
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Generic high-capacity protein capture and release by pH control†
G.
Ferrand-Drake del Castillo
 ,
R. L. N.
Hailes
,
Z.
Adali-Kaya
,
T.
Robson
and
Andreas
Dahlin
*
,
R. L. N.
Hailes
,
Z.
Adali-Kaya
,
T.
Robson
and
Andreas
Dahlin
*
Department of Chemistry and Chemical Engineering, Chalmers University of Technology, Gothenburg, Sweden. E-mail: adahlin@chalmers.se
First published on 29th April 2020
Abstract
Techniques for immobilization and release of proteins are of general interest but challenging to develop. Here we show a new method for high-capacity (several μg cm−2) immobilization of proteins in polyelectrolyte brushes by multivalent hydrogen bonds. Upon increasing pH, the proteins are fully released with preserved structure and activity.
Immobilizing large amounts of proteins without causing denaturation is broadly interesting as it relates to many areas including purification, bioanalytics and enzymatic catalysis.1 Subsequently releasing the captured proteins creates opportunities for new lab-on-a-chip,2 separation3 or drug-delivery4 technologies. Proteins, in particular antibodies, now constitute most new therapeutic drugs,5 which calls for new methods to achieve efficient immobilization with the possibility of subsequent release.
However, proteins are generally not compatible with existing capture-release designs as they require gentle chemistry to preserve structure and biological activity.6,7 Covalent binding to polymers is one option,8–10 but even if this preserves structure it is not compatible with subsequent release in a simple manner. One gentle immobilization method is the use of specific receptors designed to capture a particular protein by non-covalent interactions.11 Unfortunately, such affinity-based methods have extreme requirements for the receptor,3 which must keep the target proteins securely bound and be immobilized at high density whilst preserving activity etc.
Here we present a new and generic method for reversible protein immobilization based on polyacidic brushes. Our concept differs from the established method of using polyelectrolyte brushes to bind proteins by electrostatic attraction.9,12–15 Instead, we utilize the hydrogen bonding ability of carboxylic acid groups in their protonated state.16 As the pKa of polyacidic brushes is higher than the same polymers in solution,17,18 we can immobilize proteins at the fairly mild pH of 5. Proteins become bound in multilayers inside the brush and remained captured when exposing the surface to serum at physiological ionic strength. Since the interaction is not based on electrostatic attraction, the presence of salt does not limit the binding. Upon active buffering to a pH higher than the protein pI, the proteins are fully released.
We prepared poly(acrylic acid) (PAA) and poly(methacrylic acid) (PMAA) brushes on gold surfaces by activator-regenerated atom transfer radical polymerization (ATRP) from initiators on the surface.19 Throughout this study we used surface sensitive techniques, in particular surface plasmon resonance (SPR) and quartz crystal microbalance (QCM). The polyacidic brushes had dry thicknesses in the range of tens of nm as determined by SPR.20 We analysed the effective pKa of the brushes using SPR (Fig. 1A, data in Fig. S1, ESI†), which yields results identical to those obtained from analysis by infrared spectroscopy.20,21 QCM titration17 gave similar pKa values (±0.3). An example of QCM data of brush pH-responsiveness is shown in Fig. 1B. The frequency and dissipation changes mainly originate from changes in degree of hydration.17,20 The pKa values of the polyacidic brushes are in agreement with previous reports17,18 as is the higher pKa for PMAA compared to PAA.21 Note that we did not determine the grafting density as commonly done in grafting-to approaches.22,23 More importantly, we know the monomer density inside the brushes from the ratio of dry and wet heights.20 For instance, there is ∼80% water in PMAA at pH 5 (Fig. S2, ESI†).
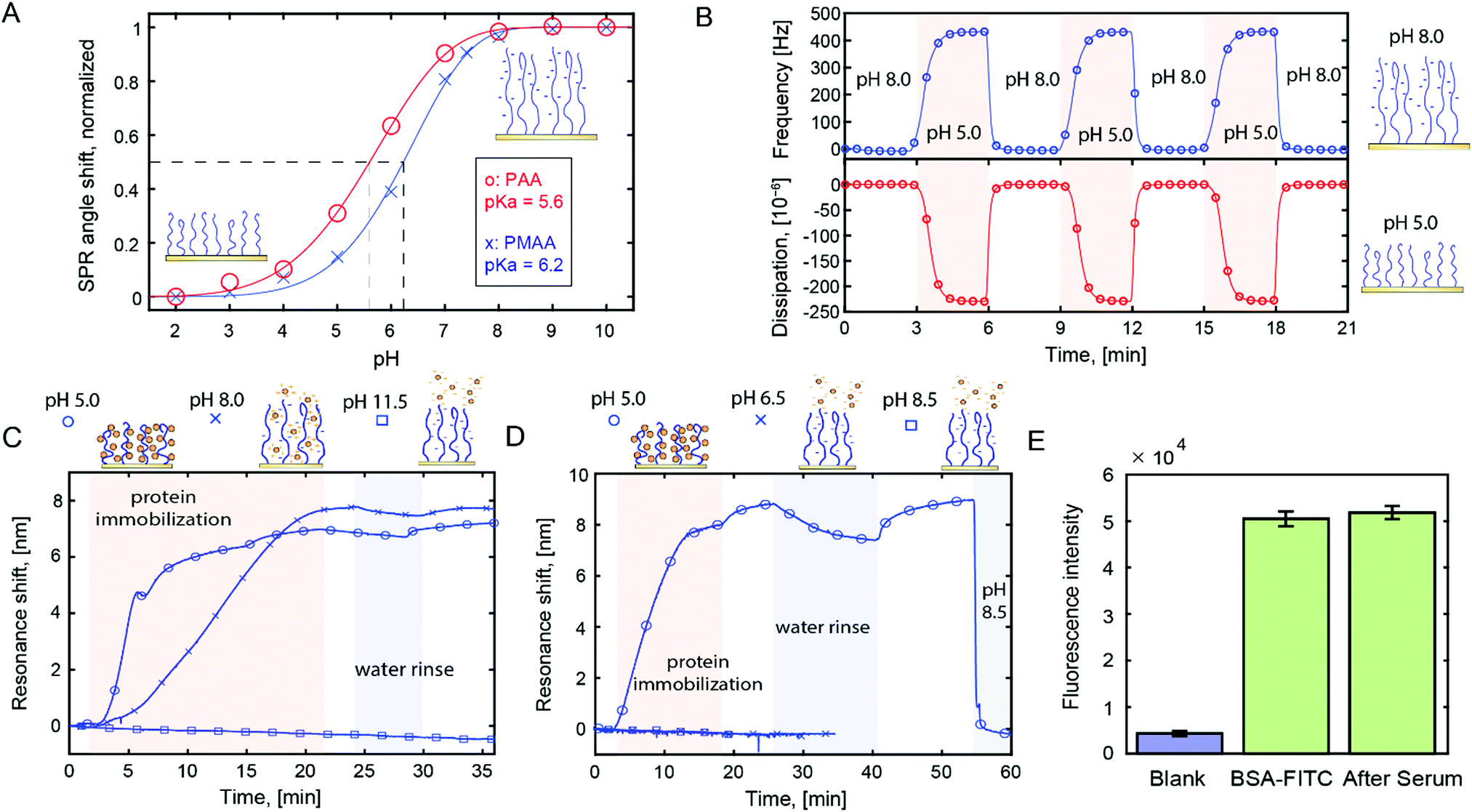 | ||
| Fig. 1 (A) Brush titration using SPR. (B) QCM with dissipation monitoring analysis of a PMAA brush (20 nm dry) when switching pH above or below the pKa. (C) Monitoring avidin immobilization in PMAA brush by electrostatic or hydrogen bond interactions using plasmonic nanohole arrays (response is linear to coverage). (D) Similar data for IgG antibodies, including release of immobilized proteins by rinsing with high pH buffer. (E) Fluorescence intensity from labelled BSA inside PMAA before and after exposure to serum for 30 min. | ||
We evaluated the affinity between polymer brushes and proteins at different pH, while always maintaining physiological ionic strength. We observed analogous protein binding behavior for both polyacids (Fig. S3, ESI†), but all results presented from here on are for PMAA. Fig. 1C shows an example of label-free detection with plasmonic nanohole arrays24 in transmission mode.25 A highly positively charged protein (avidin, pI 10) was introduced to PMAA brushes at different pH.
As expected, at pH 8, electrostatic interactions provide high binding (∼3000 ng cm−2). However, at pH 5, where the brush is close to neutral (∼90% protonated), binding is faster and reaches the same response. Also, there are notable differences in binding kinetics for the neutral and charged polymer brushes, suggesting different binding mechanisms.
For proteins with more regular pI values, the binding behavior changed drastically. For example, IgG (pI ∼ 7) showed no detectable binding to PMAA at pH 6.5 (>50% deprotonated brush, Fig. 1D). Again, at pH 5, binding was very high (∼4000 ng cm−2). This trend was consistent: at pH 5 we observed high binding capacity for all proteins (13 different types tested), while in the pH range 6–8 binding was only efficient for the proteins that had very high pI. These results agree in part with previous work by Delcroix et al. who detected more protein adsorption to PAA at lower pH.22 On the other hand, that work detected little binding at physiological ionic strength and pH 5. We attribute this difference to the fact that our brushes are prepared by grafting-from, which typically makes them much thicker and denser. We also observed binding at lower pH, but using PMAA at pH 5 is the best strategy to avoid the risk of denaturation. At pH 5 the protein structure is generally preserved or at least not permanently altered.26 (PAA cannot hydrogen bond equally well at pH 5 as it is more charged.)
When pH > pI, protein binding was not detected and any bound proteins were desorbed (Fig. 1D). Notably, this desorption required an actively buffered liquid (here 10 mM phosphate). No release was observed upon rinsing with water and not even when exposing the surface to biological solutions (tested up to ∼1 h). Fig. 1E shows the lack of change in fluorescence intensity from immobilized, labelled BSA before and after exposure to serum (including washing). This shows that other proteins do not replace the already immobilized ones, although additional proteins can be co-immobilized if they have a higher affinity for the brush (Fig. S4, ESI†). Most likely, the inherent buffering capacity of the –COOH groups contributes to keeping the proteins bound, especially when pH is close to pKa.
Our method clearly differs from the most common use of polyelectrolyte brushes for protein immobilization by electrostatic attraction. Here the polymers are almost fully neutral (pH < pKa) during binding and provide repulsion in their charged state (at least when pH > pI). Our results may seem to contradict previous studies in which proteins bind to charged polymers, sometimes even when carrying the same net charge (attributed to local “patches” on the surface).12,14,27 This disparity is to some extent explained by the fact that unlike previous studies,9,13 all of our measurements were performed at physiological ionic strength, where screening strongly limits all electrostatic attraction. In addition, a highly hydrophilic brush like PMAA is expected to repel proteins due to conformational entropy and osmotic pressure.20,22
We attribute the high protein binding capacity at pH 5 to hydrogen bonds between carboxylic acid groups in the polymers and various acceptors on the protein surface, in analogy with intermolecular complex formation between protonated polyacids and various hydrophilic polymers.16 Although mainly studied in bulk, this phenomenon is well-known since 1959.28 We have recently confirmed such interactions between PAA brushes and PEG with SPR20 and here we also observed binding of several other polymers (Fig. S5, ESI†), in agreement with literature.16 Thus, we propose that proteins can act as hydrogen bond acceptors with similar affinity as for polymers. The primary evidence is the extremely strong pH dependence: the degree of association at equilibrium is a function of acid protonation state, exactly as for interpolymer complexation.29 Many amino acids clearly have suitable hydrogen bond acceptor groups, such as amines (lysine, arginine, etc.) or ketones (aspartic acid, glutamine). Although we cannot conclude exactly which amino acids give rise to the strongest bonds, the lowest affinity was observed for horseradish peroxidase (HRP), which has an unusually low amount of amino acids with side groups that are charged and/or polar (comparison in Table S1, ESI†).
The immobilized amount of various proteins in PMAA is presented in Fig. 2A. The quantification was done by fitting Fresnel models to SPR spectra recorded in the dry state.15 This method gives highly accurate results since the protein refractivity is very similar to that of the polymers.15 A wide range of proteins with different molecular weights (M) and pI were tested, resulting in high surface coverage in all cases. Since the proteins are bound in multilayers, the PMAA brush thickness influences the loading capacity. In Fig. 2A the dry brush thickness was kept approximately the same (∼20 nm) to give a fair comparison and to show the absolute coverage. Naturally, the thicker brushes can be made to store even more proteins.30 In this work we kept the hydrated thickness comparable to the evanescent field in SPR to enable height probing.20 As expected from multivalent interactions bridging several chains and overcoming the entropic penalty of insertion,31 protein immobilization led to a compression of the brush (Fig. S2, ESI†). This agrees with the QCM results which consistently showed a dissipation decrease upon binding (example in Fig. S6, ESI†).
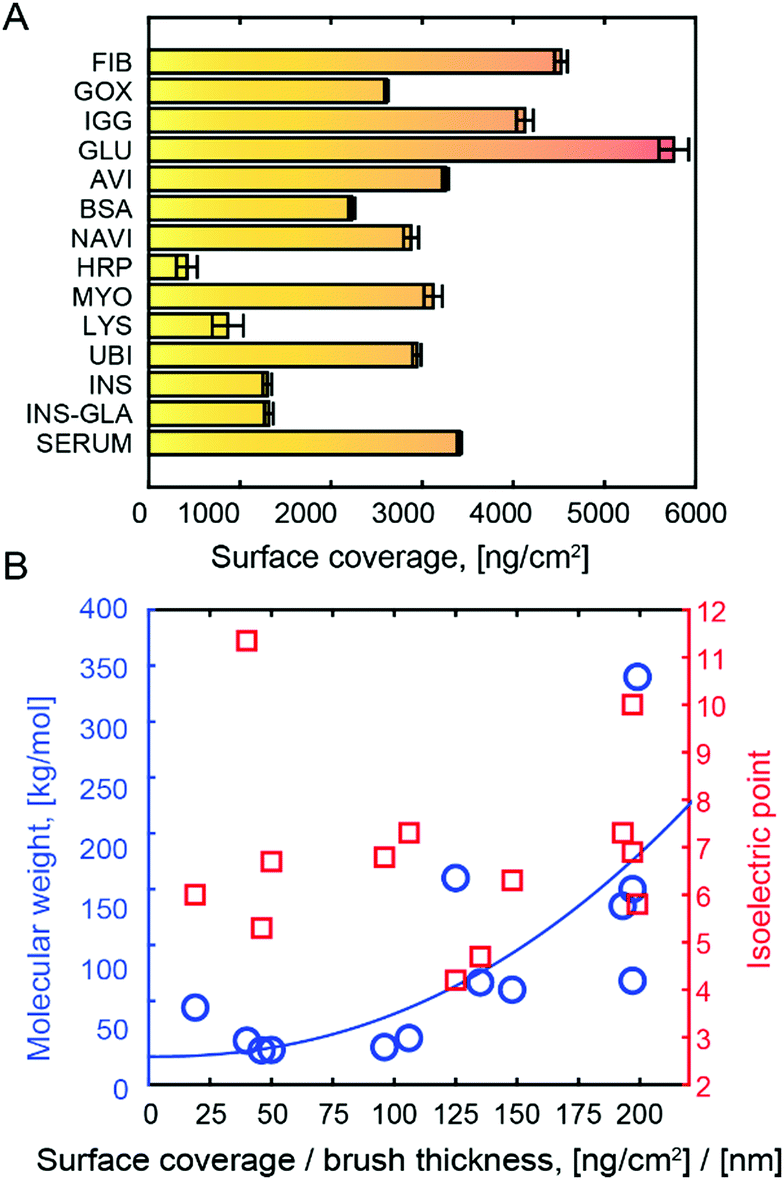 | ||
| Fig. 2 (A) Typical surface coverage after immobilization in ∼20 nm (dry thickness) PMAA at pH 5. Protein amounts were calculated from Fresnel models of SPR spectra in the dry state. (B) Plot of molecular mass and isoelectric point vs. immobilized protein amount normalized to dry brush thickness. The line shows a fit to an M1/3 dependence. | ||
When normalizing protein coverage to the coverage of PMAA, the protein amount correlated with M (p < 0.05) but not pI (Fig. 2B). This confirms that the binding is not due to electrostatic attraction. The trend of higher surface coverage with higher M can be understood from a simple scaling argument: given that the polymers interact with the surface of a protein with size R, the amount of polymer in contact with the protein is proportional to R2, while M is proportional to R3. Hence the mass of immobilized protein per mass of available polymer should scale with M1/3. Our results do not follow this law precisely (Fig. 2B), which is expected as the hydrogen bond acceptor groups also matter, but we could confirm the trend of increased normalized surface coverage with M. This strongly suggests that the polymers wrap around the hydrophilic protein exterior. Even for HRP, the amount (417 ng cm−2) corresponds to more than a monolayer.
All immobilized proteins were fully released by increasing the solution pH from 5 to above the protein pI. Fig. 3A shows SPR spectra after multiple capture-release cycles on the same surface. There was no detectable change in storage capacity or release efficiency (tested up to 10 cycles). We performed Michaelis–Menten analysis of glucose oxidase15 (Fig. 3B, calibration in Fig. S7, ESI†) as well as circular dichroism on BSA and IgGs (Fig. 3C). The results showed minimal influence on protein structure and activity after capture and release. This preservation is a very important feature of the method, yet not surprising given that the interactions with the polymer occur on the hydrophilic protein exterior. This is in contrast to immobilization based on hydrophobic interactions and release by surfactants.3 Still, it should be noted that for a few proteins with extremely high pI (>10), denaturation may occur due to the high pH at release.26
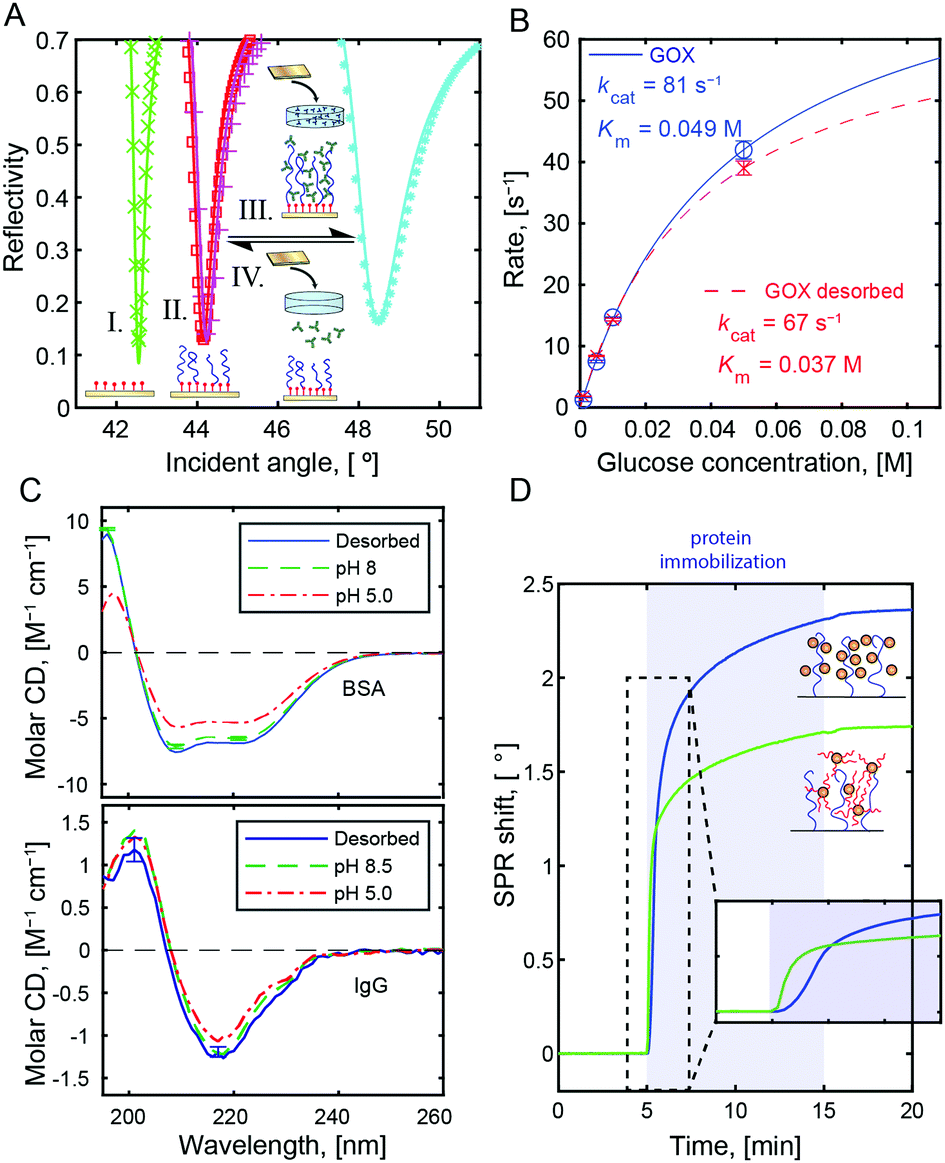 | ||
| Fig. 3 (A) SPR spectra before (I) and after (II) polymerization of PMAA as well as after capture (III) and release (IV) of BSA in repeated cycles. (B) Michaelis–Menten analysis of GOX activity before immobilization and after release. (C) Circular dichroism spectra before immobilization and after release for BSA and IgG. (D) Example of SPR data showing binding of PEG-modified BSA vs. native BSA to a PMMA brush at pH 5. | ||
In addition to proteins, other compounds with good hydrogen bond acceptor groups could be bound to PMAA at pH 5. One example of technological interest is PEG-modified proteins. Normally, PEG-modification of a compound reduces its interactions with other molecules, but upon conjugation to BSA (Fig. S8, ESI†) we observed binding of an amount comparable to the native protein (Fig. 3D). Even if PMAA cannot reach the protein surface, it can form hydrogen bonds with the PEG.16 At the same time, no binding was detected to the brushes when introducing macromolecules such as hyaluronic acid, dextran, DNA, or oxytocin (examples in Fig. S5, ESI†). This could be useful in analytical processes where one wants to separate the protein content of a biological sample from other molecules.
In conclusion, we have shown that polyacidic brushes are excellent for protein immobilization in their protonated state. We attribute this effect to multivalent hydrogen bonds. In contrast to electrostatic interactions, the binding is generic and large amounts are captured for all water-soluble proteins tested, even at physiological ionic strength. Due to the shifted pKa of the brush (compared to polymers in solution), pH 5 is sufficient to induce binding. The proteins remain securely bound in physiological environments but can still be fully released if the pH is increased. Although brushes are surface anchored, these finding may also have implications for coacervate formation in bulk.32 The method also has potential applications in several bioanalytical devices or drug delivery systems where reversible immobilization of proteins is needed.
Conflicts of interest
There are no conflicts to declare.References
- P. Jonkheijm, D. Weinrich, H. Schroder, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 9618–9647 CrossRef CAS PubMed.
- D. L. Huber, R. P. Manginell, M. A. Samara, B. I. Kim and B. C. Bunker, Science, 2003, 301, 352–354 CrossRef CAS PubMed.
- G. Di Palma, A. M. Kotowska, L. R. Hart, D. J. Scurr, F. J. Rawson, S. Tommasone and P. M. Mendes, ACS Appl. Mater. Interfaces, 2019, 11, 8937–8944 CrossRef CAS PubMed.
- E. Katz, J. M. Pingarron, S. Mailloux, N. Guz, M. Gamella, G. Melman and A. Melman, J. Phys. Chem. Lett., 2015, 6, 1340–1347 CrossRef CAS PubMed.
- A. C. Anselmo, Y. Gokarn and S. Mitragotri, Nat. Rev. Drug Discovery, 2019, 18, 19–40 CrossRef CAS PubMed.
- J. N. Talbert and J. M. Goddard, Colloids Surf., B, 2012, 93, 8–19 CrossRef CAS PubMed.
- K. Takasu, K. Kushiro, K. Hayashi, Y. Iwasaki, S. Inoue, E. Tamechika and M. Takai, Sens. Actuators, B, 2015, 216, 428–433 CrossRef CAS.
- S. P. Cullen, X. Liu, I. C. Mandel, F. J. Himpsel and P. Gopalan, Langmuir, 2008, 24, 913–920 CrossRef CAS PubMed.
- J. H. Dai, Z. Y. Bao, L. Sun, S. U. Hong, G. L. Baker and M. L. Bruening, Langmuir, 2006, 22, 4274–4281 CrossRef CAS PubMed.
- R. Dong, S. Krishnan, B. A. Baird, M. Lindau and C. K. Ober, Biomacromolecules, 2007, 8, 3082–3092 CrossRef CAS PubMed.
- A. Shastri, L. M. McGregor, Y. Liu, V. Harris, H. Q. Nan, M. Mujica, Y. Vasquez, A. Bhattacharya, Y. T. Ma, M. Aizenberg, O. Kuksenok, A. C. Balazs, J. Aizenberg and X. M. He, Nat. Chem., 2015, 7, 447–454 CrossRef CAS PubMed.
- O. Hollmann, T. Gutberlet and C. Czeslik, Langmuir, 2007, 23, 1347–1353 CrossRef CAS PubMed.
- A. Kusumo, L. Bombalski, Q. Lin, K. Matyjaszewski, J. W. Schneider and R. D. Tilton, Langmuir, 2007, 23, 4448–4454 CrossRef CAS PubMed.
- X. Xu, S. Angioletti-Uberti, Y. Lu, J. Dzubiella and M. Ballauff, Langmuir, 2018, 35, 5373–5391 CrossRef PubMed.
- G. Ferrand-Drake del Castillo, M. Koenig, M. Muller, K.-J. Eichhorn, M. Stamm, P. Uhlmann and A. Dahlin, Langmuir, 2019, 35, 3479–3489 CrossRef CAS PubMed.
- V. V. Khutoryanskiy and G. Staikos, Hydrogen-bonded interpolymer complexes: formation, structure and applications, World Scientific, Singapore, 2009 Search PubMed.
- N. Schuwer and H.-A. Klok, Langmuir, 2011, 27, 4789–4796 CrossRef CAS PubMed.
- T. Wu, P. Gong, I. Szleifer, P. Vlcek, V. Subr and J. Genzer, Macromolecules, 2007, 40, 8756–8764 CrossRef CAS.
- S. Gam-Derouich, M. N. Nguyen, A. Madani, N. Maouche, P. Lang, C. Perruchot and M. M. Chehimi, Surf. Interface Anal., 2010, 42, 1050–1056 CrossRef CAS.
- G. Ferrand-Drake del Castillo, G. Emilsson and A. Dahlin, J. Phys. Chem. C, 2018, 122, 27516–27527 CrossRef CAS.
- R. Dong, M. Lindau and C. K. Ober, Langmuir, 2009, 25, 4774–4779 CrossRef CAS PubMed.
- M. F. Delcroix, S. Demoustier-Champagne and C. C. Dupont-Gillain, Langmuir, 2014, 30, 268–277 CrossRef CAS PubMed.
- N. R. Hollingsworth, S. I. Wilkanowicz and R. G. Larson, Soft Matter, 2019, 15, 7838–7851 RSC.
- A. B. Dahlin, Analyst, 2015, 140, 4748–4759 RSC.
- A. B. Dahlin, S. Chen, M. P. Jonsson, L. Gunnarsson, M. Kall and F. Hook, Anal. Chem., 2009, 81, 6572–6580 CrossRef CAS PubMed.
- L. Konermann, eLS, John Wiley & Sons, 2012 DOI:10.1002/9780470015902.a0003004.pub2.
- A. B. Kayitmazer, D. Seeman, B. B. Minsky, P. L. Dubin and Y. Xu, Soft Matter, 2013, 9, 2553–2583 RSC.
- K. L. Smith, A. E. Winslow and D. E. Petersen, Ind. Eng. Chem., 1959, 51, 1361–1364 CrossRef CAS.
- Y. Osada and M. Sato, J. Polym. Sci., Part C: Polym. Lett., 1976, 14, 129–134 CAS.
- S. P. Cullen, X. Liu, I. C. Mandel, F. J. Himpsel and P. Gopalan, Langmuir, 2008, 24, 913–920 CrossRef CAS PubMed.
- C. Gu, R. D. Coalson, D. Jasnow and A. Zilman, J. Phys. Chem. B, 2017, 121, 6425–6435 CrossRef CAS PubMed.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, 8 figures, one table. See DOI: 10.1039/d0cc01250e |
| This journal is © The Royal Society of Chemistry 2020 |