
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShort and modular synthesis of tetraarylsalicylaldehydes†
David
Tejedor
*a,
Samuel
Delgado-Hernández
ab,
Blanca
Santamaría-Peláez
a and
Fernando
García-Tellado
 *a
*a
aInstituto de Productos Naturales y Agrobiología, CSIC, Astrofísico Francisco Sánchez 3, 38206 La Laguna, Tenerife, Spain. E-mail: fgarcia@ipna.csic.es; dtejedor@ipna.csic.es
bDoctoral and Postgraduate School, Universidad de La Laguna, Avda. Astrofísico Francisco Sánchez s/n, 38200 La Laguna, Tenerife, Spain
First published on 5th March 2020
Abstract
In this study, we describe a novel strategy that allows the obtention of all 15 possible substitution geometries of perarylated salicylaldehydes with total control of the regioselectivity. This strategy entitles the formation of the salicylaldehyde core via a Claisen rearrangement of propargyl vinyl ethers, followed by bromination and Pd-catalyzed aryl–aryl cross-coupling reactions.
Multiply arylated π-electron systems constitute important molecular components in materials science.1 Propeller-shaped hexaarylbenzenes (HABs)2 constitute a good example of this class of molecular structures, with important applications in the field of molecular-scale devices.3 The regioselective access to fully arylated π-electron systems featuring tailor-made peripheral substitution patterns constitutes a current challenge in organic synthesis. Significant progress to this end has been achieved by the groups of Yamaguchi4a and Jux4b with the synthesis of perarylated benzenes endowed with six different aryl substituents. These methodologies have allowed the exploration of large areas of the chemical space of HABs hitherto inaccessible by classical methodologies.2 The chemical space of perarylated π-extended systems based on ring cores other than benzene systems remains scarcely explored. The access to these systems should allow the mapping of their chemical spaces, bringing new opportunities to the design and applications of novel molecular devices. A handful number of synthetic methodologies to access perarylated nicotinates,5a indazoles,5b pyridines5c–e and indoles5f have been recently reported.5 They use either consecutive sp2–sp2 Suzuki cross-coupling reactions on the core ring,5c a combination of condensation/halogenation/cross-coupling reactions,5a,b,d an enamine–ketone condensation5e or a programmed coupling/ring transformation strategy.5f The condensation reaction allows using two arylated fragments to generate the core ring and install the peripheral aromatic rings at specific positions of the ring. A similar strategy has been recently reported for the synthesis of multiarylated salicylaldehyde,6 through the Rh(III)-catalyzed C–H [4+2]C annulation of vinyl enaminones with diaryl alkynes (Scheme 1a). The reaction manifold generates the salicylaldehyde ring endowed with three peripheral aromatic rings, two identical (Ar1) and one distinct (Ar2). Although the reaction is efficient and selective, it delivers salicylaldehydes endowed with a limited peripheral diversity and a diminished reactivity toward halogenation for the subsequent introduction of the fourth aryl substituent (perarylation). The only available position for halogenation is electronically deactivated, i.e. it is meta to the hydroxyl group. The importance of the salicylaldehyde motif in different areas of chemistry and related sciences,7 and the absence of a real-world methodology for the perarylation of these motifs prompted us to face the challenge. At this end, we designed the strategy outlined in Scheme 1b, which took advantage of our experience in the modular synthesis of salicylaldehydes8 to get a short (5 steps from the ketone 1) and regioselective access to all 15 possible substitution geometries of the salicylaldehyde core, spanning from the all-the same A4-TASA to the all-different ABCD-TASA (Fig. 1). In this study, we describe this strategy using cheap and commercially available benzylarylketones 1, arylacetylenes 2, methyl propiolate (3) and aryl boronic acids 7 as the building blocks (Scheme 1c). Last but not least, the strategy was designed to be independent of the electronic nature of the aromatic substituents, with the only restrictions coming from the own reactivity demands of the aromatic bromination9 or Suzuki coupling.10
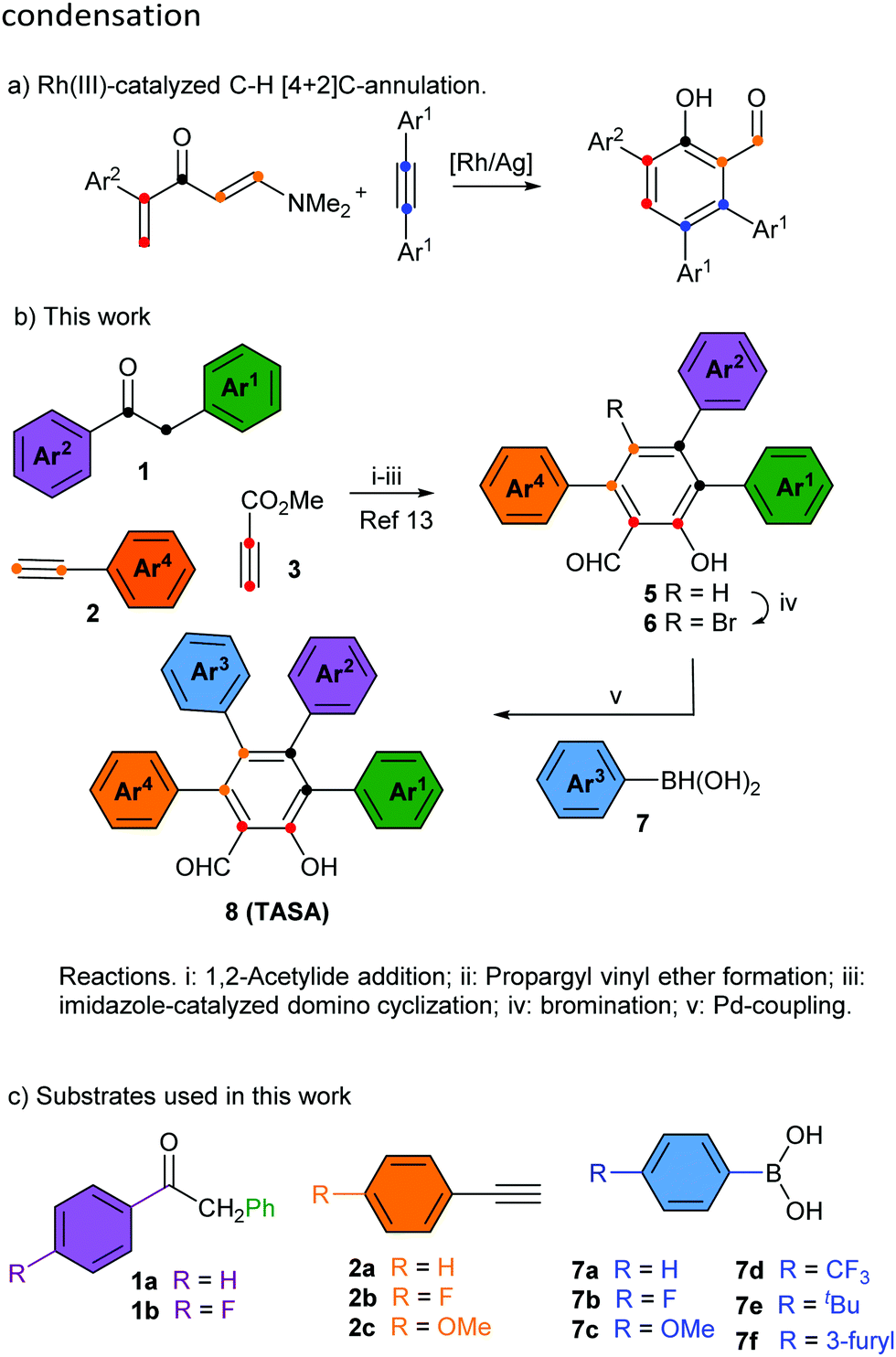 | ||
| Scheme 1 Strategy for the synthesis of tetraarylated salicylaldehydes (TASAs). | ||
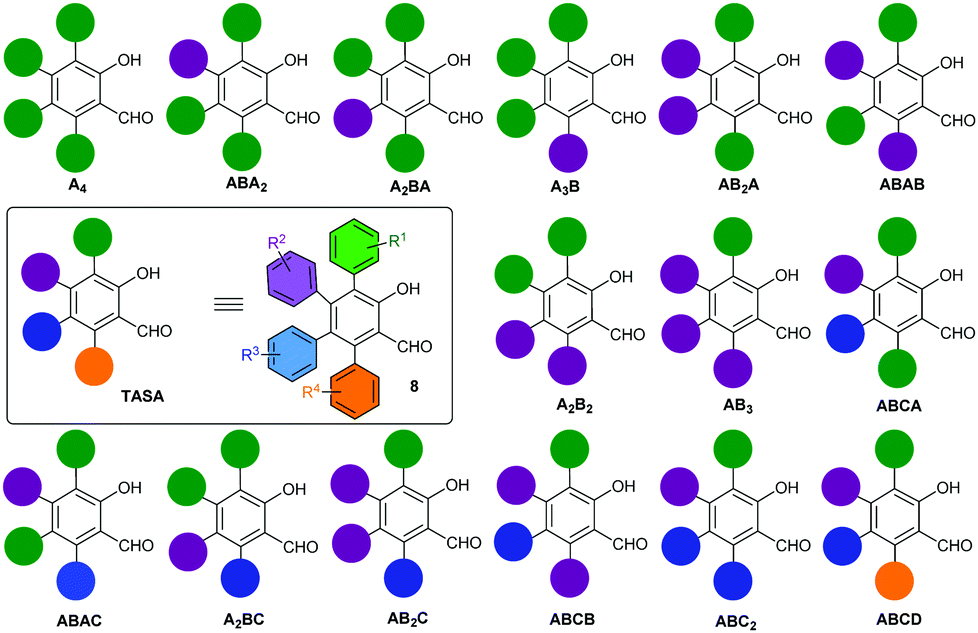 | ||
| Fig. 1 All possible substitution geometries of tetraarylsalicylaldehydes. The ABCD nomenclature follows an anti-clockwise orientation starting from the hydroxyl group and in the order of A to D. | ||
As previously identified by others, one of the main difficulties associated with the construction of perarylated aromatic ring systems is regioselectivity.2 It is a real challenge to achieve the regioselective installation of an aromatic group independent of the stereoelectronics of the other existing peripheral substituents. Our strategy overcomes this difficulty by relying on a modular approach for the construction of the salicylaldehyde ring, which utilizes a non-symmetrical ketone (aryl and benzyl substituents) and a terminal arylacetylene. Additionally, the key to the success of this strategy is that the appending of the last aryl group takes place at the only unsubstituted position of the ring which is conveniently located at the para position with respect to the hydroxyl group (and meta with respect to the formyl group).
We began this study investigating the synthesis of the triphenyl salicylaldehyde derivative 5aa (Ar1 = Ar2 = Ar3 = Ph), because although we had previous experience synthesizing di- and trisubstituted salicylaldehydes,8 we had never tackled specifically the synthesis of the triarylated substrate (Table 1). Fortunately, we were able to obtain the desired product 5aa with a slight modification of our previous synthetic protocol (see the ESI† for full details). Thus, 2-phenylacetophenone (1a) reacted with phenylacetylene (2a) under standard reaction conditions to deliver the corresponding propargylic alcohol. This was subsequently treated with methyl propiolate (3) and a catalytic amount of DABCO to obtain the propargyl vinyl ether (PVE) 4aa.11 Because of the high propensity of highly substituted PVEs to rearrange at room temperature (propargyl Claisen rearrangement),12 product 4aa was not isolated and it was engaged in the next reaction. In this manner, the reaction mixture was heated under microwave irradiation to successfully deliver the desired 3,4,6-triphenyl salicylaldehyde (5aa) (50% yield in 3 steps) (Table 1, entry 1).13 Following this standardized reaction sequence, we were able to prepare the five trisubstituted salicylaldehyde derivatives 5 needed for the purpose of completing all possible substitution geometries of TASAs (8) (Scheme S1, ESI;† and Table 1, entries 1, 5, 9, 12 and 15).
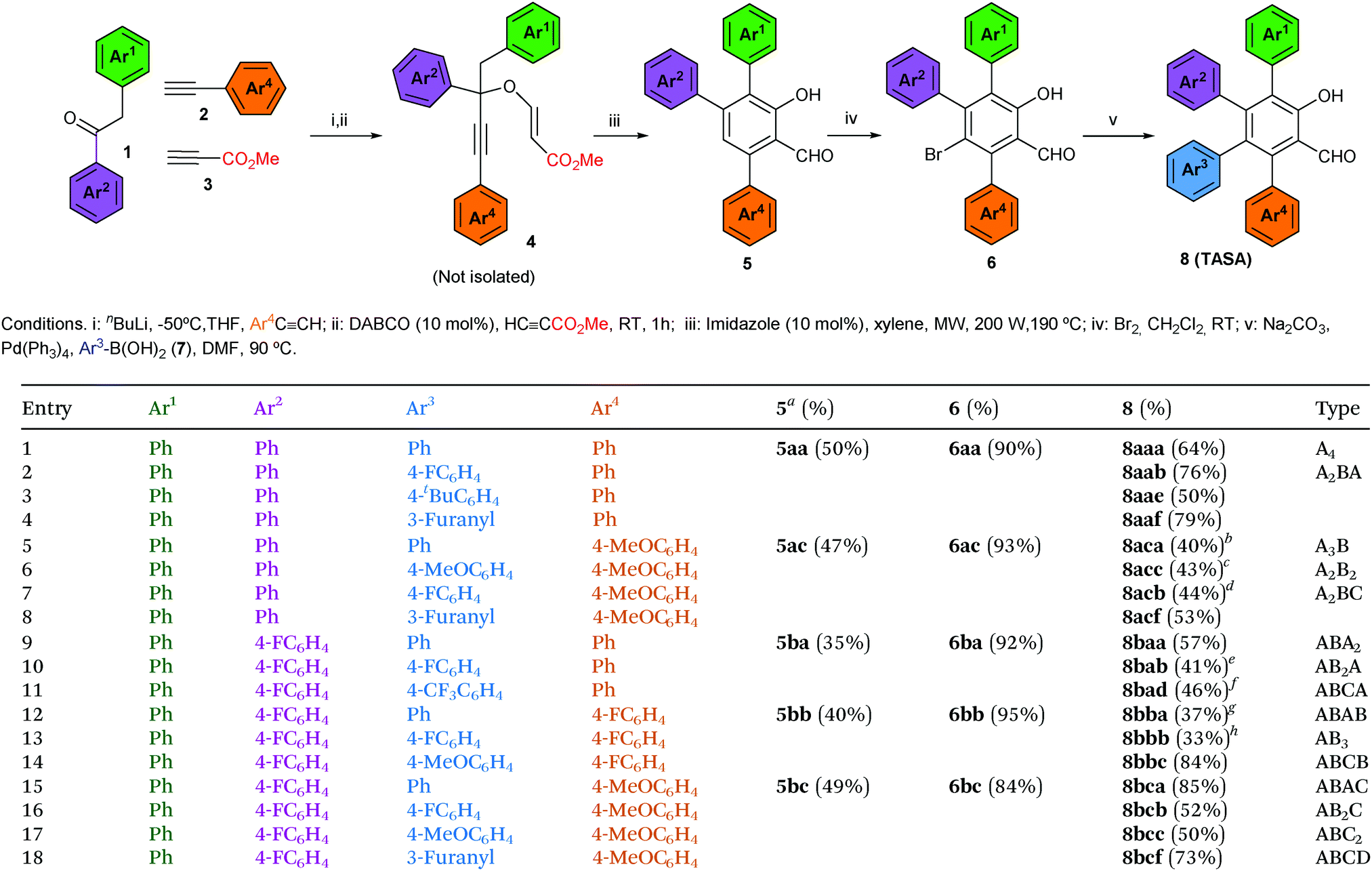
We next explored the bromination step, taking into account that we anticipated that the desired salicylaldehyde ring would be more prone to electrophilic aromatic substitution than the appended aromatic rings. Indeed, while bromination of 5aa delivered bromine 6aa in 90% yield (Table 1, entry 1), the remaining triarylated salicylaldehydes were brominated with yields ranging from 84% to 95% (Table 1, entries 5, 9, 12 and 15). We could only appreciate overbromination of the electron-rich aromatic substituents when larger excesses of Br2 were used (see the ESI† for details).
Finally, we explored the Suzuki coupling of substrate 6aa with different boronic acids using typical coupling conditions (Pd(Ph3)4 as the catalyst and Na2CO3 as the base in dimethylformamide at 90 °C). To our delight, the first A4-TASA (8aaa) was easily isolated in a satisfactory 64% yield when using phenyl boronic acid (7a) as the coupling partner (Table 1, entry 1).14 Similarly, to prove the robustness of the coupling step, three more boronic acids were used to prepare three different A2BA-TASAs (8aab, 8aae and 8aaf) in moderate to good yields (76%, 50% and 79%, respectively) (Table 1, entries 2–4).14 Overall, 3,4,6-triphenyl salicylaldehyde (5aa) enabled us to form 2 of the 15 possible substitution geometries of TASAs depicted in Fig. 1.
Next, we continued with the Suzuki coupling of substrate 6ac which would allow us to prepare three more substitution geometries of TASAs. By choosing the appropriate aryl boronic acids, TASAs A3B (8aca), A2B2 (8acc) and two A2BC (8acb and 8acf) were easily prepared under the same reaction conditions (Table 1, entries 5–8). Likewise, the brominated salicylaldehyde 6ba gave rise to three substitution geometries: ABA2 (8baa), AB2A (8bab) and ABCA (8bad) (Table 1, entries 9–11). The brominated derivative 6bb gave rise to the geometries: ABAB (8bba), AB3 (8bbb) and ABCB (8bbc) (Table 1, entries 12–14). Finally, 6bc delivered the last set of possible geometries including ABAC (8bca), AB2C (8bcb), ABC2 (8bcc) and the all-different ABCD (8bcf) (Table 1, entries 15–18). This last example is worth mentioning as it highlights that the all-different ABCD-TASA was synthesized as any other member of the set of TASAs in five synthetic steps using the commercially available building blocks.
The solutions of these perarylated salicylaldehydes 8 exhibited yellow-green fluorescence (486–573 nm) under visible irradiation (403–424 nm) (see the ESI† for the emission spectra). Because the strategy depicted in this study allows access to an immense number of perarylated derivatives, multiple batteries of potential emitters for use in materials science should be available by design. In addition, the presence of fluorine atoms in the peripheral aromatic rings should allow further diversification by reaction with different nucleophiles (SNAr), which in turn, should allow the chemomodulation of the optical properties of these π-extended motifs.
In summary, we have developed a modular and regioselective synthetic strategy to access all 15 different possible geometrical variants of perarylated salicylaldehydes 8 (TASAs) using cheap and commercially available benzylarylketones 1, arylacetylenes 2, methyl propiolate (3) and aryl boronic acids 7 as the building blocks. This strategy entitles the formation of the salicylaldehyde core via a Claisen rearrangement of the corresponding propargyl vinyl ether, followed by bromination and Suzuki cross-coupling reactions. To demonstrate the efficacy of this strategy we have synthesised at least one example of each one of the 15 possible substitution geometries using the minimum number of benzylarylketone and arylacetylene building blocks, and only five key 3,4,6-triarylated salicylaldehyde intermediates. In all, we built a small library of 18 different TASAs by using a small set of representative aryl boronic acids for the last Suzuki coupling step.
This research was supported by the European Regional Development Funds (ERDF) and the Spanish Ministry of Economy and Competitiveness (MINECO; CTQ2015-63894-P), the Spanish Ministry of Science, Innovation and Universities (MICINN) and the Agencia Estatal de Investigación (AEI) (PGC2018-094503-B-C21). The authors acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI). The authors thank Ms Estefanía Gámez and Mr Jesús Peyrac for experimental assistance and La Laguna University for the use of SEGAI. S. D. H. thanks La Laguna University and Cajasiete for a pre-doctoral contract. The authors thank Dr Verónica Pino for her assistance in the fluorescence experiments, and Dr Romen Carrillo and Prof. Uwe Pitschel for critical comments on this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected examples, see: (a) X. Wang, S. Wang, J. Lv, S. Shao, L. Wang, X. Jing and F. Wang, Chem. Sci., 2019, 10, 2915 RSC; (b) B. Traber, J. J. Wolff, F. Rominger, T. Oeser, R. Gleiter, M. Goebel and R. Wortmann, Chem. – Eur. J., 2004, 10, 1227 CrossRef CAS PubMed; (c) Z. Zeng, Z. Guan, Q. H. Xu and J. Wu, Chem. – Eur. J., 2011, 17, 3837 CrossRef CAS PubMed; (d) G. Kodis, Y. Terazono, P. A. Liddell, J. Andrèasson, V. Garg, M. Hambourger, T. A. Moore, A. L. Moore and D. Gust, J. Am. Chem. Soc., 2006, 128, 1818 CrossRef CAS PubMed; (e) K. R. J. Thomas, M. Velusamy, J. T. Lin, S. S. Sun, Y. T. Tao and C. H. Chuen, Chem. Commun., 2004, 2328 RSC.
- V. Vij, V. Bhalla and M. Kumar, Chem. Rev., 2016, 116, 9565 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Geng, A. Fechtenkötter and K. Müllen, J. Mater. Chem., 2001, 11, 1634 RSC; (b) J. D. Badjic, V. Balzani, A. Credi, J. N. Lowe, S. Silvi and J. F. Stoddart, Chem. – Eur. J., 2004, 10, 1926 CrossRef CAS PubMed; (c) V. Balzani, M. Clemente-Leon, A. Credi, J. N. Lowe, J. D. Badjic, J. F. Stoddart and D. J. Williams, Chem. – Eur. J., 2003, 9, 5348 CrossRef CAS PubMed; (d) S. Hiraoka, T. Nakamura, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2010, 132, 13223 CrossRef CAS PubMed; (e) S. Hiraoka, Y. Hisanaga, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2010, 49, 1669 CrossRef CAS PubMed; (f) M. Steeger and C. Lambert, Chem. – Eur. J., 2012, 18, 11937 CrossRef CAS PubMed; (g) Y. Tanaka, T. Koike and M. Akita, Chem. Commun., 2010, 46, 4529 RSC; (h) R. Shukla, S. V. Lindeman and R. Rathore, J. Am. Chem. Soc., 2006, 128, 5328 CrossRef CAS PubMed.
- (a) S. Suzuki, Y. Segawa, K. Itami and J. Yamaguchi, Nat. Chem., 2015, 7, 227 CrossRef CAS PubMed; (b) D. Lungerich, D. Reger, H. Hölzel, R. Riedel, M. M. J. C. Martin, F. Hampel and N. Jux, Angew. Chem., Int. Ed., 2016, 55, 5602 CrossRef CAS PubMed.
- (a) S. Hirai, Y. Horikawa, H. Ashara and N. Nishiwaki, Chem. Commun., 2017, 53, 2390 RSC; (b) O. S. Kim, J. H. Jang, H. T. Kim, S. J. Han, G. C. Tsui and J. M. Joo, Org. Lett., 2017, 19, 1450 CrossRef CAS PubMed; (c) T. Asako, W. Hayashi, K. Amaike, S. Suzuki, K. Itami, K. Muto and J. Yamaguchi, Tetrahedron, 2017, 73, 3669 CrossRef CAS; (d) C. Doebelin, P. Wanger, F. Bihel, N. Humbert, C. A. Kenfack, Y. Mely, J.-J. Bourguignon and M. Schmitt, J. Org. Chem., 2014, 79, 908 CrossRef CAS PubMed; (e) M. Arita, S. Yokoyama, H. Asahara and N. Nishiwaki, Eur. J. Org. Chem., 2020, 466 CrossRef CAS; (f) S. Suzuki, T. Asako, K. Itami and J. Yamaguchi, Org. Biomol. Chem., 2018, 16, 3771 RSC; (g) R. Ronsi, F. Bellina, M. Lessi, C. Manzini and L. A. Perego, Synthesis, 2014, 283 Search PubMed.
- Y. Zhao, Q. Zheng, C. Yu, Z. Liu, D. Wang, J. You and G. Gao, Org. Chem. Front., 2018, 5, 2875 RSC.
- For selected reviews, see: (a) A. Sebastian, V. Srinivasulu, I. A. Abu-Yousef, O. Gorka and T. Al-Tel, Chem. – Eur. J., 2019, 25, 15710 CrossRef CAS PubMed; (b) L. Pu, Acc. Chem. Res., 2017, 50, 1032 CrossRef CAS PubMed; (c) C. R. Nayar and R. Ravikumar, J. Coord. Chem., 2014, 67, 1 CrossRef CAS; (d) M. Bazarnik, B. Bugenhagen, M. Elsebach, E. Sierda, A. Frank, M. H. Prosenc and R. Wiesendanger, Nano Lett., 2016, 16, 577 CrossRef CAS PubMed; (e) C. J. Whiteoak, G. Salassa and A. W. Kleij, Chem. Soc. Rev., 2012, 41, 622 RSC; (f) S. J. Wezenberg and A. W. Kleij, Angew. Chem., Int. Ed., 2008, 47, 2354 CrossRef CAS PubMed; (g) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS PubMed.
- (a) D. Tejedor, L. Cotos, D. Márquez-Arce, M. Odriozola-Gimeno, M. Torrent-Sucarrat, F. P. Cossío and F. García-Tellado, Chem. – Eur. J., 2015, 21, 18280 CrossRef CAS PubMed; (b) D. Tejedor, G. Méndez-Abt, L. Cotos, M. A. Ramírez and F. García-Tellado, Chem. – Eur. J., 2011, 17, 3318 CrossRef CAS PubMed; (c) D. Tejedor, S. López-Tosco, G. Méndez-Abt, L. Cotos and F. García-Tellado, Acc. Chem. Res., 2016, 49, 703 CrossRef CAS PubMed.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837 CrossRef CAS PubMed.
- J. J. Lennox and J. G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412 RSC.
- D. Tejedor, S. J. Álvarez-Méndez, J. M. López-Soria, V. S. Martín and F. García-Tellado, Eur. J. Org. Chem., 2014, 198 CrossRef CAS.
- D. Tejedor, G. Méndez-Abt, L. Cotos and F. García-Tellado, Chem. Soc. Rev., 2013, 42, 458 RSC.
- See Scheme S2 in the ESI† for the synthesis of propargyl vinyl ethers 4 from terminal alkynes 2 and ketones 1, and their microwave-assisted imidazole-catalyzed domino transformation into salicylaldehydes 5.
- Because the aim of this communication was to show the power of this methodology to delivery each one of the 15 different peripheral geometries, the yields of the Suzuki couplings were found convenient at this stage and they were not optimized.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data for all new compounds 5–8. See DOI: 10.1039/d0cc00738b |
| This journal is © The Royal Society of Chemistry 2020 |