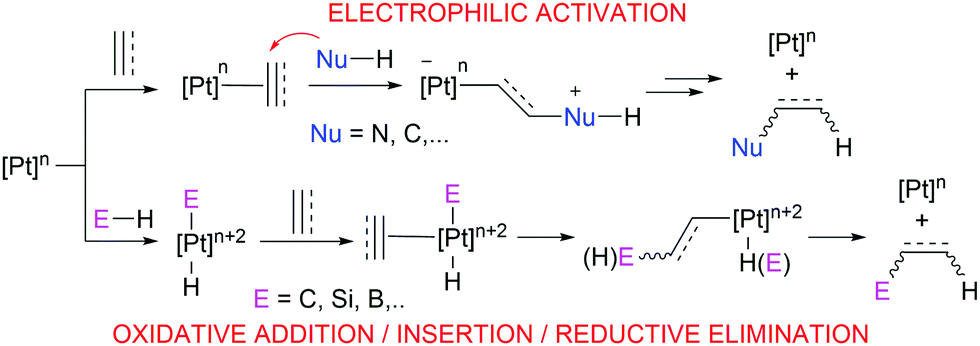
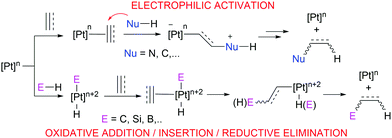
Pt-Catalysed olefin hydrosilation usually takes place via either Chalk–Harrod or modified Chalk–Harrod mechanisms, which include oxidative addition and reductive elimination steps, as it also happens in CH bond functionalisation reactions (Scheme 1). Nonetheless, the Lewis acidity of Pt(II) and its soft character open the door to different types of reactivity involving electrophilic activation of olefins or alkynes for example (Scheme 1), in comparison to other group 10 metals. As a matter of fact, cationic or low-electron count Pt derivatives are especially relevant in this type of reactions since coordination of the olefin to the metal fragment renders the multiple bond electrophilic, making it susceptible to nucleophilic attack. This is partially due to the decreased (although not negligible) degree of backbonding from the Pt center to the π* orbital of the olefin.8 Indeed, the increase in the positive charge on the metal complex seems to be a key factor in alkene activation. This has been shown experimentally9 and theoretically.10 For instance, cationic monoolefinic complexes of Pt tend to give addition reactions with oxoanions such as hydroxide, alkoxide or carboxylate groups,11 whereas neutral Pt derivatives tend to undergo substitution reactions.12 However, DFT studies have revealed that olefin fragments bound to cationic metal complexes in η2 coordination modes still remain inactive towards attack by nucleophiles; instead, a slippage movement of the alkene to η1 coordination is required for the electrophilic activation to occur.10 This phenomenon would be a plausible explanation to the usual Markovnikov regioselectivity observed in monosubstituted aliphatic and aromatic olefins (see below), and it also highlights the importance of the coordination mode of certain ligands and how their resulting geometry affects their reactivity.
 |
| | Scheme 1 | |
Therefore, platinum complexes can be regarded as versatile catalysts which can lead to catalytic processes involving oxidative addition/reductive elimination events or as Lewis acids involving electrophilic activation reactions with no changes in its oxidation state. It is the aim of this feature article to show how it is possible to modulate the catalytic performance of well-defined platinum systems to enhance its chemical reactivity leading to more efficient processes using either mono- or di-cationic, or low-coordinate platinum complexes, or systems that can behave in a similar way. Thus, either ligand design or structure design of the metal complexes is a key feature in the reactivity they exhibit. This is not an exhaustive review-type article but an account through some selected and important reaction processes involving this type of platinum complexes.13
Hydroarylation of C–C multiple bonds
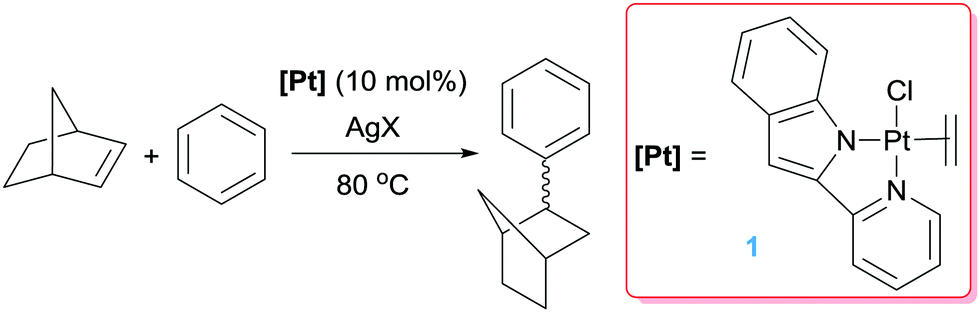
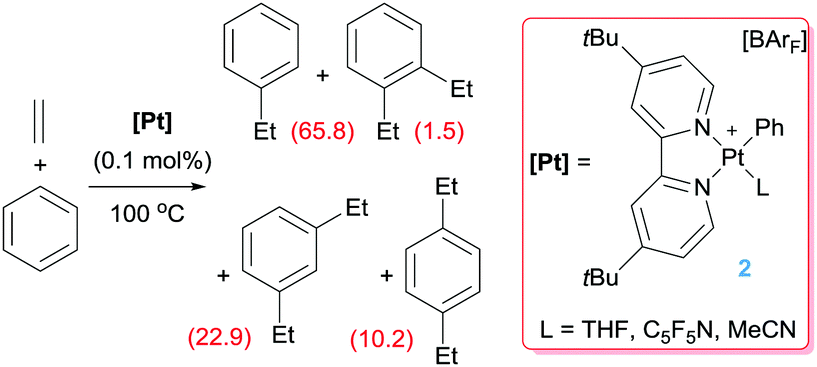
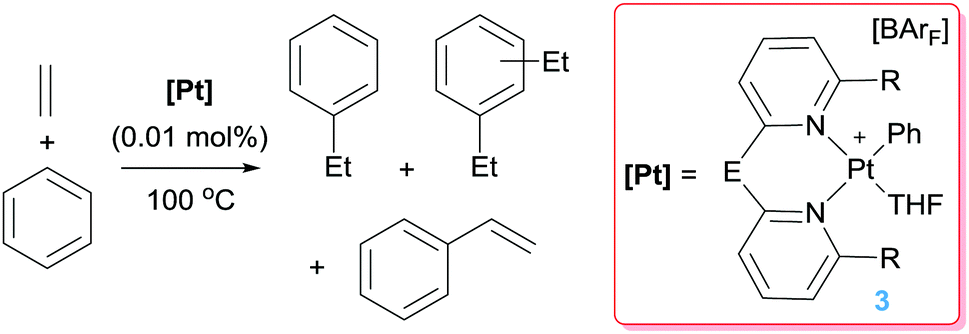
Platinum complexes have played a major role in the study of C–H bond activation and functionalisation reactions of hydrocarbons. The discovery first by Garnett and Hodges,14 followed by Shilov and co-workers6 that platinum salts can cleave carbon–hydrogen bonds of hydrocarbons provided the basis to new opportunities in which the design of new architectures based on platinum complexes led to key discoveries and understanding of the critical steps involved in the activation of C–H bonds of hydrocarbons. The readers can find excellent reviews in the literature covering most of the important findings in the field, some of which are quite recent.15 It is not the scope of this feature article to duplicate such information but to focus on well-defined platinum complexes which are electrophilic or low-coordinate, or that can lead to transient species of this nature. In this sense, one particular process that has benefited for these type of species is the formal addition of a C–H bond of an aromatic compound into a multiple bond, a process known as hydroarylation. Early studies by Fujiwara et al.16 showed that platinum salts in combination with silver salts are amenable systems for the hydroarylation of alkynes through electrophilic metalation of C–H bonds of aromatic compounds. Thereafter, some efforts were geared at developing platinum systems that could provide access to low-coordinate, 14-electron Pt(II) species since they have been proposed as key intermediates in the activation of C–H bonds of hydrocarbons,15,17 an important fundamental elementary step in hydroarylation reactions. Indirect evidence for the participation of these species in these processes has been provided in several examples,15c but our group reported the first evidence in which an isolable Pt(II) 14-electron compound can intermolecularly activate C–H bonds of aromatic compounds.18 From a catalytic perspective in the arena of hydroarylation reactions, one of the first examples was developed by Tilley, Bell and co-workers.19 Complex 1 in Scheme 2, together with a silver salt to generate a vacant site, proved to be active in the hydroarylation of norbornene with benzene, although with comparable activity than other platinum complexes such as Zeise's dimer activated by silver salts (ca. 90% yield in 2 h). In search for new catalysts, Gunnoe et al. found that the cationic bipyridine Pt(II) complex 2 having labile ligands such as THF, C5F5N or MeCN that can de-coordinate from the metal, hydroarylates benzene and other benzene-substituted organic compounds.20 The reaction of benzene and ethylene catalysed by complex 2 leads to ethylbenzene accompanied by 20–25% of diethylbenzenes (Scheme 3). The ratio of the ortho-, meta- and para-diethylbenzene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15.3:13.6, respectively) formed in the reaction is in accordance with a catalytic system working via a platinum-mediated C–H bond activation process instead of the typical Friedel–Crafts reaction pathway usually observed in hydroarylation reactions. The same conclusion can be drawn from the fact that the reaction of propylene with benzene produces certain amounts of the linear n-propylbenzene derivative, and has been supported by kinetic experiments and DFT calculations.20a Optimization of the reaction conditions lead to a system operating at 120 °C (0.025 mol% of 2) yielding 89% of alkyl-arene products.20a The fact that the catalyst is not working under Friedel–Crafts conditions but by insertion of olefins into Pt–aryl bonds is very attractive, since selectivities can be completely different and could be controlled through modifications of the ligands around the metal centre.
:15.3:13.6, respectively) formed in the reaction is in accordance with a catalytic system working via a platinum-mediated C–H bond activation process instead of the typical Friedel–Crafts reaction pathway usually observed in hydroarylation reactions. The same conclusion can be drawn from the fact that the reaction of propylene with benzene produces certain amounts of the linear n-propylbenzene derivative, and has been supported by kinetic experiments and DFT calculations.20a Optimization of the reaction conditions lead to a system operating at 120 °C (0.025 mol% of 2) yielding 89% of alkyl-arene products.20a The fact that the catalyst is not working under Friedel–Crafts conditions but by insertion of olefins into Pt–aryl bonds is very attractive, since selectivities can be completely different and could be controlled through modifications of the ligands around the metal centre.
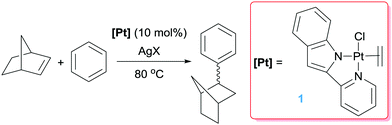 |
| | Scheme 2 Hydroarylation of norbornene catalysed by complex 1. | |
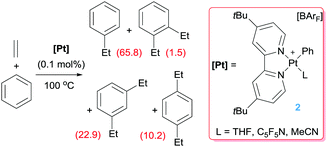 |
| | Scheme 3 Hydroarylation of ethylene catalysed by complex 2 (turnovers are given in parenthesis for each compound after 16 h of reaction). | |
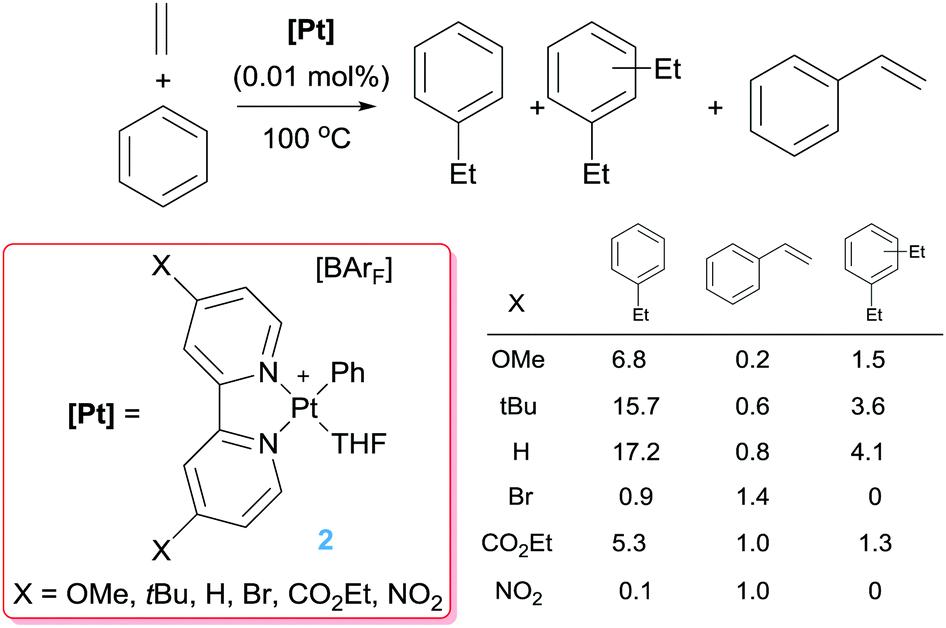
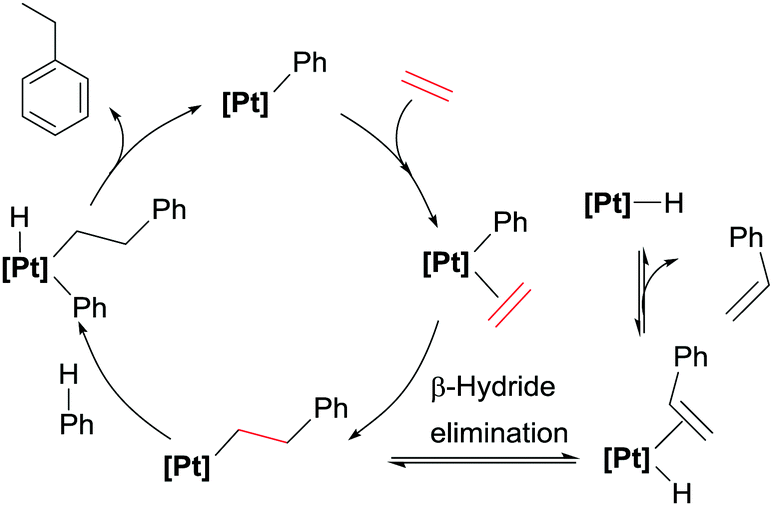
Comparison of these cationic catalysts with related neutral systems (see below) suggest that the production of diethylbenzenes might be inhibited by decreasing the electrophilic character at the platinum centre, since mechanistic studies point out that dissociation from the coordination sphere of the ethylbenzene formed initially (that can undergo a CH bond activation leading to the diethylbenzenes) is substantially more difficult in cationic systems. To understand the effect of the electronic properties of the platinum atom in the selectivity and rate of the reactions, bipyridines with substituents at the 4,4′-positions with different electronic properties have been evaluated (Scheme 4).21 Nevertheless, it has been observed that the influence of the ligand has a more important impact on the competitive formation of styrene rather than on the distribution of ethylbenzene and diethylbenzenes. In general, less-donating groups tend to increase the amounts of styrene formed during the catalytic reaction, working at higher rates than Pt complexes bearing more electron-rich bipyridines (in the series were X = H, tBu and OMe). However, the systems with electron-withdrawing groups (Br, CO2Et, NO2) are more prone to decomposition, something that has been linked to more rapid β-hydride elimination processes with electron-poor bipyridines leading to more unstable platinum-hydride intermediates (as shown in Scheme 5).
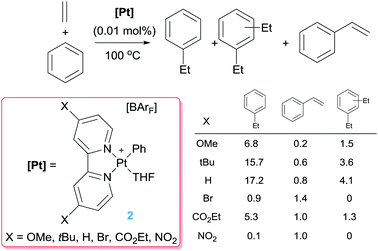 |
| | Scheme 4 Hydroarylation of ethylene catalysed by complexes 2. Table shows product distribution (turnovers) after 4 h of reaction. | |
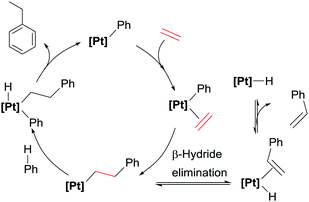 |
| | Scheme 5 Proposed catalytic cycle for hydroarylation process and competitive β-hydride elimination reaction leading to styrene. | |
On the other hand, the hydroarylation of benzene with propene by complexes 2 lead to mixtures of the branched (cumene) or linear (n-propylbenzene) products (3:1 ratio respectively). Bipy ligands with electron-poor substituents at the 4,4′ position originate mixtures in which the cumene/n-propylbenzene is higher (branched to linear product ratios ranging from 3.8 to 4.6).22 The stability of the platinum catalyst is an important issue in order to develop more efficient catalytic systems. A further step to improve the longevity of the catalyst was made by changing the bipy-type ligands by dipyridyls, such as 2,2-dipyridylmethane (dpm) (Scheme 6).23 Bipy and dpm have rather close electronic properties, but different steric properties. It has been shown that complexes of type 3 bearing a dpm ligand (E = CH2, R = H in Scheme 6) are about 5.6 times more active than tBubipy systems of type 2 under the same reaction conditions (Scheme 3). In addition, whereas type 2 complexes deactivate after a period of 24 h, complexes 3 remain active for more than 4 days (leading to turnover numbers up to 469). According to mechanistic studies and DFT calculations, entropic factors arising from an increase on the ring size from 5 to 6-membered rings (going from tBubipy ligand to dpm) are responsible for these beneficial effects. However, the efficacy of the dpm-based platinum complex is lower (TO 11.8 in 4 h) than that of the tBubipy (TO 33.5 in 4 h) in the hydroarylation of propene and benzene, presumably due to steric effects.22 With respect to the selectivity of the processes, although styrene is a minor component, diethylbenzenes account for about 20% of the final products. Attempts to modify the dipyridyl ligand by means of different E or R groups (E = CH2CH2, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, NH, O; R = Me) did not lead to improvement of the catalyst activity, due in part to catalyst deactivation, although some benefits in selectivity were observed when Me groups are at the 6,6′ position of the pyridyl units (R = Me).24
O, NH, O; R = Me) did not lead to improvement of the catalyst activity, due in part to catalyst deactivation, although some benefits in selectivity were observed when Me groups are at the 6,6′ position of the pyridyl units (R = Me).24
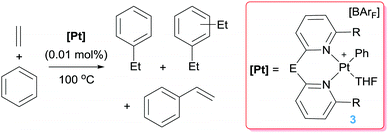 |
| | Scheme 6 Hydroarylation of ethylene catalysed by complexes 3. | |
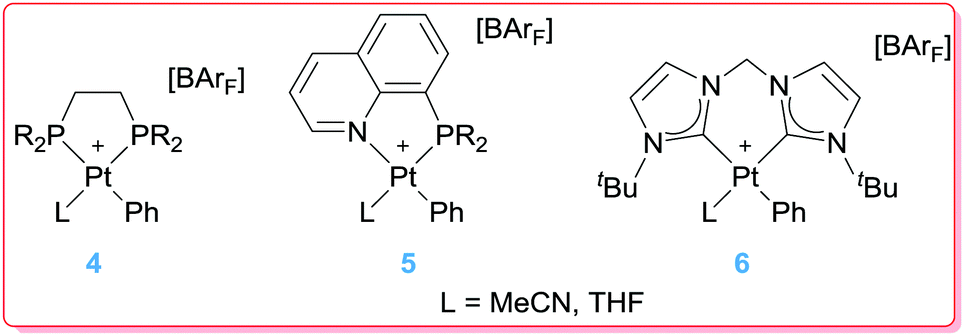
Having in mind that platinum complexes bearing bipyridine ligands with electron-withdrawing groups at the 4,4′ position increase the amount of styrene in the reaction of benzene with ethylene catalysed by complexes 2, the next step was to evaluate the effect of incorporating in the structure of the catalyst more electron rich ligands such as phosphines or N-heterocyclic carbenes such as those in Fig. 1.25 Contrary to initial expectations, the reaction with the platinum complexes bearing di-phosphines gave rise to styrene as a major product with very low efficiency (less than 2 turnovers after 4 h at 100 °C), whereas systems incorporating mixed phosphine–pyridine units or bis-NHCs (4–6 in Fig. 1) were completely ineffective.
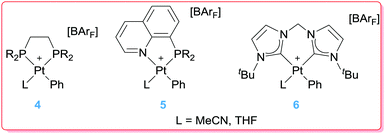 |
| | Fig. 1 Phosphine and NHC-based Pt complexes. | |
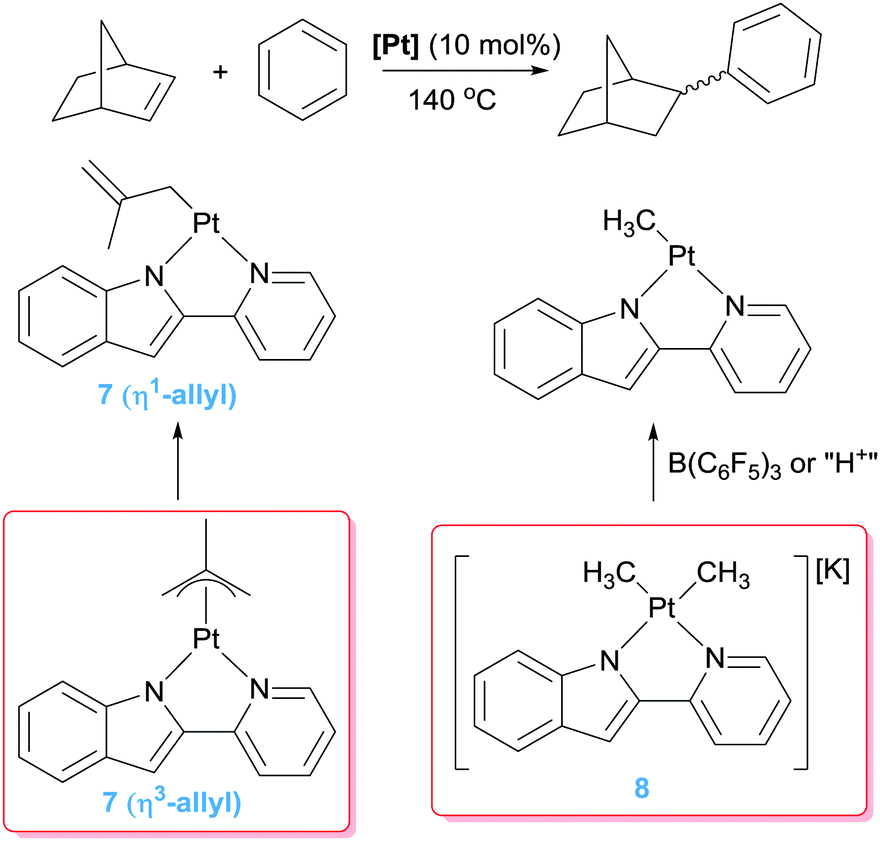
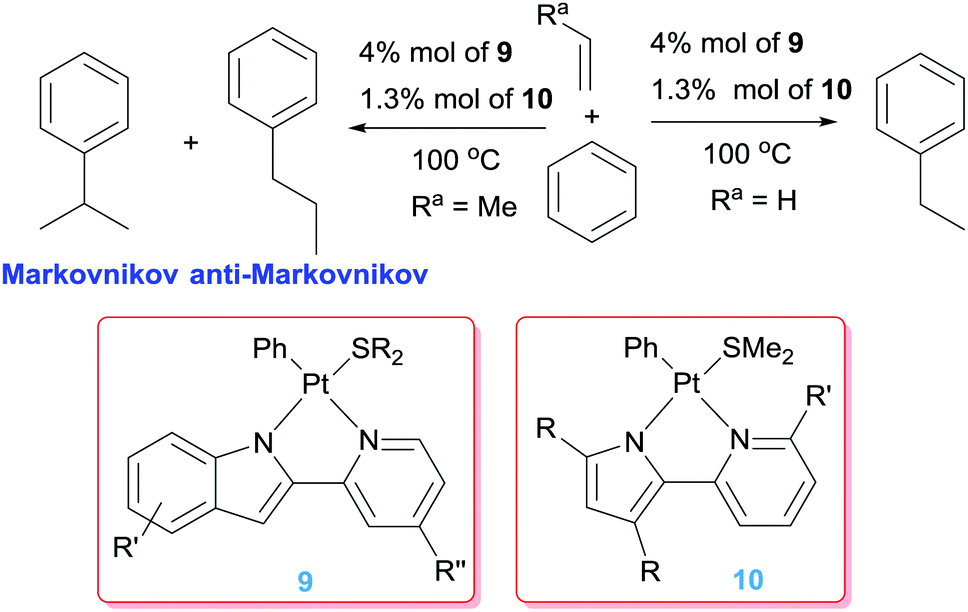
Besides the use of cationic platinum complexes for the hydroarylation of alkenes, the groups of Goldberg and Tilley, independently, have explored platinum derivatives with monoanionic ligands as a source of transient (and competent), neutral 14-electron platinum(II) intermediates. In 2006 Tilley et al. hypothesised that complexes 7 (Scheme 7) could provide an entry to 14-electron Pt(II) species through a change in the coordination mode of the allyl ligand from η3 to η1. Alternatively, analogous low-coordinated derivatives could be targeted by methyl abstraction or protonation of complexes of type 8.26 It turned out that these kind of platinum complexes are active in hydroarylation of benzene, although with important limitations since, besides relatively moderate conversions (38%), the process works only with norbornene. A few years later, Tilley and co-workers synthesised a series of platinum complexes with 2,2′-pyridyl-indolate ligands (9 in Scheme 8) with different electronic and steric properties and evaluated them as catalysts in the hydroarylation of alkenes.27 As expected from previous results from Goldberg's group using related platinum catalysts (see below), the reaction proceeds with the nearly exclusive formation of ethylbenzene (turnovers ranging from 7.4 to 25.2), with only traces of over-alkylated products such as diethylbenzenes being detected (less than 1 turnover in all cases) (Scheme 8). However, it was not easy to rationalise the effect of the substituents on the ligand on the activity of the catalysts. One important remark from these systems is that their efficiency is controlled by catalyst decomposition through, mainly, β-hydride elimination processes (Scheme 5) leading to unstable platinum-hydride species that can undergo elimination of the ligand from the coordination sphere giving rise to platinum black (generally after 24 h). Thus, once again, as in some of the systems reported by Gunnoe, an important issue to take into account in the catalytic hydroarylation of alkenes is formation of unstable platinum-hydride complexes.
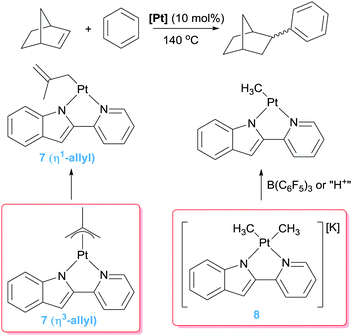 |
| | Scheme 7 Hydroarylation of norbornene catalysed by complexes 7 and 8. | |
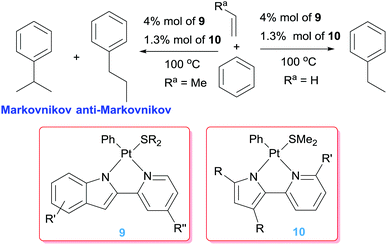 |
| | Scheme 8 Hydroarylation of olefins catalysed by complexes 9 and 10. | |
A few years before, Goldberg et al. observed that during the thermolysis of the 16-electron Pt(IV) complex [PtMe3(dmpp)] (dmpp = 3,5-dimethyl-2-(2-pyridyl)pyrrolide) in C6D6 in the presence of ethylene certain amounts of ethylbenzene were detected consistent with catalytic activity in hydroarylation of ethylene.28 Since it was known that thermolysis of related Pt(IV) complexes undergo reductive elimination processes of ethane to generate transient Pt(II) 14-electron species of the type [PtMeL2], complexes of the type 10 (Scheme 8) were targeted as potential catalysts for the hydroarylation of propene and other substituted olefins.29 It was found that all of them yield mixtures of both Markovnikov and anti-Markovnikov additions with little formation of styrene products derived from β-hydride elimination processes, with the exception of the pyridine methyl-substituted system (R′ = Me; R = Me in complexes 10) for which this process is prevalent. The catalyst with substituents at the pyrrolide (R = Me in complexes 10) gave higher amounts of the branched products (Markonikov/anti-Markovnikov ratios up to 85:15 with TONs up to 17.9). However, the complexes without Me groups at the pyrrolide fragment (R = H and R′ = H in complexes 10) are less efficient (TONs up to 10.7), but a significant increase of the linear products is observed (leading to nearly 1:1 ratios of the products). The decrease in activity for this latter system has been justified by the less basic nature of the unsubstituted pyrrolide ligand, which translates into a lower propensity of the platinum centre to activate C–H bonds through an oxidative addition mechanism. Higher selectivities toward anti-Markovnikov products were observed when bulkier alkenes such as neohexene were used (up to 94:6 selectivities).
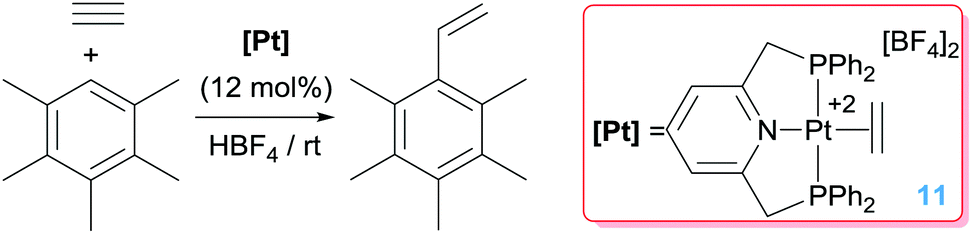
The hydroarylation of alkynes is also a challenging reaction, in which the control of the selectivity is an important issue as it is the finding of efficient catalytic systems.30 Some examples on the catalytic activity of platinum salts in this relevant process have been reported,16 but it has been shown that some well-defined platinum complexes are indeed very active. Hahn et al. reported that dicationic Pt(II) complexes activate alkynes once bound to the metal centre, to the point that they can undergo nucleophilic addition of electron-rich arenes under very mild reaction conditions through a Friedel–Crafts type process (as in Scheme 1).31 Under acidic conditions it is possible to cleave the generated [Pt]–C bond releasing the hydroarylated product and regenerating the catalyst (Scheme 9). This last step is rate-limiting since it is known that protonolysis of dicationic [Pt]–C bonds is a rather difficult process.32
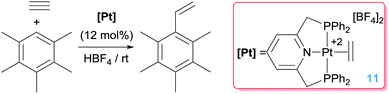 |
| | Scheme 9 Hydroarylation of acetylene catalysed by complex 11. | |
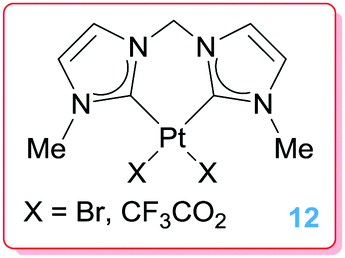
Biffis et al. explored the use of platinum bis-NHC complexes 12 (Fig. 2) in hydroarylation of activated alkynes and in the related insertion of alkynes into CH bonds of heterocyclic compounds. The use of NHC ligands would benefit from the excellent stability of their complexes, even under acidic reaction conditions. These complexes were, however, less efficient than their palladium counterparts (using catalyst loadings of 1%) both in terms of activity (conversions of 40–58%, vs. 99% for Pd derivatives) and selectivity (selectivities for Z isomers up to 68 vs. 79 for Pd catalysts).33
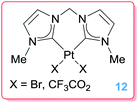 |
| | Fig. 2 Platinum bis-NHC complexes. | |
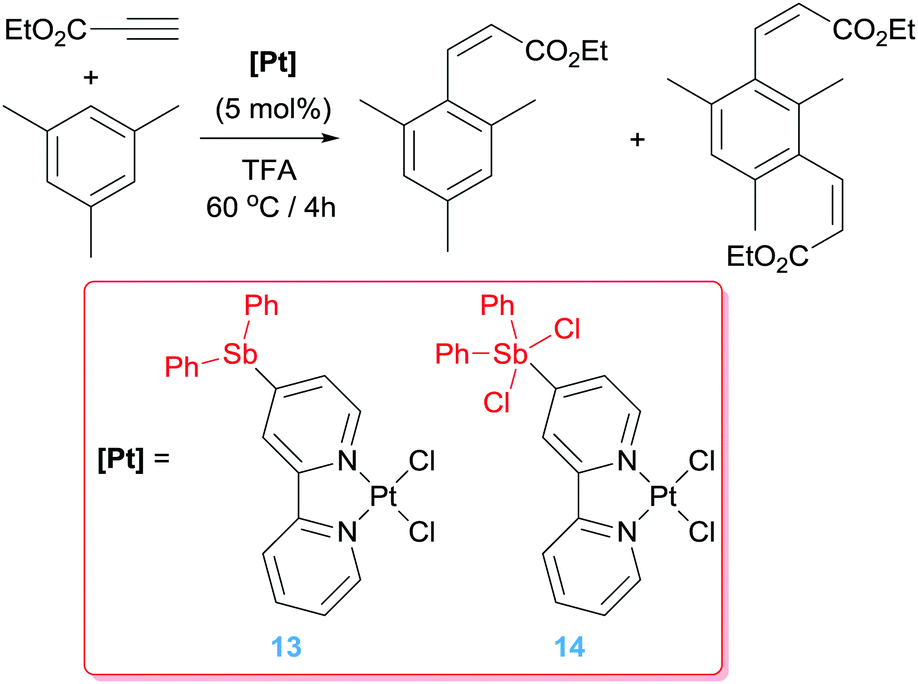
Alternative ways to increase the electrophilic character of the metal and therefore to improve catalytic efficiency has been focused on ligand modifications. In this regard, Gabbaï and co-workers developed antimony-substituted bipyridine ligands and studied the effect of the oxidation state on the Sb in their reactivity (Scheme 10),34 most particularly if long-range effects, without the Sb atom directly interacting with the metal centre,35 would have an impact in the electronic properties of platinum. Cyclic voltammetry, together with UV-vis and DFT studies indicated a decrease on electron density in system 14 lowering its LUMO. In fact, whereas complex 13 shows similar catalytic behaviour than the unsubstituted bipy complex [PtCl2(bipy)] in the hydroarylation of an activated alkyne with mesitylene (leading to yields of 55–65% and selectivities of 1:1 of mono- and di-substituted alkene) (Scheme 10), an enhanced catalytic activity is observed when complex 14 was used under identical reaction conditions (yields of 83% with selectivities of 2.9:1 of mono and di-substituted alkene, respectively).
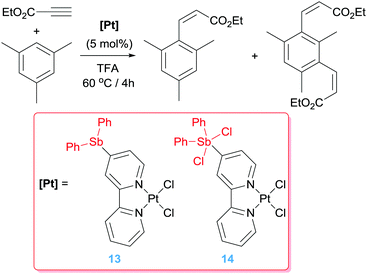 |
| | Scheme 10 Hydroarylation of electron-poor alkynes catalysed by complexes 13 and 14. | |
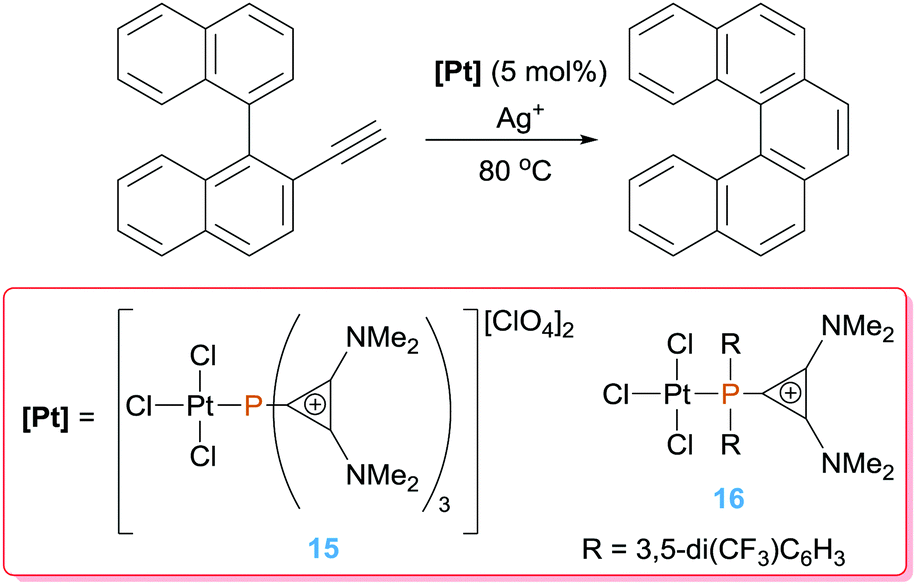
The intramolecular version of alkyne hydroarylation has been studied by Alcarazo and co-workers for the synthesis of polycyclic homo- and hetero-arenes. To increment the Lewis acidity of the platinum centre the group of Alcarazo devised the use of cationic phosphines, that according to computational studies behave as poor σ-electron donors and good π-acceptors.36 As expected, their complexes have a LUMO lower in energy than that in their corresponding species bearing neutral phosphine ligands such as PPh3. Complex 15 (activated with a silver salt), bearing a tricationic phosphine ligand, boosts the catalytic hydroarylation of ortho-biaryls substituted alkynes with full conversion in less than 20 min (Scheme 11) in comparison to other typical π-acceptor phosphines such as PF3 (600 min with <50% conversion) leading selectively to the corresponding 6-endo-dig polycyclic derivatives. A further improvement of the catalytic efficiency came with the use of complex 16, in which the phosphine ligand is now monocationic. This catalytic system is even faster than that based on complex 15, completing the reaction in ca. 2 min under identical reaction conditions, in spite of having a net neutral charge. This apparent discrepancy, in which a less electrophilic system gives better results, seems to have two possible origins: on one side the R substituents on the phosphine in complex 16 are 1,3-bis(trifluoromethyl)phenyl groups, that are effective electron-withdrawing groups, and on the other hand solubility issues might be also important, since dicationic pre-catalyst 15 has a rather low solubility in organic solvents.
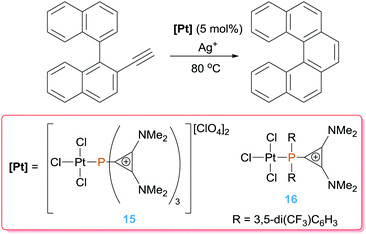 |
| | Scheme 11 Intramolecular hydroarylation of alkynes catalysed by complexes 15 and 16. | |
Olefin dimerisation
In some occasions, the binding of the olefin to a platinum centre induces a significant activation of the double bond conferring a carbocation-like reactivity of the carbon atoms (Scheme 1). Earlier reports by Vitagliano et al. showed that dicationic Pt(II) precursors have an important impact on the activation of the olefin by several orders of magnitude in comparison with monocationic or neutral complexes.37 Thus, the olefin is susceptible for nucleophilic addition of electron rich olefins leading to a co-dimerization process of ethylene/olefins as shown in Scheme 12 (95% conversion of the starting material). Importantly, a modification of the ligand in the coordination sphere at platinum can alter the outcome of the reaction promoting a cyclopropanation reaction. Both the hydrovinylation and the cyclopropanation seem to occur through a common carbocation intermediate (Scheme 12). The selectivity of the process is linked to the trans influence of the central fragment of the pincer ligand, with systems bearing low trans influence moieties, such as pyridine (18), directing the reaction towards a hydrovinylation (95:0 ratio hydrovinylated:cyclopropanated products) and the high trans influence ligands, such as phosphines (17), to cyclopropanation (0:100 ratio hydrovinylated:cyclopropanated products with 11% conversion). A similar behaviour was observed with catalyst 19, for which an excellent selectivity (>99%, 68% conversion) towards hydrovinylation is observed, although under harsher reaction conditions (Scheme 12).38
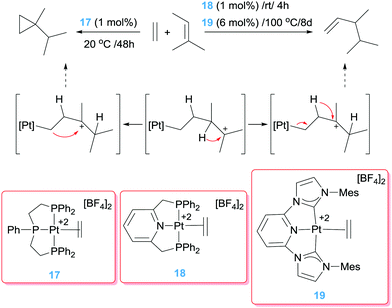 |
| | Scheme 12 Olefin dimerization and cyclopropanation catalysed by complexes 17–19. | |
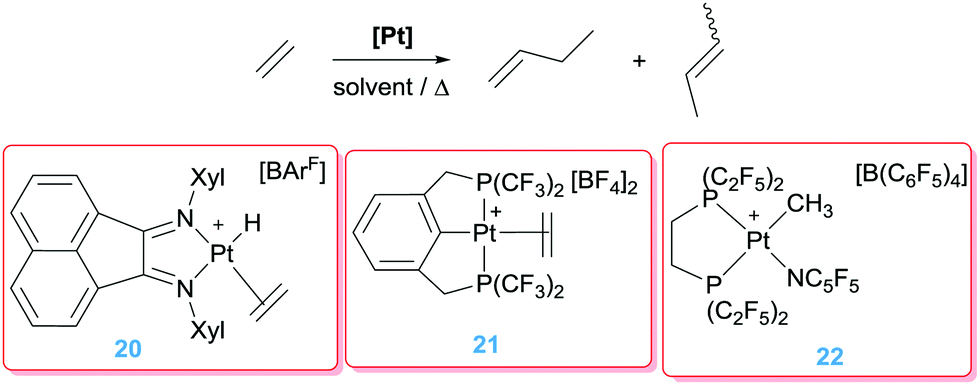
It's worth noting that ethylene itself is not involved in a dimerisation process. The catalytic system only works in the presence of a more nucleophilic alkene. However, Brookhart, Templeton and co-workers observed that the monocationic platinum catalyst 20 (Scheme 13) is involved in the dimerisation of ethylene into butenes.39 Moreover, the mechanism of the reaction involves classical hydride/ethylene and alkyl/ethylene insertion reactions rather than nucleophilic addition into a coordinated olefin as observed by Vitagliano, something likely due to the monocationic nature of complex 20. In search for transient Pt(II) 14-electron species with enhanced electrophilicity Roddick et al. reported the synthesis of platinum complexes bearing fluorinated bis-phosphine ligands (21 and 22) expecting an increased reactivity by means of the good π-acceptor properties of these phosphines. In fact, complex 22 is some orders of magnitude better than complex 20 (achieving TOF numbers up to 150 h−1 at 22 °C vs. 0.1 h−1 by 20 at 100 °C). On the contrary, the pincer-based platinum complex 21 showed an intermediate activity (TOF 7 h−1 at 20 °C).40
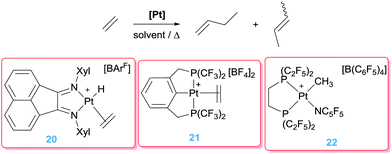 |
| | Scheme 13 Olefin dimerization and cyclopropanation catalysed by complexes 20–22. | |
Cycloisomerisation of dienes and enynes
Cycloisomerisation reactions of multiple bonds catalysed by platinum systems are, arguably, one of the most important processes in which this metal is involved. The cycloisomerisation allows for the synthesis of complex molecules with a large structural diversity under complete atom economy and mild reaction conditions.41 Platinum compounds have shown a marked propensity to activate multiple bonds facilitating further reactivity patterns that depends strongly on the nature of the reaction conditions, substrate used, ligands surrounding the metal centre, etc. The π-acidity of platinum triggers the reaction by transferring the electrophilicity of the metal to the multiple bond. Seminal works by Murai,42 followed by intense research developed by Fürstner on the use of platinum salts43 initiated a vibrant research in the field that was complemented by the development of gold catalysis.41a,c Although initial reports were based on the use of platinum salts, it was soon recognised that the electronic properties of the platinum centre can be modified through a judicious choice of ligands to increment its chemical reactivity and direct the catalytic processes to more selective ones.
Cyclisations of dienes
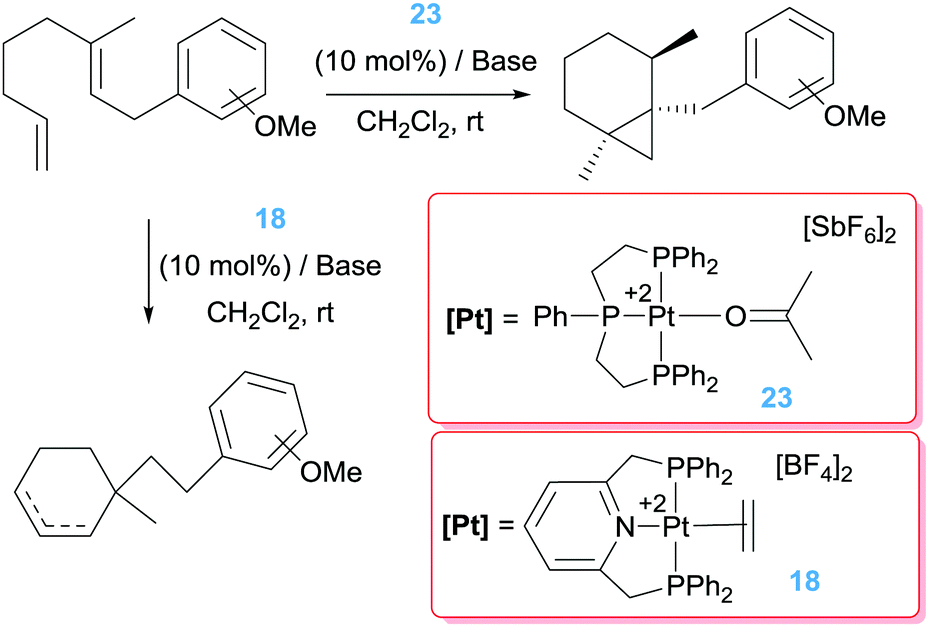
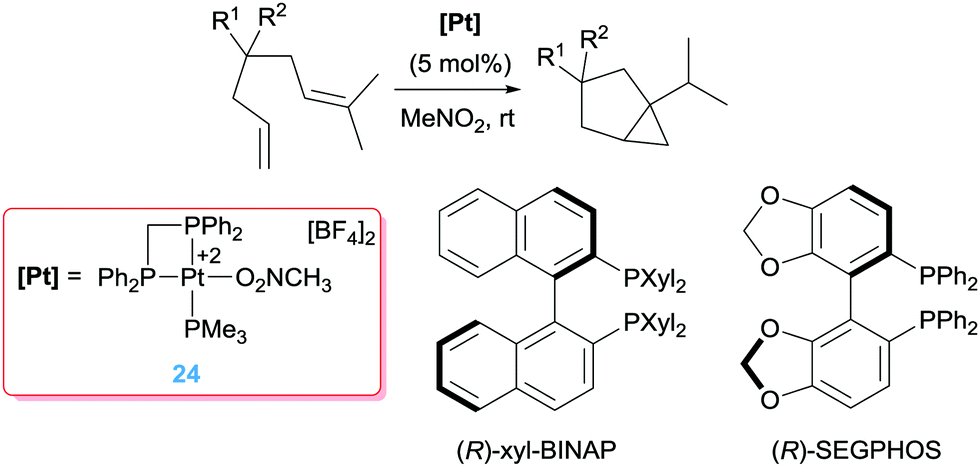
Pioneering work by Gagné, inspired by the work of Vitagliano on the dimerization of olefins,37b involved the use of well-defined dicationic platinum complexes stabilised by tridentate phosphine based ligands (bis(diphenylphosphinoethyl)phenylphosphine, triphos) (Scheme 14).44 This ligand architecture was selected, on the one hand, to stabilise the dicationic platinum centre and on the other hand, to prevent (or reduce) possible β-hydride elimination processes. To their surprise, complex 23 catalysed the isomerisation of the diene shown in Scheme 14 into a bicyclic structure containing a cyclopropane unit with diastereomeric ratios of 95:5. The reaction takes place at faster reaction rates when the counteranion is SbF6− whereas the more coordinating BF4− leads to a catalyst operating at slower rates, suggesting the necessity of generating a vacant site at the platinum centre. Interestingly, changing the nature of the pincer ligand, incorporating pyridine unit in the structure, results in catalyst 18 with a completely different outcome of the reaction (Scheme 14), in line with the results observed by Vitagliano in the previous section.37a The chiral version of this catalytic reaction is challenging as a consequence of the difficulties in synthesising chiral tridentate phosphine ligands. To circumvent this problem, the same group explored other type of platinum complexes based on mixtures of diphosphine/monophosphine ligands (Scheme 15).45 In spite of the possibility of having side reactions arising from β-hydride elimination processes from some intermediates (due to easiness of ligand dissociation of the monophosphine ligand in comparison to tridentate ligands), no such a process is observed. In fact, this had led to a catalyst that is 20 times faster than the tridentate analogue in the same reaction, operating at lower temperatures (rt instead of 40 °C).46 One of the reasons for having a more reactive system seems to be due to the use of a bis(diphenyl)phosphinomethane (dppm) ligand, in which the smaller bite angle opens more room facilitating the rate-limiting step of the process. Diphosphine ligands with bigger bite angles led to inferior catalytic activities. Varying the dppm ligand by chiral diphosphine versions (Scheme 15) resulted in the catalytic conversion of the dienes into bicyclopropane products with enantioselectivities up to 96%. Somehow surprisingly, it has been observed that the synthesis of bicyclopropanes can be carried out with platinum complexes bearing formally one diphosphine ligand ([(R-BINAP)Pt][BF4]2) raising the question about the role of the third phosphine ligand in precluding β-hydride elimination competing processes.
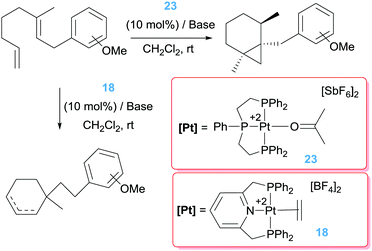 |
| | Scheme 14 Isomerisation of dienes catalysed by complexes 23 and 18. | |
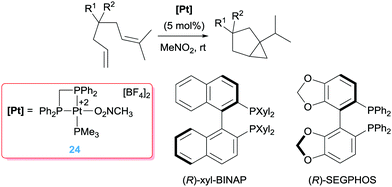 |
| | Scheme 15 Cycloisomerisation of dienes catalysed by complex 24 and chiral phosphines leading to best enantiomeric excess. | |
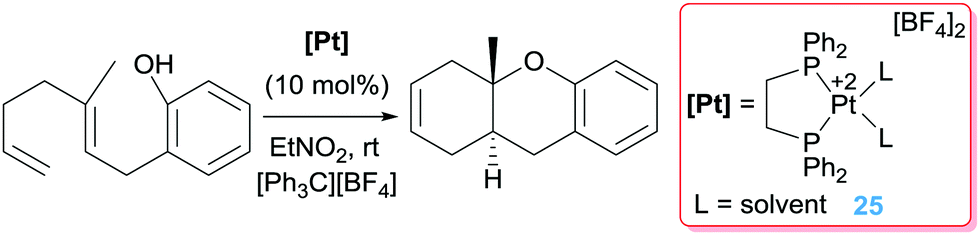
In other occasions, the β-hydride elimination pathway is desirable for the construction of polycyclic structures through polyene cascade reactions catalysed by electrophilic platinum complexes. To this end, Gagné and co-workers devised the possibility of using the dicationic platinum system [Pt(dppe)][BF4]2 (dppe = 1,2-bis(diphenylphosphino)ethane) in combination with a hydride abstractor [Ph3C][BF4] to initiate a cascade reaction leading to polycyclic structures in yields between 45 to 90% (Scheme 16).47
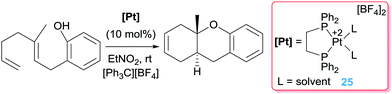 |
| | Scheme 16 Polycyclic synthesis catalysed by complex 25. | |
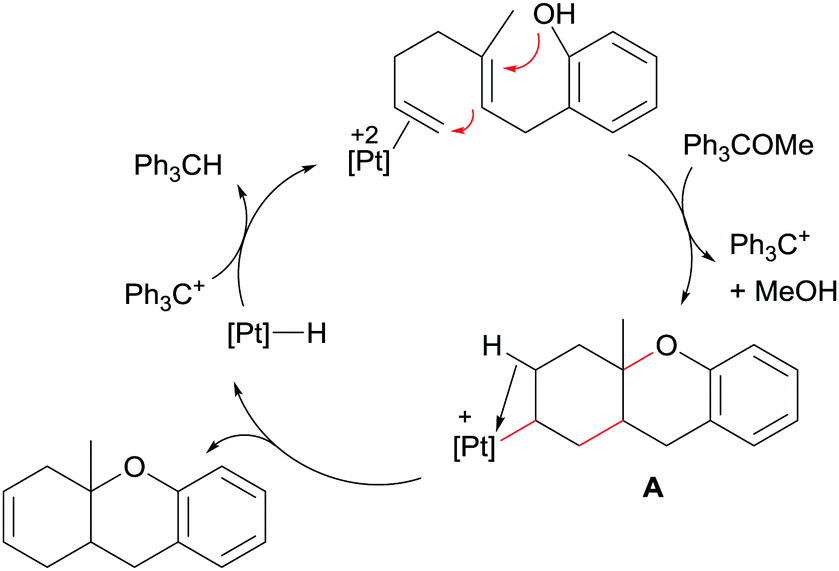
A key point in the catalytic process, besides using a very electrophilic platinum dication, is the use of a hydride abstractor (in the form of Ph3C+, generated in situ from Ph3COMe)48 to regenerate the active catalytic species from the monocationic platinum-hydride intermediate, [Pt(H)(dppe)][BF4], generated after β-hydride elimination from the carbocyclic alkyl intermediate formed in the cyclisation event (Scheme 17). It is worth mentioning that the monocationic platinum hydride species [Pt(H)(dppe)][BF4] is not able to catalyse the reaction itself, demonstrating the need of a more electrophilic dicationic species with two formally vacant sites to promote the whole catalytic cycle.
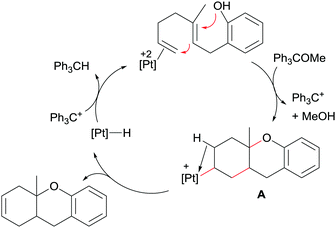 |
| | Scheme 17 Proposed mechanism for the cyclisation of dien-ols. | |
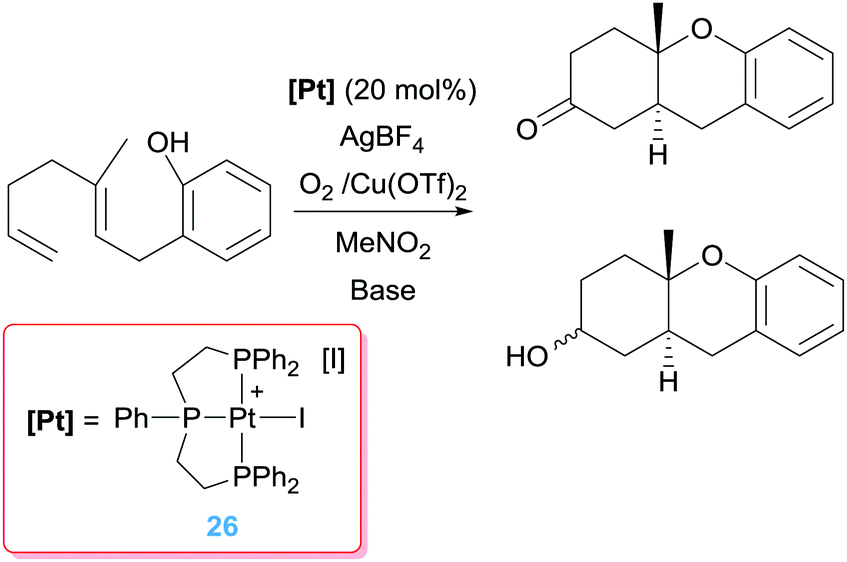
Intermediate A in Scheme 17 has been observed by NMR spectroscopy during the catalytic cycle in both the symmetric and the enantioselective version of the process.49 As mentioned above, the catalytic system comprises a platinum derivative with a diphosphine ligand in order to favour the β-hydride elimination process. If, on the contrary, a tridentate ligand such as triphos is used, the catalytic reaction is blocked since the carbocyclic alkyl platinum species A (Scheme 17) is not able to undergo the β-hydride elimination reaction. Nonetheless, Gagné et al. demonstrated that it is possible to cleave the Pt–C bond by means of oxidations processes, using oxidants (O2 combined with Cu(II) salts) that are compatible with the regenerated Pt catalyst (Scheme 18), although high catalytic loadings are required to achieve moderate to good yields (46–64%) of the mixture products (alcohol:ketone).50
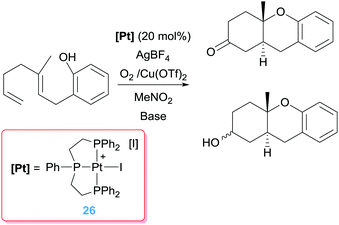 |
| | Scheme 18 Tandem cyclisation/C–O bond formation mediated by complex 26. | |
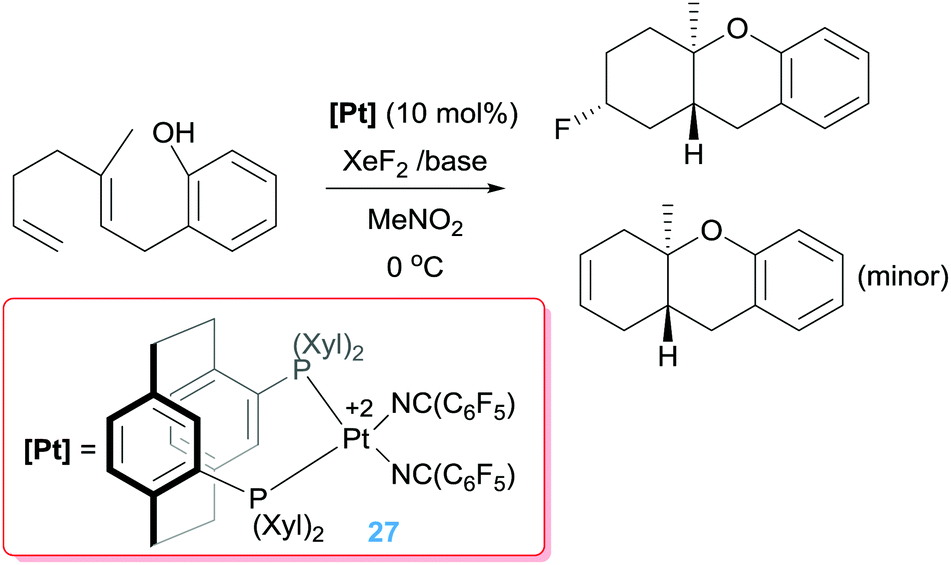
Other oxidants, such as XeF2, seem to be compatible with the catalytic process to create C–F bonds.51 Although initial reports were carried out under stoichiometric conditions using the platinum dication [Pt(triphos)][BF4]2 as a platform,51b the reaction can be carried out under catalytic conditions with diphosphine-stabilised platinum dications yielding fluorinated polycycles (Scheme 19) in good yields (49–80%).51a Remarkably, the generated Pt–alkyl intermediates (of type A in Scheme 17) are intercepted before suffering a β-hydride elimination process, undergoing a faster oxidation by XeF2 but, still, by-products arising from this competitive reaction pathway are formed as minor components (between 0% and 24%). It has been reasoned that the C–F coupling event takes place very fast as a result of steric crowding between the supporting ligands around the metal centre and the “F” and “alkyl” fragments in the putative Pt(IV) intermediates.51a Notably, the fluorinated reagent reacts only with the Pt–alkyl intermediates and not with the dicationic platinum catalyst, making possible the regeneration of the catalyst.
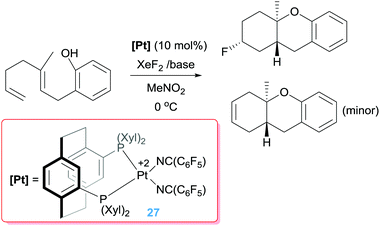 |
| | Scheme 19 Tandem cyclisation/C–F bond forming process catalysed by 27. | |
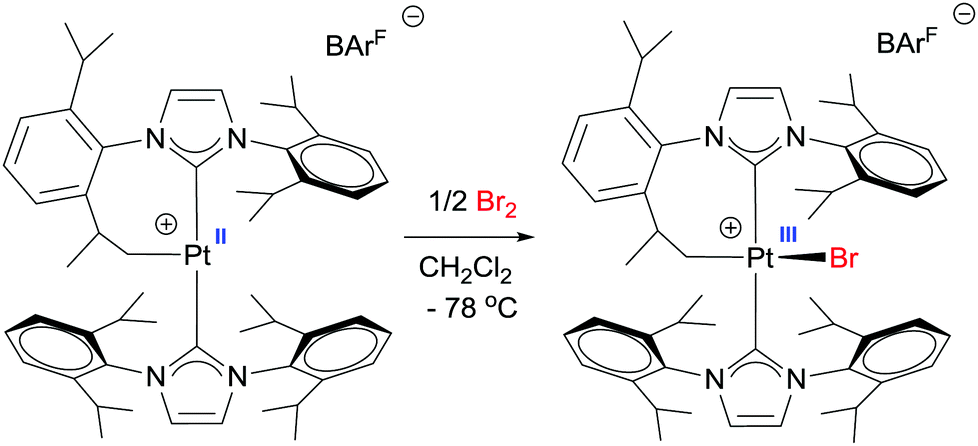
In some of these oxidations processes, the authors invoked the participation of Pt(III)–alkyl intermediates to explain the 1:1 ketone/alcohol ratio. Such species are likely formed through a one-electron oxidation of the cationic Pt(II)–alkyl by the Cu(II) salts. Paramagnetic Pt(III)–alkyl compounds have been suggested (and sometimes EPR detected) as key intermediates in platinum-mediated oxidation reactions of alkyls, but they are usually too reactive to be observed. Nevertheless, in 2012 our group isolated the first cationic Pt(III)–alkyl derivative by means of an oxidation reaction of a cyclometallated, low-electron count, Pt(II) cationic species stabilised by bulky N-heterocyclic carbene ligands (Scheme 20).52 Although unstable at temperatures above −20 °C, the X-ray structure revealed a very unusual seesaw structure. This compound is involved in the stoichiometric formation of C–Br bonds.
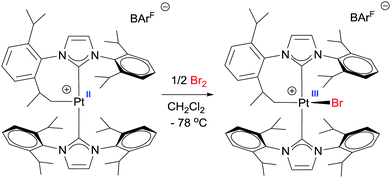 |
| | Scheme 20 Synthesis of a mononuclear, paramagnetic Pt(III) complex. | |
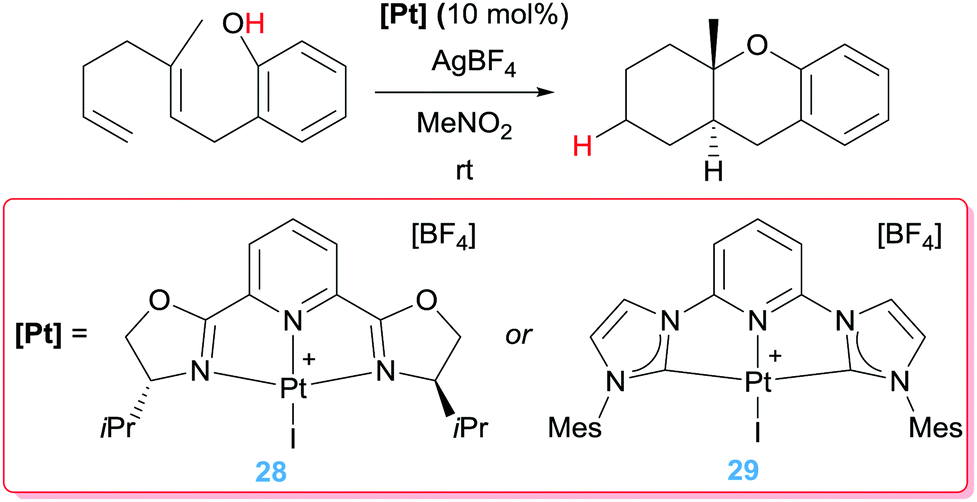
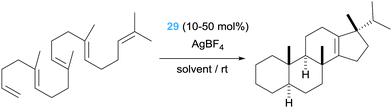
Coming back to cycloisomerization of dienes bearing alcohol units, we have already mentioned that the key point in directing the type of products formed depends strongly on the ability of the Pt–alkyl system to undergo a β-hydride elimination process. In the previously mentioned catalytic processes, the proton generated from the OH functionality is removed from the media to avoid side reactions. However, this proton could, in principle, protonate the Pt–alkyl intermediate (intermediate A in Scheme 17), provided that the β-hydride elimination process is sufficiently slow. Nevertheless, protonation of cationic Pt–alkyl compounds is usually challenging, but tuning the electronic properties of the ligands around the metal centre could bypass this obstacle. At the same time, the ligand of choice should, ideally, confer electrophilic characteristics to the metal centre to initiate the cyclisation reaction. To this end, two types of pincer ligands have been devised (Scheme 21). One of them is based on nitrogen-based pincer ligands, among which 2,6-bis[(4R)-isopropyl-2-oxazolin-2-yl]pyridine (PyBOX), which additionally could allow for enantio-differentiation of the new- generated stereogenic centres, is the most effective one, even in the absence of added bases.53 Nevertheless, although high conversions (49 to 93%) are obtained with diastereoselectivities above 20:1, low enantiomeric excesses (up to 38%) are generated. The mismatch between a hard ligand (N-based) and a soft metal (Pt(II)), might be behind the excellent activities.
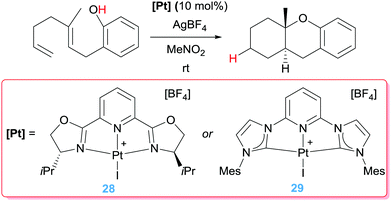 |
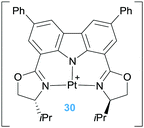
| | Scheme 21 Cycloisomerisation of dien-ols mediated by complexes 28 and 29. | |
The fact that no alkene derived from a β-hydride elimination process is observed indicates that the protodemetallation is very fast. If, on the other hand, a more electron donating anionic N-based ligand is used as a supporting ligand, as that in Fig. 3, no cycloisomerisation is observed revealing that the electrophilic character of this monocationic platinum system (generated in situ from the corresponding iodine derivative) is not enough to trigger the cyclisation process.
 |
| | Fig. 3 Mono-cationic platinum system. | |
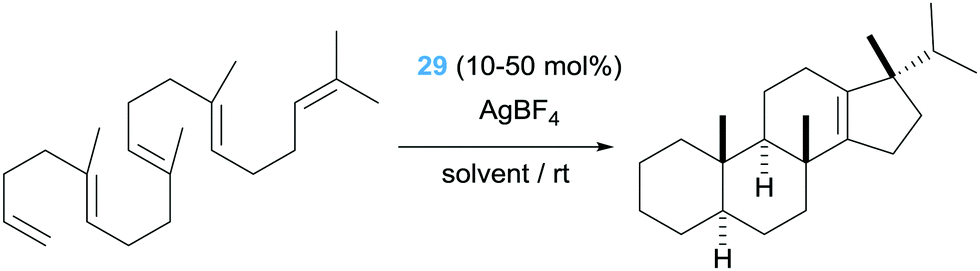
The second type of pincer ligand was based on the excellent sigma-donor electron properties of N-heterocyclic carbene ligands (NHCs). The authors conceived that the NHC ligands should be in the lateral positions of the pincer ligand, whereas a less electron donor pyridine fragment should be located at the central position in order to be in a trans position of the incoming alkene substrate. This ligand design proved to be efficient in the cycloisomerisation of dien-ols (complex 29, Scheme 21), working under very mild conditions (rt) generating the final products in yields up to 88%.54
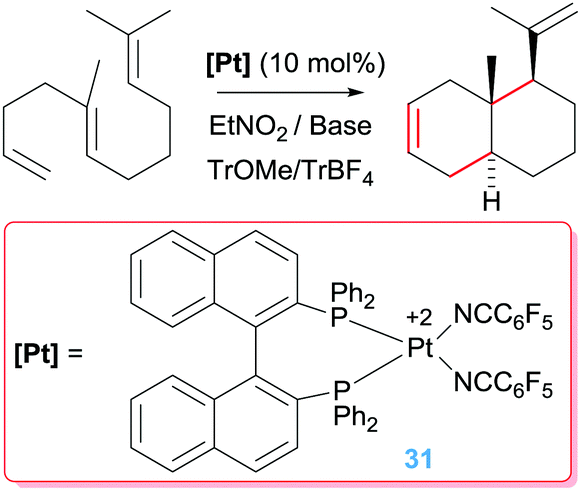
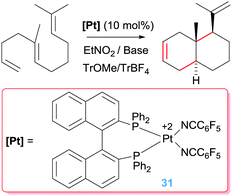
The catalytic activity of the bis-NHC pincer complex is not limited to dien-ols but it is also efficient for the diastereoselective cascade cyclization reactions of other polyenes lacking acidic OH functional groups that are usually more difficult to cyclise (Scheme 22).55 This system allows the synthesis of polycycles of up to four fused cycles as exemplified in Scheme 22. It is worth noting once again the lack of products arising from β-hydride elimination events. If, however, a bidentate diphosphine ligand is used instead of the pincer bis-NHC above mentioned the cascade cyclization reaction leads, eventually, to the alkene products (Scheme 23).56
 |
| | Scheme 22 Cycloisomerisation of polyenes catalysed by complex 29. | |
 |
| | Scheme 23 Cycloisomerisation of polyenes catalysed by complex 31. | |
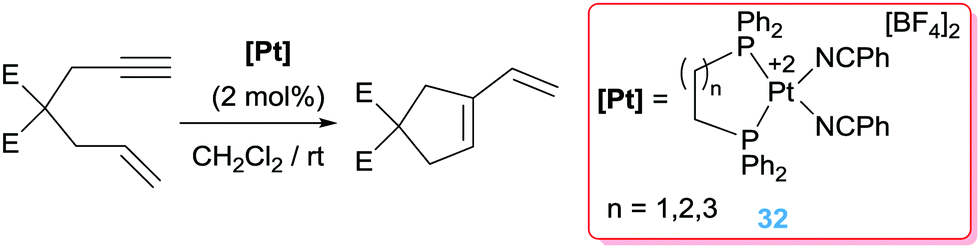
Cyclisations of enynes
Besides the cyclisation reactions involving dienes, enynes are also prone to undergo cyclisation processes mediated by electrophilic compounds. Earlier reports by Inoue, Oi and co-workers (based on previous works by Murai using platinum salts)42 showed that dicationic platinum complexes bearing specific phosphine ligands catalyse efficiently the cycloisomerisation of 1,6-dienes into 1-vinylcyclopentenes under mild conditions.57 It was found that chelating phosphines such as bis(diphenyl)phosphinoethane (dppe) are effective ligands (with yields of the cycles up to 100%), whereas monodentate such as PPh3 are ineffective (Scheme 24). Additionally, coordinating solvents such as THF or acetonitrile inhibit the reaction, suggesting the need of vacant sites available at the platinum centre for the reaction to proceed.
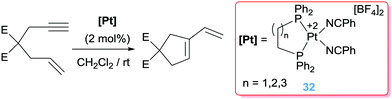 |
| | Scheme 24 Cyclisation of en-ynes mediated by complex 32. | |
A few years later, Fensterbank, Gimbert and co-workers carried out a detailed mechanistic study that shed light to some important points regarding the nature of the catalyst.58 When comparing the lower activity of PtCl2 with respect to complex [Pt(dppp)(NCPh)2][BF4]2 it was actually found that coordination of water, present in the reaction media, to the platinum centre led to an active catalytic species [Pt(dppp)(H2O)][BF4]2 which played an important role in facilitating the cycloisomerisation process. A key feature is that during the first step of the reaction coordination of the alkyne to the metal centre is not symmetrical, but unsymmetrical leading to a “slipped” η1-alkyne complex (eqn (1)),10,59
| | 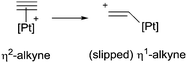 | (1) |
something not observed when PtCl
2 is used as a catalyst. It seems that this mode of coordination of the alkyne enhances its reactivity towards nucleophiles decreasing the energetic barriers considerably. In addition, the water molecule hinders coordination of the C
C double bond favouring nucleophilic attack to the coordinated alkyne.
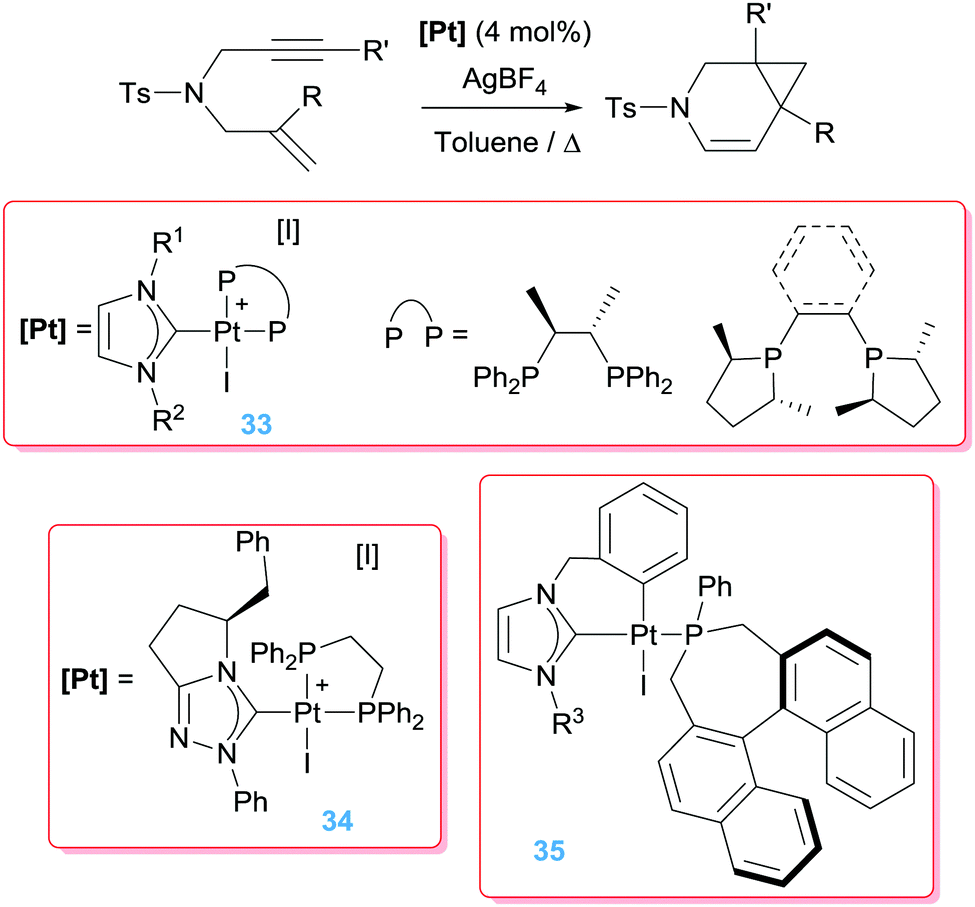
Marinetti et al. have explored the asymmetrical version of the cycloisomerisation of 1,6-enynes. To this end, they combined NHCs and phosphines in the coordination sphere of the platinum atom to stabilise dicationic complexes (Scheme 25).60 The catalytic reaction leads in this case to 6-endo-dig products (azabicyclo[4.1.0]heptene derivatives) with relatively low enantiomeric excess (up to 56%), although the catalytic activity was slightly lower than that previously reported using simple PtCl2 salts (operating temperature of 80 °C instead of 90 °C).61 The role of the NHC was also investigated and found to be essential since pre-catalyst [PtI2(chiraphos)] (chiraphos = 2,3-bis(diphenylphosphino)butane), in combination with AgBF4 to generate the vacant site, yielded only trace amounts of the azabicyclo[4.1.0]heptane compounds under identical reaction conditions.62
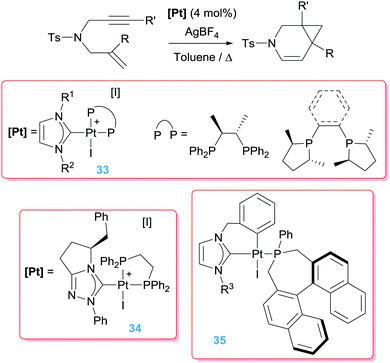 |
| | Scheme 25 Cyclisation of en-ynes mediated by complexes 33–35. | |
A further improvement of the catalytic system was made by means of a cyclometallated NHC ligand together with monodentate chiral phosphines (35 in Schemes 25), for which ee of 88–97% were achieved at 60 °C.63 It's noteworthy that at variance to all previously mentioned dicationic platinum systems, the catalytically competent cyclometallated version reported by Marinetti is mono-cationic (generated by reaction of 35 with a silver salt) yet still rather active, in contrast to the observations reported by Gagné on the use of the mono-cationic catalyst shown in Fig. 3. Although the electrophilic activation of alkynes by mono-cationic platinum-based systems is less common than with dicationic systems, some other research groups have made use of them for catalysing reactions involving alkynes and alkenes. For example, Iwasawa et al. reported a [3+2]-cycloaddition of acyclic γ,δ-ynones and vinyl ethers making use of diphosphine complexes of the type [PtCl2(PR3)2] (in combination with AgSbF6 to generate the cationic platinum active species).64 In a similar way, related enyne substrates can undergo cycloisomerisation processes leading to [4.1.0] bicycles mediated by monocationic Pt species. Echavarren reported that the cyclometallated platinum(II) complex in Scheme 26 works under milder reaction conditions (rt) than the platinum salt PtCl2 (requiring 50–80 °C).65 Significantly, the authors conceived that the more electrophilic nature of the platinum catalyst might facilitate a ring expansion process from the putative carbene intermediate B in Fig. 4 leading formally to a 7-endo-dig (seven membered) cycle, a process that does not take place using PtCl2.66 Although they succeeded (in some specific cases) in reaching this goal, low yields (∼17%) of the desired products were obtained. Nevertheless, this is a good example of how it is possible to modify a platinum system making it more electrophilic for undergoing unusual reactivities.
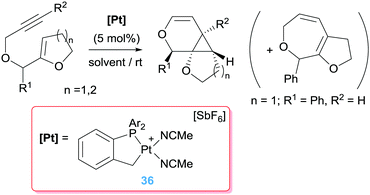 |
| | Scheme 26 | |
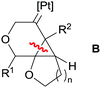 |
| | Fig. 4 Route to ring expansion from intermediate B. | |
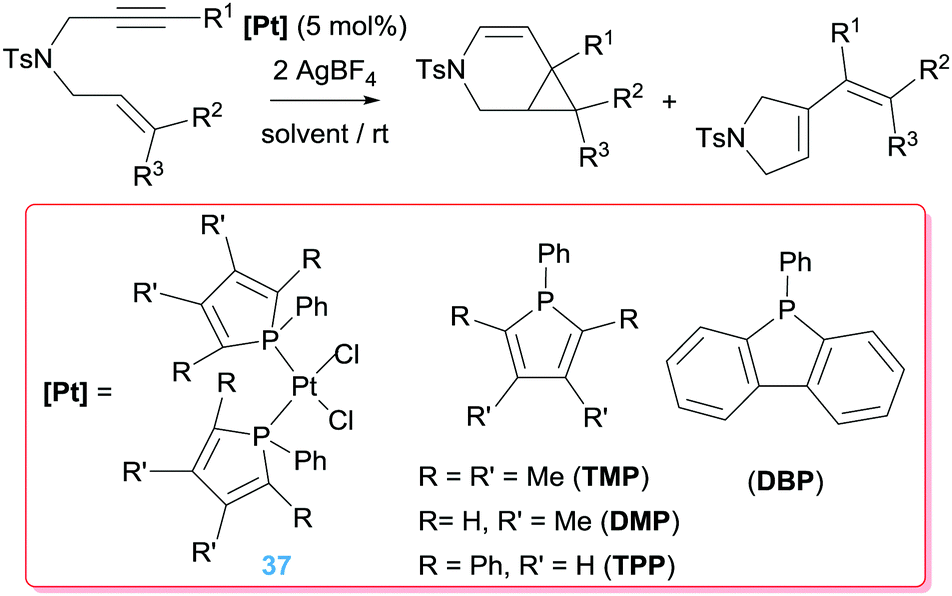
There are in the literature other ligands with different architectures that have been explored as potential catalysts. In this sense the steric and electronic properties of phosphole-based ligands can be fine-tuned to control the reactivity of the complexes they form.67 Gouygou and co-workers have prepared a series of Pt complexes bearing phospholes in their coordination sphere and studied their catalytic performance in the cyclisation of enynes (Scheme 27).68 Although some of them were able to catalyse the process, even at rt, diverse mixtures of 6-endo-dig and 5-exo-dig products have been observed that, on the other hand, can be directed to one or another through modifications of the reaction conditions (solvent, time and temperature). The more basic phosphole ligand TMP is the most active among the series studied with conversions of 96% (1 h), whereas bulkier phosphole ligands were considerably less efficient (37% conversion after 3 h). This effect has been attributed to a very favourable trans disposition of the phosphole ligands in the catalytically active species.
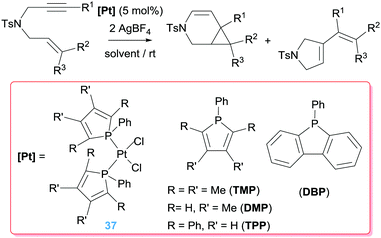 |
| | Scheme 27 Cyclisation of en-ynes mediated by complexes 37. | |
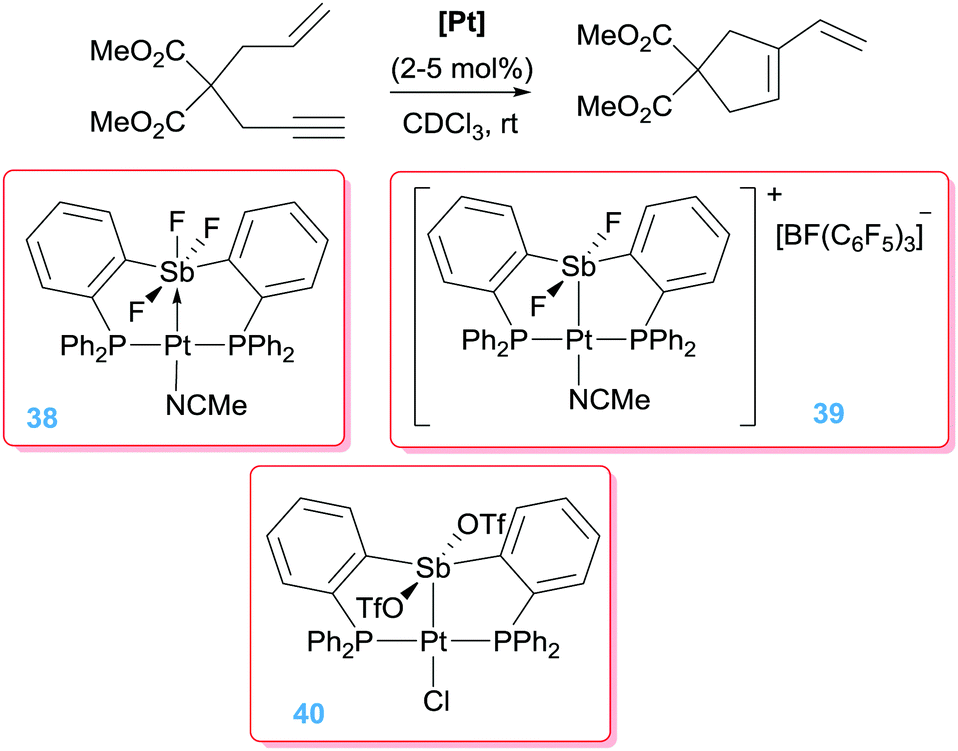
In recent years, some research activity has been headed to the use of non-innocent ligands that, under certain conditions, can alter the electronic properties of the metal centre. One original system that has been developed by Gabbaï et al. makes use of the properties that Z-type ligands impart to the metal centre. This type of ligands act as σ-accepting systems modifying the electronic properties of a complex by depleting electron density from the metal.69 As such, the metal centre becomes more electrophilic and can undergo reactivity characteristic of, for example, cationic systems. With this idea in mind a bis(phosphine) ligand system incorporating an antimony Lewis acid in its structure was developed (38, Schemes 28).70 The design allows for a direct interaction of the platinum atom with the antimony fragment that, on the other hand, can be modulated by means of the different coordination modes available at the antimony atom. It turned out that in spite of having a Z-type interaction between the Pt and Sb atoms in complex 38, the platinum centre is still too electron rich to promote cycloisomerisation reactions of 1,6-enynes. However, the authors considered the possibility of increasing the Lewis acid properties of the Sb atom by removing one F atom (complex 39). In doing so, the system became more electrophilic through a stronger Pt → Sb interaction. NBO analysis of this interaction indicated that it is covalent in nature and cannot be described as a donor–acceptor interaction between the two atoms, which translates in an increased σ-acceptor properties of the Sb atom. This new system catalysed the cycloisomerisation of 1,6-enynes efficiently (100% conversion within 4 h), thus providing a new way to activate the metal centre for this kind of transformations.
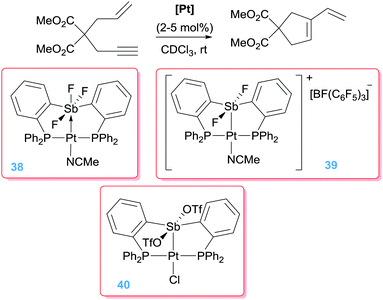 |
| | Scheme 28 Cyclisation of en-ynes mediated by complexes 38–40. | |
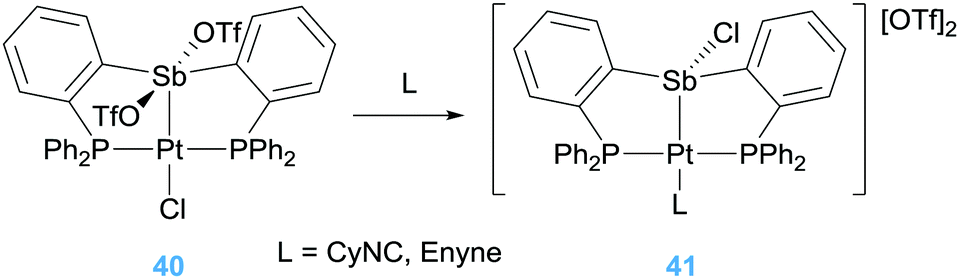
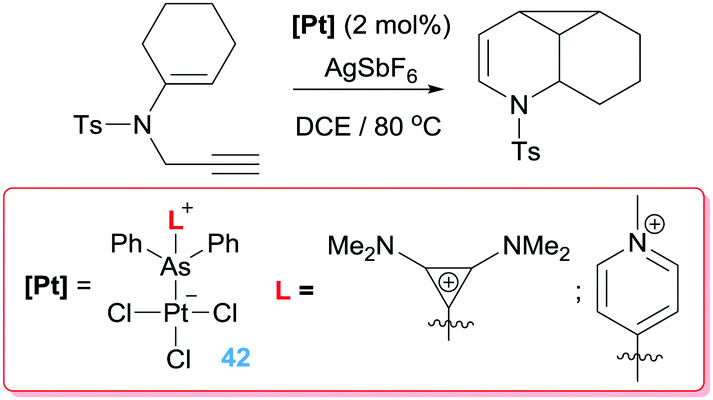
Alternatively, Gabbaï et al. also reported what they termed a “self-activated catalyst” based on complex 40 in Scheme 29.71 In this particular complex, the authors speculated that the Lewis acidic Sb atom is able to abstract the chlorine atom from the Pt–Cl unit in the presence of the 1,6-enyne substrate, leading to a highly active catalyst 41 (Scheme 29). This assumption was built on the reactivity of complex 40 with other nucleophiles such as CyNC (Scheme 29). In this latter structure, NBO analysis supports that the Sb atom is acting as a π-acceptor ligand retrieving electron density from d orbitals of platinum. This electronic situation is, to some extent, reminiscent to that induced by the cationic phosphine ligands.72 In fact, Alcarazo et al. have reported that cationic phosphine ligands bind to platinum centres in a way in which the σ-donation from the ligand to the metal is smaller than back-donation from the metal to the cationic phosphine ligand leading to depletion of electron density from the metal. This unusual bonding situation is also found in analogous arsine-based ligands. The introduction of a positive charge on the As atom reduces its HOMO diminishing its σ-donor ability in comparison to neutral arsines such as AsPh3.73 However, the nature of the cationic substituent on the As atom has little influence of their σ-donor properties but a rather high impact on the π-acceptor ones, providing a way for fine-tuning the electronic properties of the platinum centre. Therefore, the platinum atom in some of these complexes (specifically the one bearing a cyclopropenium substituent) is carbophilic enough to be active in cycloisomerisation of enynes with full conversion of the starting material in 20 min (Scheme 30). Nevertheless, the arsine bearing the more π-acceptor pyridinium fragment proved to be inefficient in the process that, according to the authors, seems to be due to an excessive π-acidic properties of the platinum system that lead to side reactions in addition to catalyst degradation. In any case, this report constitutes a nice example in which the tuning of the ligand properties leads to a platinum catalyst that surpasses any other system previously reported for this particular transformation.
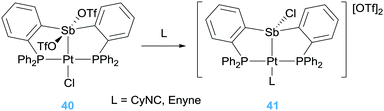 |
| | Scheme 29 Self-activation of catalyst 40 into 41. | |
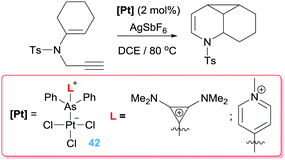 |
| | Scheme 30 Cyclisation of en-ynes mediated by complexes 42. | |
Hydroamination of C–C multiple bonds
Hydroamination is an industrially valuable process that consists of the addition of a nucleophilic N–H functionality across a C–C multiple bond, giving rise to the corresponding C–N and C–H fragments.74 Whereas this reaction has been widely studied with several catalytic systems,75–77 there exist drawbacks arising from their sensitivity to oxygen and moisture and limited functional group compatibility. In light of this situation, late transition metal-based complexes have appeared as an efficient alternative in homogeneous catalysis.78 Among them, Platinum is one of the metals of choice for this transformation,79 for which anionic,80 neutral81 and cationic species have been reported. Given the scope of this feature article, only the latter class will be described in more detail.
Intermolecular hydroamination reactions
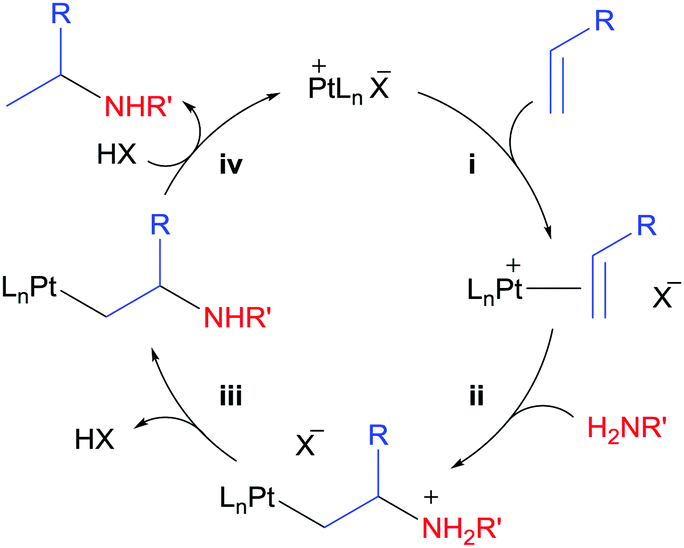
The field of intermolecular hydroamination catalysed by electron-deficient Pt complexes started to develop in 2005.82 The same year, Tilley et al. reported the catalytic olefin hydroamination by arylsulfonamides and weakly basic anilines.83 Such reaction was catalysed by the electrophilic complex (COD)Pt(OTf)2 or Zeise's dimer activated by AgBF4 (10 mol% each). As a result, yields greater than 95% were obtained, and short reaction times and mild reaction conditions (down to 75 °C and 2 hours) were employed. Mechanistic investigations suggest olefin coordination to the electrophilic Pt centre followed by Markovnikov addition of the N–H group, as depicted in Scheme 31. Whereas NMR and Mass Spectrometry characterization of the intermediate [(COD)Pt(norbornene)2][OTf]2 supports the former step (step (i) in Scheme 31), kinetic studies point to step (ii) as the rate-determining step in the cycle. Then, release of acid would yield the corresponding β-aminoalkyl Pt complex (iii), which can release the hydroamination product after further protonation (iv). No catalysis was observed in the presence of the hindered base 2,6-di-tert-butylpyridine, which supports the involvement of an acidic species during the catalytic cycle. As one can deduce from the mechanism, basic amines (pKa of the conjugated acid >4, according to the authors) can either coordinate to the metal centre or they might not be acidic enough to undergo proton-transfer (step (iii)), which narrows the library of possible amines available for this reaction.83 Therefore, ligand design is crucial in order to solve this kind of problems by tuning the steric and electronic properties of the catalysts.
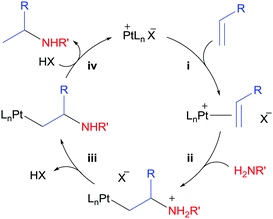 |
| | Scheme 31 Mechanistic proposal for the outer-sphere hydroamination of alkenes. | |
Although this is the general mechanism for the olefin catalytic hydroamination catalysed by electrophilic, cationic Pt complexes, the Tilley group reported an alternative mechanism involving proton transfer to the olefin substrate followed by amine attack in the case of norbornene.84 The strategy of using Pt chloride complexes as pre-catalysts in combination with halide scavengers was also used by Widenhoefer so as to carry out the catalytic hydroamination of monosubstituted allenes by secondary alkylamines. This led to the selective formation of the E-diastereomer with yields between 73–99% when using dppf as supporting ligand after screening through a selection of phosphines. Again, a mechanism involving coordination of the allene followed by outer-sphere attack of the amine was suggested by the authors.85
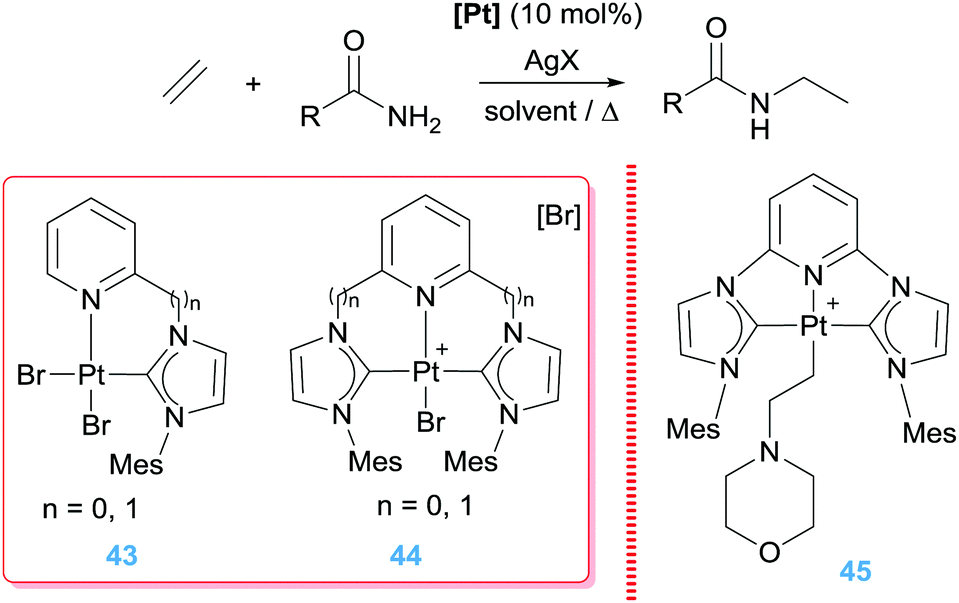
In a similar way, Limbach and co-workers carried out the hydroamination of unactivated olefins such as ethylene, propylene or styrene with weak bases like carboxamides and sulphonamides (Scheme 32).86 Very high selectivity towards Markovnikov products (up to 10:1) was obtained by using NHC-stabilized bi- and tri-dentate ligands (Scheme 32) in combination with AgBF4. When using one of the tridentate complexes and morpholine (pKa of conjugated acid >8)87 the cationic β-aminoalkyl Pt complex 45 (Scheme 32) obtained after step iii (Scheme 31) could be isolated and characterized by X-ray crystallography, confirming once again this type of mechanism.86
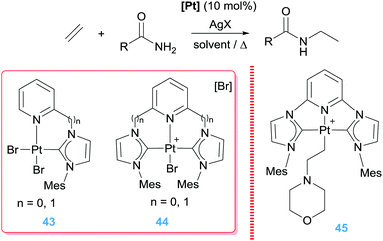 |
| | Scheme 32 Hydroamination of alkenes by complexes 43–45. | |
Intramolecular hydroamination reactions
Platinum complexes have also proven effective in the cyclisation of aminoalkenes, as described in Scheme 33. In fact, Stradiotto et al. performed a screening of different catalysts and concluded that complexes PtCl2 and PtCl2(COD) are indeed competent pre-catalysts for the cyclohydroamination of secondary alkyl/arylamines tethered to α-alkenes. However, the zwitterionic complex 46 gave comparable conversion values (ca. 90%) under the same conditions (110 °C).88
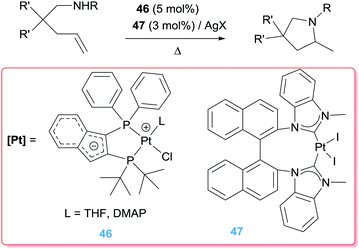 |
| | Scheme 33 Intramolecular hydroamination of alkenes catalysed by complexes 46–47. | |
Similarly to Limbach and co-workers, the group of Xu and Shi employed NHC-stabilized Pt complexes (47, Scheme 33) in combination with silver salts to afford cationic catalysts able to carry out the cyclisation of olefins containing secondary alkylamines in excellent yields (>90%; racemates were obtained in all cases).89 This methodology tolerates a wide range of functional groups such as bromo, cyano, nitro, ester or methoxy fragments. Unlike the previous example,88 the authors claim that no reaction takes place in the absence of a halide scavenger, and they propose a mechanism similar to that observed for the intermolecular hydroamination reaction (i.e. coordination of the olefin to the cationic complex followed by outer-sphere amine attack).89
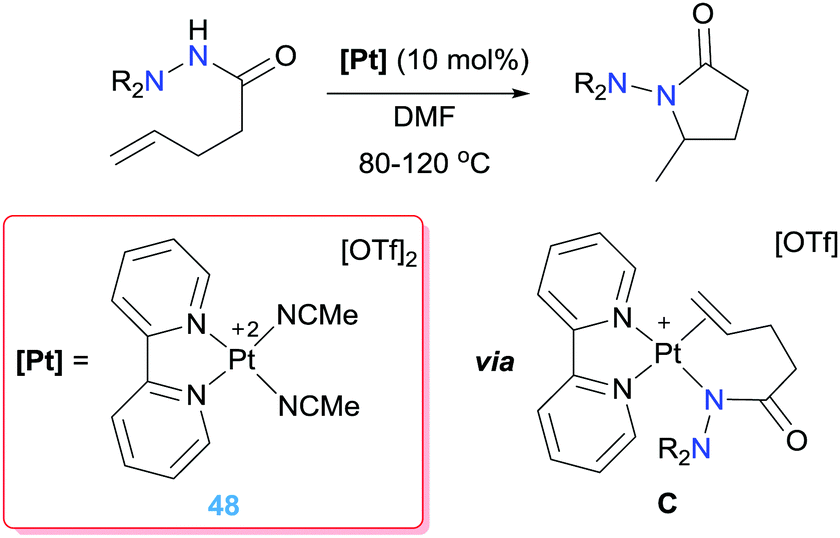
Nonetheless, a different mechanism was proposed by Mayer and Michael for the intramolecular hydrohydrazination when using dicationic (bpy)Pt(II) complexes (obtained again by halide abstraction) (Scheme 34).90 Instead, alkene insertion into a Pt–N bond via intermediate C (Scheme 34) is proposed based on the results obtained in stoichiometric reactions. In addition, kinetic studies rule out a nucleophilic attack mechanism, given that no rate dependence is observed upon addition of Brønsted acid or base (the protonolysis step is carried out by the own hydrazide). As a result, the 5-exo cyclization product is selectively obtained, without detection of the 6-endo derivative.
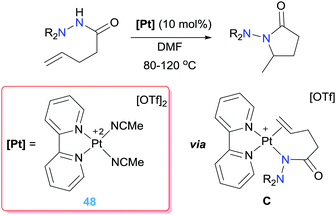 |
| | Scheme 34 Intramolecular hydroamination of alkenes catalysed by complexes 48. | |
Functionalisation of E–H bonds (E = B, Si)
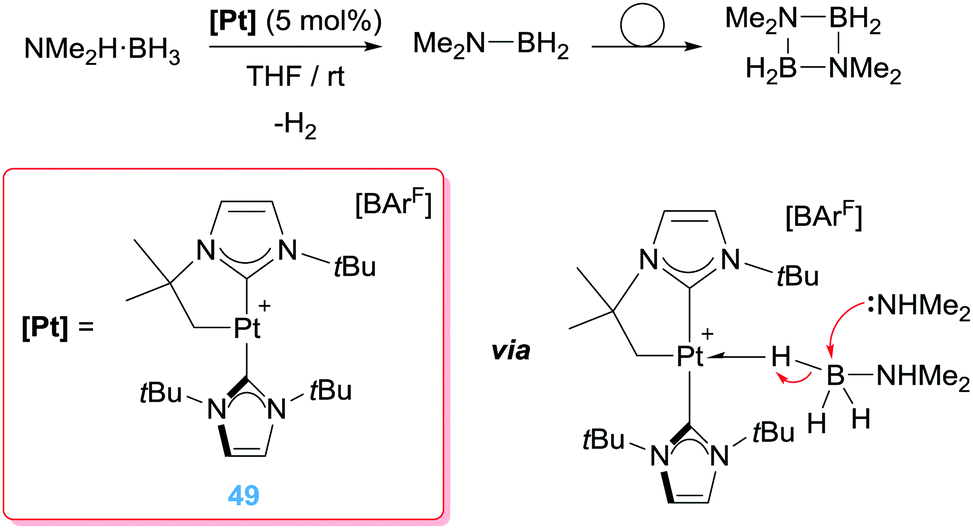
All the examples described in the previous sections deal with activation of organic molecules in different ways (C–H bond activation, electrophilic activation of alkenes or alkynes). However, our group has focused its research on the activation of BH and SiH bonds of boranes and silanes by electrophilic platinum complexes and implementing this activation in catalytic processes. To this aim, we have synthesised a series of 14-electron Pt(II) complexes stabilised by N-heterocyclic carbene ligands. Although this type of compounds are often postulated as transient species, as in some of the examples previously described, the excellent electron donor properties of NHCs, together with the easiness to tune the steric environment around the metal centre have allowed us to isolate and characterise a series of these compounds.18,91 Some of them exhibit a highly electrophilic character and can activate B–H and Si–H bonds of boranes and silanes, respectively, in a different way than other previously described systems. In 2013, we reported that the 14-electron Pt(II) complex 49 is an active catalyst in the dehydrocoupling of amine-boranes, most particularly NMe2H·BH3 (Scheme 35).92 Initial investigations indicated that this platinum complex catalyses efficiently the dehydrocoupling of NMe2H·BH3 into dimethylamino-borane NMe2BH2 (that subsequently undergoes a dimerization process; full conversion in less than 20 min). Intriguingly, the mechanism by which this process takes place occurred through a distinct reaction pathway never previously described by any other metal catalyst. An in-depth mechanistic study supported that the reaction involves, in a first step, coordination of the amine–borane leading to a Shimoi-type (or η1-BH) complex (Scheme 35 bottom right). Although these species were observed by NMR at low temperatures, X-ray diffraction studies on the homologue complex bearing a pyridine borane C5H5N·BH3 (that cannot undergo a dehydrocoupling reaction) confirmed the postulated structure.93 This coordination mode enhanced the electrophilic character of the boron atom so that it can undergo a nucleophilic addition of an external nucleophile leading to a net transfer of a hydride from the borane to the metal centre. As a result, a neutral platinum hydride complex and a boronium cation are formed. The latter contains an acidic NH proton that under specific reaction conditions can react with the Pt-hydride releasing the dimethylamino-borane and dihydrogen. In this last step, the catalyst is regenerated. Therefore, at variance to other metal catalyst the B–H bond activation process is not taking place through a common oxidative addition process. In our case, no changes in the oxidation state are taking place at the Pt(II) centre, and therefore the platinum system is behaving as a mere Lewis acid, as in most of the examples described in the preceding sections.
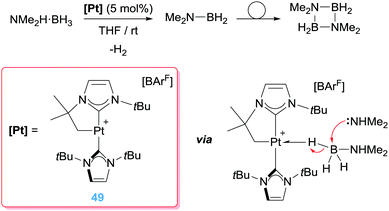 |
| | Scheme 35 Dehydrocoupling of amine–boranes mediated by complex 49. | |
If, on the other hand, an external amine is deliberately added in stoichiometric proportion with respect to the amine–borane, a double dehydrocoupling process occurs leading to diamino-boranes (Scheme 36). Complex 49 proved to be an excellent catalyst in this reaction, with full conversion using as low as 0.5% of the catalyst leading to TOF numbers up to 3692 h−1, outperforming previously described catalytic systems.93
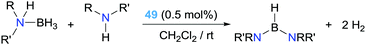 |
| | Scheme 36 Dehydrocoupling of amine–boranes and amines mediated by complex 49. | |
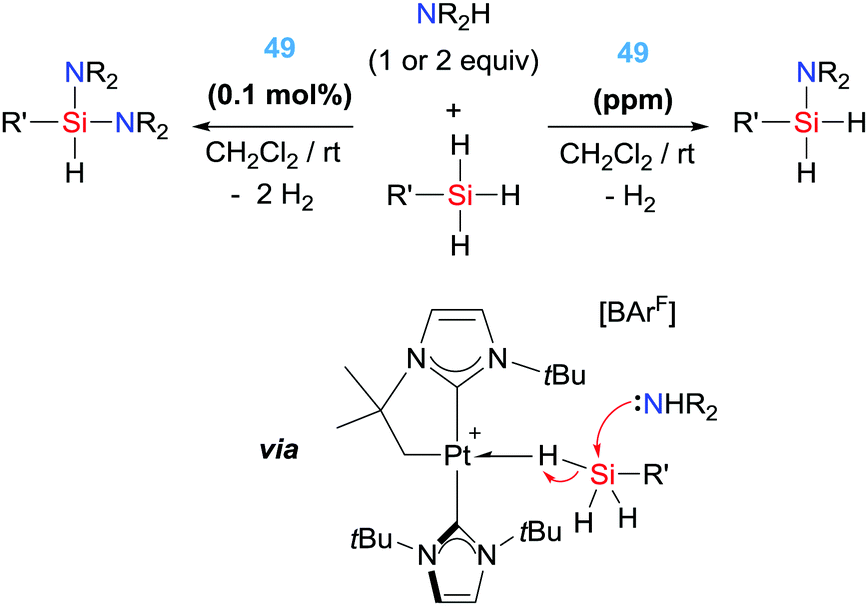
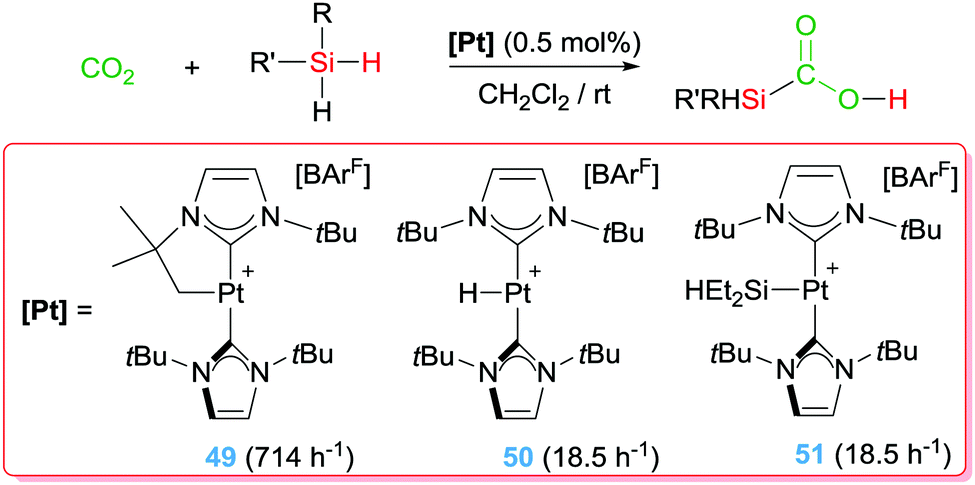
With these precedents in mind, we have also explored the catalytic activity of complex 49 in the dehydrocoupling of silanes and amines. In this particular reaction it's challenging to achieve good activities and good selectivities with the same catalytic system when primary silanes are used. However, platinum complex 49 showed an extraordinary activity and selectivity, leading to TON and TOF numbers up to 98000 and 330000 h−1, respectively, that are two orders of magnitude higher than any other catalytic system reported (yields above 86%) (Scheme 37).94 In addition, the system is completely selective towards the formation of mono-aminosilanes using catalyst loadings at the ppm level (up to 10 ppm). However, by increasing the catalyst loading (0.1%) it was possible to generate bis-aminosilanes through a double dehydrocoupling reaction. Thus, the selectivity of the process can be controlled by means of the catalyst loading. Mechanistic studies suggest that the reaction takes place in a very similar way to the dehydrocoupling of dimethylamine–borane.92 In this case, the first step involves the coordination of the silane to the metal centre to form a σ-SiH complex. This coordination enhances the electrophilicity at the silicon atom95 making it susceptible for nucleophilic attack by the amine. In other words, an electrophilic transfer from Pt to Si is occurring. It proved difficult to ascertain the nature of the interaction of the silane with the platinum centre, particularly having in mind that there were no reports on the detection or isolation of this highly reactive species. Although the interaction of the silane with the Pt was observable by NMR at very low temperatures, it was not possible to distinguish between two of the possible coordination modes of the silane to transition metals, either the pervasive η2-SiH or the extremely rare η1-SiH.96 According to DFT calculations carried out on the interaction of complex 49 with silanes, the most stable coordination mode is the unusual η1-SiH.97 Only very recently, we have succeeded in the isolation of closely related species of this type confirming the predicted η1-SiH coordination.98 It is likely that this mode of coordination of the silane to the cationic platinum centre enhances even more the reactivity of the silicon atom, since π-back donation from the metal to the σ*-Si–H bond is expected to be low.96 In fact, it is likely that this electrophilic character is also relevant in the selective catalytic hydrosilylation of carbon dioxide (Scheme 38) to the formic acid level by complex 49 and the related complexes 50 and 51, a reaction taking place at rt with catalyst loadings leading to TONs and TOFs up to 200 and 714 h−1, respectively.97 Nevertheless, according to DFT calculations all three complexes 49–51 lead to η1-SiH complexes, but their catalytic performance is rather different, with complex 49 being the most active. Therefore, other parameters (electronic and steric) may be also important in the electrophilic activation of the silane.
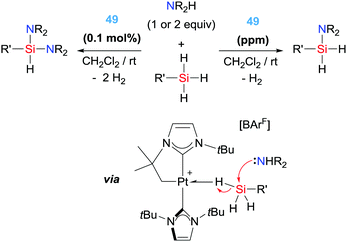 |
| | Scheme 37 Dehydrocoupling of silanes and amines catalysed by complex 49. | |
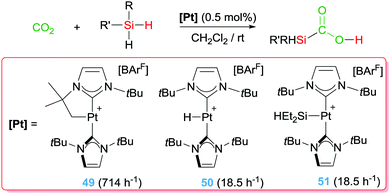 |
| | Scheme 38 Hydrosilylation of CO2 mediated by complexes 49–51 (TOFs given in brackets). | |
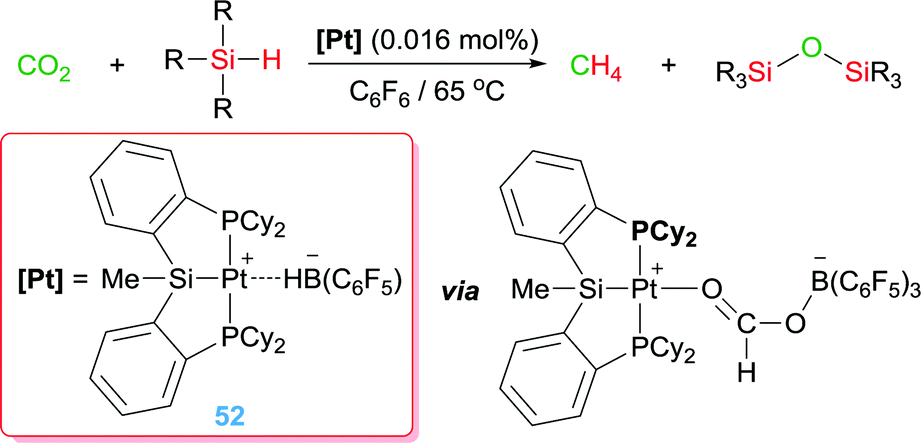
Turculet et al. used complex 52 for the reduction of CO2 to CH4 using hydrosilanes (Scheme 39). The combination of a platinum centre with the borane-based Lewis acid B(C6F5)3 give rise to a very efficient system, working with a catalyst loading as low as 0.016 mol% (TONs up to 2156 after 16 h), able to activate a molecule of CO2 leading to a formaborate Pt complex (Scheme 39) that undergoes a reduction process mediated by both the Pt centre and the borane Lewis acid.99
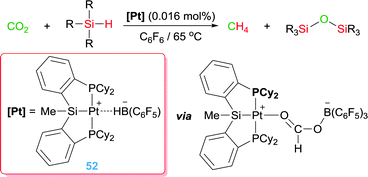 |
| | Scheme 39 Hydrosilylation of CO2 mediated by complex 52. | |
Conclusions
Well-defined electrophilic or low-electron count platinum complexes are very versatile catalysts for a variety of chemical transformations. Rational modulation of its coordination environment and electronic properties can lead to very efficient and selective catalytic reactions that have mainly been explored in C–H bond functionalization reactions, dimerisation of olefins, cycloisomerisation processes, hydroamination and more recently in dehydrocoupling reactions involving boranes and silanes and CO2 reduction. One key feature of these platinum systems is their ability to undergo processes through either oxidative addition/reductive elimination or insertion processes (commonly found in other late transition metals), but also through their excellent behaviour as a Lewis acid. Interestingly, most of the reactions described in this feature article are catalysed by platinum complexes in a +2 oxidation state but still to develop are electrophilic systems based on a +4 oxidation state. Moreover, although mononuclear stable complexes in oxidation states +1100 and +352,101 in platinum are still rare, they might find new applications in this field. In this sense, the role of Z-type ligands might be relevant to uncover new ways to increase the reactivity of platinum complexes, and in stabilising unusual oxidation states.71 Moreover, it is clear that other catalytic process involving an electrophilic transfer from the platinum centre to the incoming substrate are still to come beyond those related to carbophilic activation, as we have observed in the dehydrocoupling of silanes and amines. Further developments are still needed to increase efficiency, particularly in reactions such as hydroarylation (increasing turn over numbers and rates) and in terms of catalytic loadings in reactions involving electrophilic activations, although as we have reported the right set of ligands can promote the electrophilic activation of silanes using as low catalytic loadings as ppm. Finally, a rational design of the platinum environment can lead to unusual coordination modes of the substrates enhancing their reactivity, as in the examples of systems favouring transient or stable η1 coordination of olefins or alkynes and silanes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the Spanish MINECO RED2018-102387-T and CTQ2016-76267-P, FEDER support. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Notes and references
- V. P. Ananikov, D. G. Musaev and K. Morokuma, Organometallics, 2005, 24, 715 CrossRef CAS.
- S. A. Macgregor, G. W. Neave and C. Smith, Faraday Discuss., 2003, 124, 111 RSC.
- V. P. Ananikov, ACS Catal., 2015, 5, 1964 CrossRef CAS.
- A. R. Chianese, S. J. Lee and M. R. Gagné, Angew. Chem., Int. Ed., 2007, 46, 4042 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15 CrossRef CAS PubMed.
- N. F. Goldshleger, M. B. Tyabin, A. E. Shilov and A. A. Shteinman, Russ. J. Phys. Chem., 1969, 43, 1222 Search PubMed.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521 CrossRef CAS PubMed.
- C. Hahn, Chem. – Eur. J., 2004, 10, 5888 CrossRef CAS PubMed.
- L. Maresca and G. Natile, Comments Inorg. Chem., 1994, 16, 95 CrossRef CAS.
-
(a) S. Sakaki, K. Maruta and K. Ohkubo, Inorg. Chem., 1987, 26, 2499 CrossRef CAS;
(b) O. Eisenstein and R. Hoffmann, J. Am. Chem. Soc., 1981, 103, 4308 CrossRef CAS.
- F. P. Fanizzi, F. P. Intini, L. Maresca, G. Natile and F. Gasparrini, J. Chem. Soc., Dalton Trans., 1990, 1019 RSC.
- F. R. Hartley and J. J. Perié, Nature, 1975, 256, 636 CrossRef CAS.
- We have not considered the combination in situ of PtX2 (X = Cl, I) or PtCl4 salts with ligands since the nature of the active species is unknown.
- J. L. Garnett and R. J. Hodges, J. Am. Chem. Soc., 1967, 89, 4546 CrossRef CAS.
-
(a) J. A. Labinger, Chem. Rev., 2017, 117, 8483 CrossRef CAS PubMed;
(b) A. N. Vedernikov, Curr. Org. Chem., 2007, 11, 1401 CrossRef CAS;
(c) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS PubMed;
(d) U. Fekl and K. I. Goldberg, Adv. Inorg. Chem., 2003, 54, 259 CrossRef CAS;
(e) A. E. Shilov and G. B. Shul’pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed.
- C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS PubMed.
- M. A. Ortuño, S. Conejero and A. Lledós, Beilstein J. Org. Chem., 2013, 9, 1352 CrossRef PubMed.
- O. Rivada-Wheelaghan, M. A. Ortuño, J. Díez, A. Lledós and S. Conejero, Angew. Chem., Int. Ed., 2012, 51, 3936 CrossRef CAS PubMed.
- D. Karshtedt, A. T. Bell and T. D. Tilley, Organometallics, 2004, 23, 4169 CrossRef CAS.
-
(a) B. A. McKeown, H. E. Gonzalez, M. R. Friedfeld, T. B. Gunnoe, T. R. Cundari and M. Sabat, J. Am. Chem. Soc., 2011, 133, 19131 CrossRef CAS PubMed;
(b) B. A. McKeown, N. A. Foley, J. P. Lee and T. B. Gunnoe, Organometallics, 2008, 27, 4031 CrossRef CAS.
- B. A. McKeown, H. E. Gonzalez, M. R. Friedfeld, A. M. Brosnahan, T. B. Gunnoe, T. R. Cundari and M. Sabat, Organometallics, 2013, 32, 2857 CrossRef CAS.
- B. A. McKeown, B. M. Prince, Z. Ramiro, T. B. Gunnoe and T. R. Cundari, ACS Catal., 2014, 4, 1607 CrossRef CAS.
- B. A. McKeown, H. E. Gonzalez, T. B. Gunnoe, T. R. Cundari and M. Sabat, ACS Catal., 2013, 3, 1165 CrossRef CAS.
- B. A. McKeown, H. E. Gonzalez, T. Michaelos, T. B. Gunnoe, T. R. Cundari, R. H. Crabtree and M. Sabat, Organometallics, 2013, 32, 3903 CrossRef CAS.
- A. M. Brosnahan, A. Talbot, B. A. McKeown, S. E. Kalman, T. B. Gunnoe, D. H. Ess and M. Sabat, J. Organomet. Chem., 2015, 793, 248 CrossRef CAS.
- D. Karshtedt, J. L. McBee, A. T. Bell and T. D. Tilley, Organometallics, 2006, 25, 1801 CrossRef CAS.
- B. A. Suslick, A. L. Liberman-Martin, T. C. Wambach and T. D. Tilley, ACS Catal., 2017, 7, 4313 CrossRef CAS.
- A. T. Luedtke and K. I. Goldberg, Angew. Chem., Int. Ed., 2008, 47, 7694 CrossRef CAS PubMed.
- M. L. Clement, K. A. Grice, A. T. Luedtke, W. Kaminsky and K. I. Goldberg, Chem. – Eur. J., 2014, 20, 17287 CrossRef CAS PubMed.
-
(a) A. Biffis, C. Tubaro and M. Baron, Chem. Rec., 2016, 16, 1742 CrossRef CAS PubMed;
(b) T. Kitamura, Eur. J. Org. Chem., 2009, 1111 CrossRef CAS.
- C. Hahn, M. Miranda, N. P. B. Chittineni, T. A. Pinion and R. Perez, Organometallics, 2014, 33, 3020 CrossRef.
- M. Elena Cucciolito, A. D’Amora, A. Tuzi and A. Vitagliano, Organometallics, 2007, 26, 5216 CrossRef.
-
(a) G. Buscemia, A. Biffisa, C. Tubaro, M. Basato, C. Graiff and A. Tiripicchio, Appl. Organomet. Chem., 2010, 24, 285 Search PubMed;
(b) A. Biffis, C. Tubaro, G. Buscemi and M. Basato, Adv. Synth. Catal., 2008, 350, 189 CrossRef CAS.
- Y.-H. Lo and F. P. Gabbaï, Organometallics, 2018, 37, 2500 CrossRef CAS.
- H. Yang and F. P. Gabbaï, J. Am. Chem. Soc., 2015, 137, 13425 CrossRef CAS PubMed.
-
(a) A. Kozma, T. Deden, J. Carreras, C. Wille, J. Petuskova, J. Rust and M. Alcarazo, Chem. – Eur. J., 2014, 20, 2208 CrossRef CAS PubMed;
(b) J. Carreras, M. Patil, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2012, 134, 16753 CrossRef CAS PubMed.
-
(a) M. E. Cucciolito, A. D’Amora and A. Vitagliano, Organometallics, 2005, 24, 3359 CrossRef CAS;
(b) C. Hahn, M. E. Cucciolito and A. Vitagliano, J. Am. Chem. Soc., 2002, 124, 9038 CrossRef CAS PubMed.
- D. Serra, P. Cao, J. Cabrera, R. Padilla, F. Rominger and M. Limbach, Organometallics, 2011, 30, 1885 CrossRef CAS.
- M. Shiotsuki, P. S. White, M. Brookhart and J. L. Templeton, J. Am. Chem. Soc., 2007, 129, 4058 CrossRef CAS PubMed.
-
(a) J. J. Adams, N. Arulsamy and D. M. Roddick, Organometallics, 2009, 28, 1148 CrossRef CAS;
(b) S. Basu, N. Arulsamy and D. M. Roddick, Organometallics, 2008, 27, 3659 CrossRef CAS;
(c) S. White, B. L. Bennett and D. M. Roddick, Organometallics, 1999, 18, 2536 CrossRef CAS.
-
(a) J. L. Mascareñas, I. Varela and F. López, Acc. Chem. Res., 2019, 52, 465 CrossRef PubMed;
(b) R. J. Felix, C. Munro-Leighton and M. R. Gagné, Acc. Chem. Res., 2014, 47, 2319 CrossRef CAS;
(c) P. Y. Toullec and V. Michelet, Curr. Org. Chem., 2010, 14, 1245 CrossRef CAS;
(d) A. M. Echavarren and C. Nevado, Chem. Soc. Rev., 2004, 33, 431 RSC.
- N. Chatani, N. Furukawa, H. Sakurai and S. Murai, Organometallics, 1996, 15, 901 CrossRef CAS.
- A. Fürstner, Acc. Chem. Res., 2014, 47, 925 CrossRef PubMed.
- W. D. Kerber, J. H. Koh and M. R. Gagné, Org. Lett., 2004, 6, 3013 CrossRef CAS PubMed.
- J. A. Feducia, A. N. Campbell, M. Q. Doherty and M. R. Gagné, J. Am. Chem. Soc., 2006, 128, 13290 CrossRef CAS PubMed.
- W. D. Kerber and M. R. Gagné, Org. Lett., 2005, 7, 3379 CrossRef CAS PubMed.
- C. A. Mullen and M. R. Gagné, J. Am. Chem. Soc., 2007, 129, 11880 CrossRef CAS PubMed.
- N. A. Cochrane, M. S. Brookhart and M. R. Gagné, Organometallics, 2011, 30, 2457 CrossRef CAS.
- C. A. Mullen, A. N. Campbell and M. R. Gagné, Angew. Chem., Int. Ed., 2008, 47, 6011 CrossRef CAS PubMed.
- M. J. Geier and M. R. Gagné, Organometallics, 2013, 32, 380 CrossRef CAS PubMed.
-
(a) N. A. Cochrane, H. Nguyen and M. R. Gagne, J. Am. Chem. Soc., 2013, 135, 628 CrossRef CAS PubMed;
(b) S.-B. Zhao, J. J. Becker and M. R. Gagné, Organometallics, 2011, 30, 3926 CrossRef CAS PubMed.
- O. Rivada-Wheelaghan, M. A. Ortuño, J. Díez, S. E. García-Garrido, C. Maya, A. Lledós and S. Conejero, J. Am. Chem. Soc., 2012, 134, 15261 CrossRef CAS PubMed.
- H. Nguyen and M. R. Gagné, ACS Catal., 2014, 4, 855 CrossRef CAS.
- M. J. Geier and M. R. Gagné, J. Am. Chem. Soc., 2014, 136, 3032 CrossRef CAS PubMed.
- C. H. McCulley, M. J. Geier, B. M. Hudson, M. R. Gagné and D. J. Tantillo, J. Am. Chem. Soc., 2017, 139, 11158 CrossRef CAS PubMed.
- J. G. Sokol, N. A. Cochrane, J. J. Beckerb and M. R. Gagné, Chem. Commun., 2013, 49, 5046 RSC.
- S. Oi, I. Tsukamoto, S. Miyano and Y. Inoue, Organometallics, 2001, 20, 3704 CrossRef CAS.
- S. Baumgarten, D. Lesage, V. Gandon, J.-P. Goddard, M. Malacria, J.-C. Tabet, Y. Gimbert and L. Fensterbank, ChemCatChem, 2009, 1, 138 CrossRef CAS.
- A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC.
- D. Brissy, M. Skander, P. Retailleau and A. Marinetti, Organometallics, 2007, 26, 5782 CrossRef CAS.
-
(a) A. Fürstner, F. Stelzer and H. Szillat, J. Am. Chem. Soc., 2001, 123, 11863 CrossRef PubMed;
(b) A. Fürstner, F. Stelzer and H. Szillat, J. Am. Chem. Soc., 2000, 122, 6785 CrossRef.
- D. Brissy, M. Skander, P. Retailleau, G. Frison and A. Marinetti, Organometallics, 2009, 28, 140 CrossRef CAS.
-
(a) Y. Zhang, H. Jullien, D. Brissy, P. Retailleau, A. Voituriez and A. Marinetti, ChemCatChem, 2013, 5, 2051 CrossRef CAS;
(b) H. Jullien, D. Brissy, R. Sylvain, P. Retailleau, J.-V. Naubron, S. Gladiali and A. Marinetti, Adv. Synth. Catal., 2011, 353, 1109 CrossRef CAS;
(c) D. Brissy, M. Skander, H. Jullien, P. Retailleau and A. Marinetti, Org. Lett., 2009, 11, 2137 CrossRef CAS PubMed.
-
(a) H. Kusama, E. Watanabe, K. Ishida and N. Iwasawa, Chem. – Asian J., 2011, 6, 2273 CrossRef CAS PubMed;
(b) K. Ishida, H. Kusama and N. Iwasawa, J. Am. Chem. Soc., 2010, 132, 8842 CrossRef CAS PubMed.
- C. Ferrer, M. Raducan, C. Nevado, C. K. Claverie and A. M. Echavarren, Tetrahedron, 2007, 63, 6306 CrossRef CAS.
- C. Nevado, C. Ferrer and A. M. Echavarren, Org. Lett., 2004, 6, 3191 CrossRef CAS PubMed.
- K. Fourmy, D. H. Nguyen, O. Dechy-Cabareta and M. Gouygou, Catal. Sci. Technol., 2015, 5, 4289 RSC.
- K. Fourmy, S. Mallet-Ladeira, O. Dechy-Cabaret and M. Gouygou, Dalton Trans., 2014, 43, 6728 RSC.
-
(a) D. You and F. P. Gabbaï, Trends Chem., 2019, 1, 485 CrossRef;
(b) A. Amgouneab and D. Bourissou, Chem. Commun., 2011, 47, 859 RSC.
- D. You, H. Yang, S. Sen and F. P. Gabbaï, J. Am. Chem. Soc., 2018, 140, 9644 CrossRef CAS.
- D. You and F. P. Gabbaï, J. Am. Chem. Soc., 2017, 139, 6843 CrossRef CAS PubMed.
-
(a) Y. Canac, Chem. – Asian J., 2018, 13, 1872 CrossRef CAS PubMed;
(b) M. Alcarazo, Acc. Chem. Res., 2016, 49, 1797 CrossRef CAS PubMed.
- J. W. Dube, Y. Zheng, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2016, 138, 6869 CrossRef CAS PubMed.
- J. Huo, G. He, W. Chen, X. Hu, Q. Deng and D. Chen, BMC Chem., 2019, 13, 89 CrossRef PubMed.
- S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS PubMed.
- M. Arrowsmith, M. R. Crimmin, A. G. M. Barrett, M. S. Hill, G. Kociok-Köhn and P. A. Procopiou, Organometallics, 2011, 30, 1493 CrossRef CAS and references therein.
- E. Chong, S. Qayyum, L. L. Schafer and R. Kempe, Organometallics, 2013, 32, 1858 CrossRef CAS and references therein.
- For some representative examples, see:
(a) Z. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 1570 CrossRef CAS PubMed;
(b) X. Shen and S. L. Buchwald, Angew. Chem., Int. Ed., 2010, 49, 564 CrossRef CAS;
(c) K. D. Hesp, S. Tobisch and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 413 CrossRef CAS PubMed . See also ref. 74 for more examples.
- H. M. Senn, P. E. Blöchl and A. Togni, J. Am. Chem. Soc., 2000, 122, 4098 CrossRef CAS.
-
(a) J.-J. Brunet, N.-C. Chu and M. Rodriguez-Zubiri, Eur. J. Inorg. Chem., 2007, 4711 CrossRef CAS;
(b) M. Rodriguez-Zubiri, S. Anguille, J.-J. Brunet and J.-C. Daran, J. Mol. Catal. A: Chem., 2013, 379, 103 CrossRef CAS.
-
(a) X. Wang and R. A. Widenhoefer, Organometallics, 2004, 23, 1649 CrossRef CAS;
(b) C. F. Bender and R. A. Widenhoefer, J. Am. Chem. Soc., 2005, 127, 1070 CrossRef CAS PubMed;
(c) C. F. Bender, W. B. Hudson and R. A. Widenhoefer, Organometallics, 2008, 27, 2356 CrossRef CAS;
(d) C. F. Bender, T. J. Brown and R. A. Widenhoefer, Organometallics, 2016, 35, 113 CrossRef CAS.
- H. Qian and R. A. Widenhoefer, Org. Lett., 2005, 7, 2635 CrossRef CAS PubMed.
- D. Karshtedt, A. T. Bell and T. D. Tilley, J. Am. Chem. Soc., 2005, 127, 12640 CrossRef CAS PubMed.
- J. L. McBee, A. T. Bell and T. D. Tilley, J. Am. Chem. Soc., 2008, 130, 16562 CrossRef CAS PubMed.
- K. L. Toups and R. A. Widenhoefer, Chem. Commun., 2010, 46, 1712 RSC.
- P. Cao, J. Cabrera, R. Padilla, D. Serra, F. Rominger and M. Limbach, Organometallics, 2012, 31, 921 CrossRef CAS.
- H. K. Hall, J. Am. Chem. Soc., 1957, 79, 5441 CrossRef CAS.
- C. B. Lavery, M. J. Ferguson and M. Stradiotto, Organometallics, 2010, 29, 6125 CrossRef CAS.
- R. Zhang, Q. Xu, L.-Y. Mei, S.-K. Li and M. Shi, Tetrahedron, 2012, 68, 3172 CrossRef CAS.
- J. M. Hoover, A. DiPasquale, J. M. Mayer and F. E. Michael, J. Am. Chem. Soc., 2010, 132, 5043 CrossRef CAS PubMed.
-
(a) M. A. Ortuño, P. Vidossich, S. Conejero and A. Lledós, Angew. Chem., Int. Ed., 2014, 53, 14158 CrossRef PubMed;
(b) M. Roselló-Merino, O. Rivada-Wheelaghan, M. A. Ortuño, P. Vidossich, J. Díez, A. Lledós and S. Conejero, Organometallics, 2014, 33, 3746 CrossRef;
(c) O. Rivada-Wheelaghan, M. Roselló-Merino, J. Díez, C. Maya, J. López-Serrano and S. Conejero, Organometallics, 2014, 33, 5944 CrossRef CAS;
(d) O. Rivada-Wheelaghan, M. Roselló-Merino, M. A. Ortuño, P. Vidossich, E. Gutiérrez-Puebla, A. Lledós and S. Conejero, Inorg. Chem., 2014, 53, 4257 CrossRef CAS PubMed;
(e) O. Rivada-Wheelaghan, B. Donnadieu, C. Maya and S. Conejero, Chem. – Eur. J., 2010, 16, 10323 CrossRef CAS PubMed.
- M. Roselló-Merino, J. López-Serrano and S. Conejero, J. Am. Chem. Soc., 2013, 135, 10910 CrossRef PubMed.
- M. Roselló-Merino, R. J. Rama, J. Díez and S. Conejero, Chem. Commun., 2016, 52, 8389 RSC.
- P. Ríos, M. Roselló-Merino, O. Rivada-Wheelaghan, J. Borge, J. López-Serrano and S. Conejero, Chem. Commun., 2018, 54, 619 RSC.
- M. C. Lipke, A. L. Liberman-Martin and T. D. Tilley, Angew. Chem., Int. Ed., 2017, 56, 2260 CrossRef CAS PubMed.
- J. Yang, P. S. White, C. K. Schauer and M. Brookhart, Angew. Chem., Int. Ed., 2008, 47, 4141 CrossRef CAS PubMed.
- P. Ríos, J. Díez, J. López-Serrano, A. Rodríguez and S. Conejero, Chem. – Eur. J., 2016, 22, 16791 CrossRef PubMed.
-
(a) P. Ríos, H. Fouilloux, P. Vidossich, J. Díez, A. Lledós and S. Conejero, Angew. Chem., Int. Ed., 2018, 57, 3217 CrossRef PubMed;
(b) P. Ríos, H. Fouilloux, J. Díez, P. Vidossich, A. Lledós and S. Conejero, Chem. – Eur. J., 2019, 25, 11346 Search PubMed.
- S. J. Milton and L. Turculet, Chem. – Eur. J., 2012, 18, 15258 CrossRef.
-
(a) Y. Kratish, A. Kostenko, A. Kaushansky, B. Tumanskii, D. Bravo-Zhivotovskii and Y. Apeloig, Angew. Chem., Int. Ed., 2018, 57, 8275 CrossRef CAS;
(b) M. C. MacInnis, J. C. DeMott, E. M. Zolnhofer, J. Zhou, K. Meyer, R. P. Hughes and O. V. Ozerov, Chem, 2016, 1, 902 CrossRef CAS;
(c) T. Troadec, S.-Y. Tan, C. J. Wedge, J. P. Rourke, P. R. Unwin and A. B. Chaplin, Angew. Chem., Int. Ed., 2016, 55, 3754 CrossRef CAS PubMed.
- See: O. Rivada-Wheelaghan, M. A. Ortuño, S. E. García-Garrido, J. Díez, P. J. Alonso, A. Lledós and S. Conejero, Chem. Commun., 2014, 50, 1299 RSC and references therein.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Amor
Rodríguez
,
Amor
Rodríguez