
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA caged E3 ligase ligand for PROTAC-mediated protein degradation with light†
Cyrille S.
Kounde
 a,
Maria M.
Shchepinova
a,
Charlie N.
Saunders
a,
Marcel
Muelbaier
b,
Mark D.
Rackham
c,
John D.
Harling
d and
Edward W.
Tate
*a
a,
Maria M.
Shchepinova
a,
Charlie N.
Saunders
a,
Marcel
Muelbaier
b,
Mark D.
Rackham
c,
John D.
Harling
d and
Edward W.
Tate
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London W12 0BZ, UK. E-mail: e.tate@imperial.ac.uk
bCellzome GmbH, a GSK Company, Meyerhofstrasse 1, 69117 Heidelberg, Germany
cMerck Sharp & Dohme, 1st Floor, 2 Pancras Square, London NC1 4AG, UK
dGlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage SG1 2NY, UK
First published on 3rd April 2020
Abstract
With the intent of achieving greater spatiotemporal control of PROTAC-induced protein degradation, a light-activated degrader was designed by photocaging an essential E3 ligase binding motif in a BRD4 targeting PROTAC. Proteolysis was triggered only after a short irradiation time, the kinetics of which could be monitored by live-cell video microscopy.
Heterobifunctional degraders known as Proteolysis Targeting Chimeras (PROTACs) are well-established as powerful tools for small molecule-directed protein degradation.1,2 These two-headed molecules consist of a ligand for a protein of interest joined to an E3 ligase-recruiting motif via a linker that plays an important role in directing the formation of a non-physiological ternary complex between the E3 ligase and the protein of interest in cells.3 PROTACs thereby promote intracellular polyubiquitination and subsequent proteasome-dependent degradation of the targeted protein in a unique post-translational mode of action, which complements traditional gene and siRNA protein silencing strategies.4 Moreover, the catalytic mechanism of action,5 versatility of the approach6 and potential to degrade targets lacking enzymatic activity7 has prompted major drug discovery efforts both in academia and industry.8
We hypothesized that PROTAC-mediated protein knockdown could be achieved with an additional level of precision if placed under a conditional stimulus such as light. Since protein function is often regulated in time and space, light would represent a suitable tool to activate or deactivate degradation as light itself can be controlled both spatially and temporally. Besides, the combination of PROTACs with light to degrade a disease-relevant protein may offer some applications in the field of photopharmacology.9 Light-mediated small molecule regulation of mammalian proteins has not been explored extensively until very recently.10 For example, whilst this manuscript was in preparation, a number of reports have described the optical control of protein degradation with PROTACs bearing photoswitchable or photolabile groups.11–13 The caging approach is based on the inactivation of a PROTAC with a photocleavable appendage that prevents key binding interactions with either the protein of interest or the E3 ligase, and has been applied to both Cereblon and Von Hippel Lindau (VHL) E3 ligase recruiting PROTACs.14,15
Herein, we expand the caged PROTAC toolbox by attaching the photocleavable 4,5-dimethoxy-2-nitrobenzyl (DMNB) group to a VHL E3 ligase-recruiting ligand and produce a degrader that can be activated on demand using light (Fig. 1).
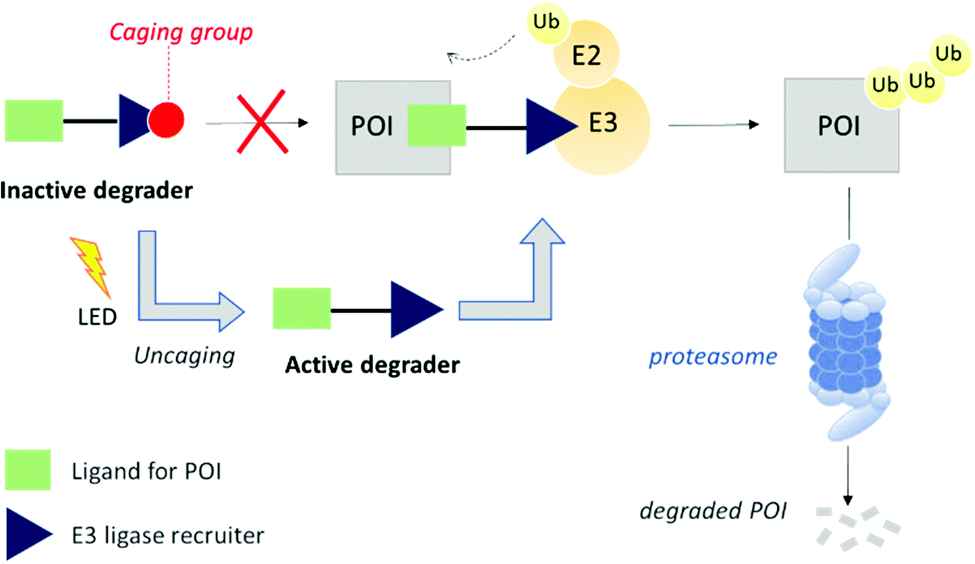 | ||
| Fig. 1 PROTAC caging concept. Attachment of a caging group leads to an inactive degrader. Post-translational modification of the protein of interest (POI) with ubiquitin (Ub) followed by subsequent degradation by the proteasome occurs only after uncaging with a light-emitting diode (LED). | ||
The optimized VHL ligand 1, which is derived from Hypoxia-Inducible Factor16 was functionalized with a DMNB moiety at the essential hydroxyl group to block the recruitment of VHL E3 ligase (Fig. 2A). We used pan-bromodomain inhibitor JQ1 as a warhead and coupled it to the caged version of 1 through a short PEG linker (Scheme S2, ESI†). The final bifunctional molecules 2 (uncaged) and 3 (caged) are structurally related to the previously described PROTAC MZ1.17 Additionally, another reported BRD4 degrader, compound 4, was included as a control for biological evaluation.18
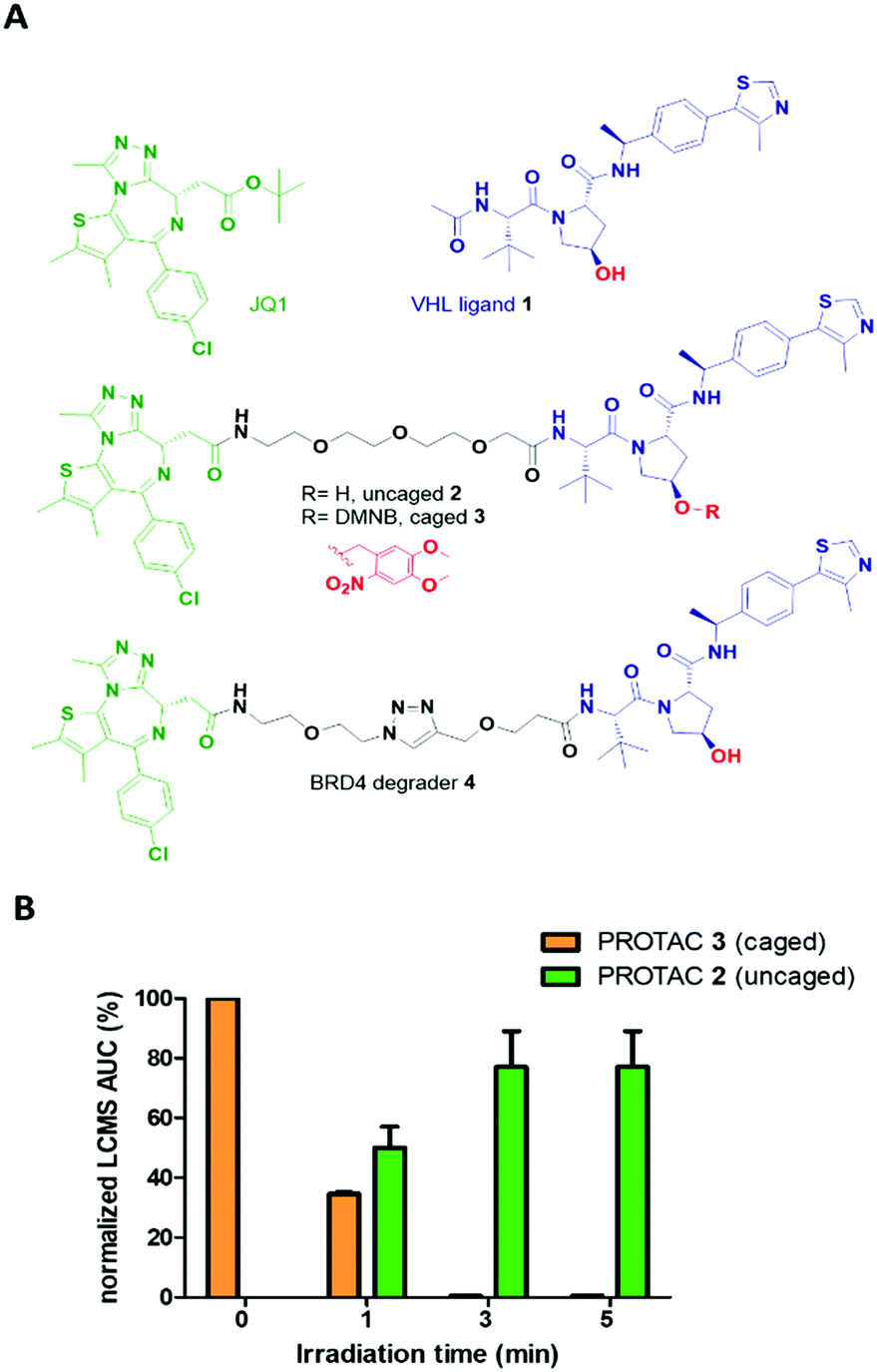 | ||
Fig. 2 Caged PROTAC design and photocharacterization. (A) Chemical structures of pan-bromodomain inhibitor JQ1, Von Hippel Lindau E3 ligase ligand 1, uncaged PROTAC 2 with free hydroxyl group, caged PROTAC 3 with 4,5-dimethoxy-2-nitrobenzyl moiety, and previously reported BRD4 degrader 4. (B) Uncaging of PROTAC 3 following irradiation with a 25 mW 365 nm LED. A 50 μM solution of 3 in acetonitrile–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was irradiated for 1, 3 or 5 min. LCMS area under the curve (AUC) was extracted from the chromatogram full UV spectrum. :1) was irradiated for 1, 3 or 5 min. LCMS area under the curve (AUC) was extracted from the chromatogram full UV spectrum. | ||
Cleavage of the DMNB caging group using a 365 nm 25 mW LED was assessed by UV-Vis spectroscopy (Fig. S1, ESI†) and monitored by LCMS (Fig. 2B). Complete uncaging of compound 3 to release PROTAC 2 was achieved after a 50 μM solution was irradiated for 180 seconds with over half of the initial concentration of 3 found as uncaged 2 in just 60 seconds (Fig. S2, ESI†). Furthermore, compound 3 showed good stability over the course of several days in solution when protected from light (Fig. S3, ESI†).
We first assessed the ability of the uncaged PROTAC 2 to degrade a bromodomain-containing protein targeted by JQ1, BRD4. Consistent with the reported degraders 4 and MZ1,17 compound 2 efficiently knocked down BRD4 at concentrations above 100 nM (Fig. 3A). Expression of the transcription factor c-Myc was also affected by BRD4 knockdown with 2 (Fig. S4, ESI†). Subsequently, we tested the ability of the caged PROTAC 3 to degrade BRD4 following activation with light. HeLa cells were incubated with 3 for 2 h prior to irradiation for 60 seconds at 80 mm from a 25 mW 365 nm LED. Dose-dependent degradation of BRD4 was observed only upon irradiation, with complete knockdown seen at 1 μM (Fig. 3B). Moreover, caged PROTAC 3 showed good stability in the cellular environment since no degradation was observed in non-irradiated cells even after a 24 h-incubation period.
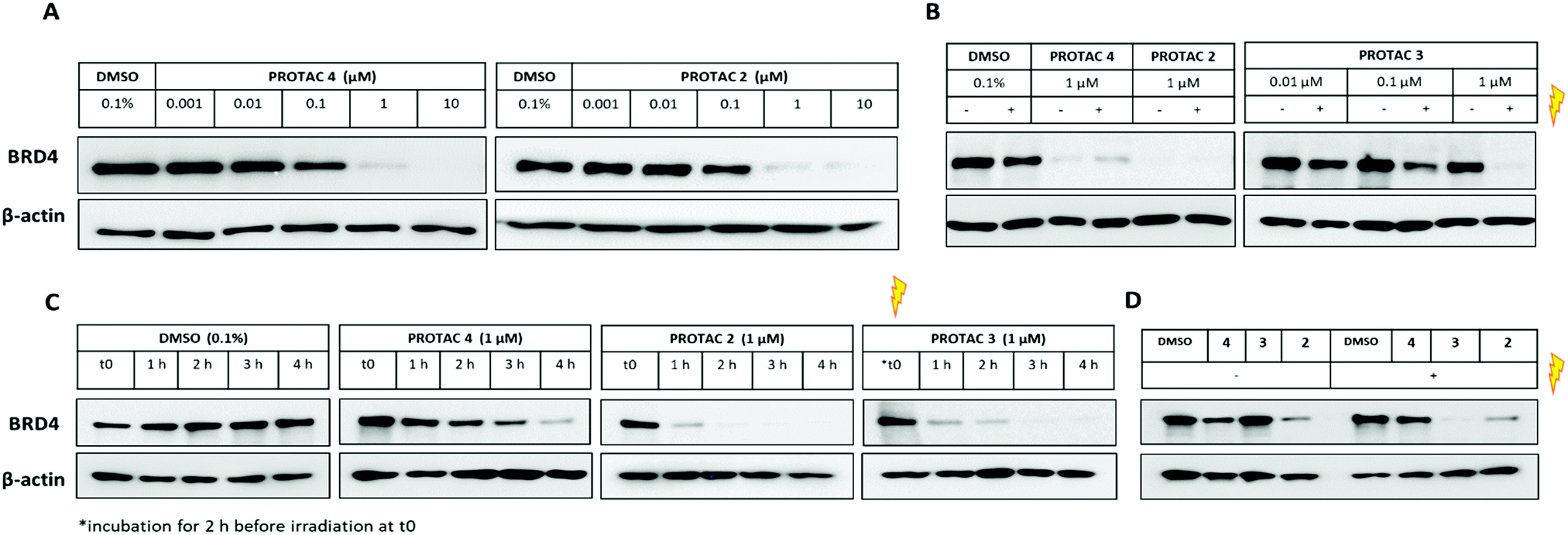 | ||
| Fig. 3 Evaluation of caged PROTAC activity. (A) HeLa cells were treated with PROTAC 4 or uncaged PROTAC 2 for 24 h. Cells were lysed, and lysates collected for western blot analysis. (B) Effect of irradiation: cells were incubated with DMSO, PROTAC 4, PROTAC 2 or PROTAC 3 for 2 h then irradiated for 1 min at 365 nm. After 22 h, lysates were collected for western blot analysis. (C) Time-course experiment: cells were lysed at 60 min intervals to evaluate onset of BRD4 degradation. For PROTAC 3, t0 represents time immediately following irradiation. (D) Washout experiment: cells were incubated with DMSO, PROTAC 4, 3 or 2 (1 μM) for 2 h, then washed 3 times with PBS before irradiation. Cells were lysed after 6 h, and lysates collected for western blot analysis. | ||
We next asked if degradation with 3 after irradiation could be observed within the same timeframe as the parent PROTAC 2, given that the caged molecule 3 requires an activation step before it can act as a degrader. BRD4 level was monitored for the first few hours following incubation with uncaged and caged PROTACs (Fig. 3C). Interestingly, while PROTAC 4 took about 4 h to fully knockdown BRD4, complete loss of the protein was seen just after 1 h for both 2 and 3. This result demonstrates that our caging strategy allows degradation to proceed within the same timeframe as for the parent uncaged molecule 2 and rules out uncaging as a rate-limiting step. Next, to ensure that caged PROTAC 3 enters the cell before being uncaged, a washout experiment was conducted (Fig. 3D). After a 2 h-incubation with compounds, HeLa cells were thoroughly washed to remove residual PROTAC then were irradiated at 365 nm for 60 seconds. Western blot analysis suggests that 3 effectively entered the cells within the 2 h-treatment period since degradation of BRD4 was still observed after irradiation. Incomplete knockdown of BRD4 was noticed with PROTAC 4 after a 2 h incubation and correlated with the slower degradation rate seen in the time-course experiment. In addition, we also confirmed that our caging strategy did not affect the mode of action of the degrader 3 which operates in an E3 ligase and proteasome-dependent manner only after being uncaged (Fig. S5, ESI†).
BRD4 inhibition is reported to significantly impact cell proliferation,19 and we therefore examined the overall phenotypic effect of 3 using live-cell imaging analysis before and after irradiation (Fig. 4 and Fig. S6, ESI†).
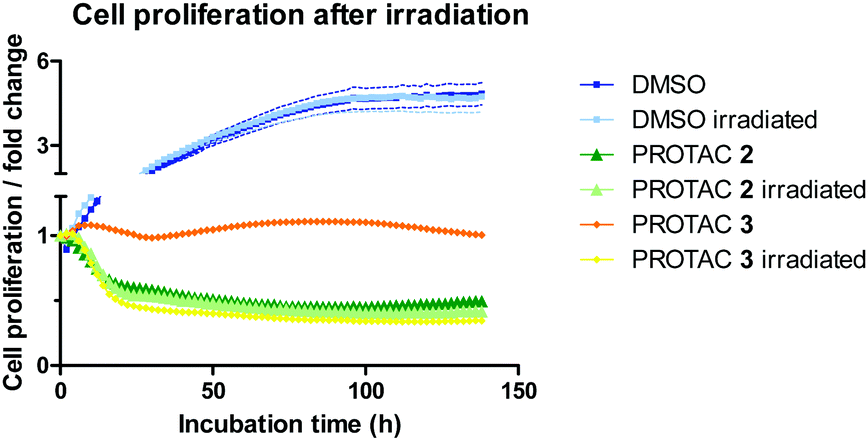 | ||
| Fig. 4 Effect of PROTACs on cell proliferation. HeLa cells were treated with DMSO, PROTAC 2 (1 μM) or 3 (1 μM) either without initial irradiation or with a 60 second irradiation time. Cell count was followed over 6 days, normalized to cell count at t0, and fold changes plotted. | ||
HeLa cells were monitored for up to 6 days after treatment with 1 μM of caged and uncaged PROTACs. The cell proliferation profile without irradiation highlights the different mode of action between PROTAC 2 and 3 (Fig. 4). While the caged compound 3 displayed a cytostatic profile over time (average proliferation fold change = 1), the uncaged PROTAC 2 reduced cell count by half (average proliferation fold change <0.5) after 48 h. Most importantly, uncaging of compound 3 was characterized by a striking change in the growth curve profile that exactly overlaid with 2, confirming mechanistically the release of the active degrader upon irradiation. We attribute the cytostatic effect of PROTAC 3 before irradiation to interaction of the JQ1 moiety with BRD4 (Fig. S6B, ESI†). An in-cell target engagement study using a cellular thermal shift assay20 (CETSA) confirmed PROTAC 3 binding to BRD4 (Fig. S7, ESI†).
Finally, to complement our previous assessment of BRD4 degradation by western blots, we investigated photoinduced BRD4 knockdown in real time using live-cell fluorescence imaging (Fig. 5).
 | ||
| Fig. 5 Light-induced GFP-BRD4 degradation by caged PROTAC measured by live-cell fluorescence imaging of HEK293 cells transfected with pEGFP-BRD4-C1 plasmid. Cells were incubated with (A) 1 μM PROTAC 3 for 1 h and UV irradiated for 60 seconds, or with (B) 1 μM PROTAC 3, (C) 1 μM PROTAC 2 or (D) DMSO vehicle (0.1% v/v), and UV irradiated for 60 seconds. GFP fluorescence was monitored over 3 h, with images taken every 20 min. Scale bar = 20 μm. (E) Quantification of GFP signal degradation over time following PROTAC (1 μM) or DMSO (0.1% v/v) treatment, with or without UV irradiation. Each data point represents background-subtracted 0 time-normalized mean fluorescence from n = 10 single cells, error bars represent SEM. | ||
For this purpose, a GFP-BRD4 expression construct was generated for transient expression in HEK293 cells. Prior to transfection, we also confirmed that PROTAC 3 degraded BRD4 in HEK293 cells when irradiated (Fig. S8, ESI†). Similar to the data gathered from HeLa cells, 1 μM compound 2 delivered significant GFP-BRD4 knockdown within 2 h as shown by the rapid decrease in fluorescence as compared to the DMSO control (Fig. 5C–E). On the other hand, PROTAC 3 had no effect on GFP-BRD4 levels until it was uncaged to deliver an active degrader that removed GFP-tagged BRD4 within the same timeframe as 2 (Fig. 5A, B and E). A comparable degradation profile was obtained with 5 μM PROTAC 3 following irradiation (Fig. S9 and S10, ESI†).
In summary, we have demonstrated conditional PROTAC-directed protein degradation using a caged VHL ligand and light as a conditional stimulus. This method relies on the straightforward attachment of the DMNB group onto a VHL E3 ligase ligand, enabling intracellular activation of the caged degrader with a short irradiation time (60 seconds) followed by VHL- and proteasome-dependent removal of BRD4. Overall, this study establishes caged VHL PROTACs as useful tools for the spatiotemporal control of protein stability, whereby E3 ligase activity is rendered conditional on light, independent of the ligand against the protein of interest. We anticipate this caging strategy will have widespread applicability considering the importance of VHL E3 ligase in the targeted protein degradation field.
This study was supported by the Engineering and Physical Sciences Research Council (EPSRC) grants EP/R512540/1 and EPSRC Centre for Doctoral Training in Chemical Biology (EP/F500416/1), the Biotechnology and Biological Sciences Research Council (BBSRC) grants BB/S001565/1 and BB/N016947/1, and GlaxoSmithKline Research and Development Ltd.
Conflicts of interest
M. M. and J. D. H. are employees and shareholders of GlaxoSmithKline (GSK). E. W. T. is a Director and shareholder of Myricx Pharma Ltd. M. D. R. is an employee of Merck Sharp & Dohme.References
- A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef CAS PubMed.
- I. Churcher, J. Med. Chem., 2018, 61, 444–452 CrossRef CAS.
- Y. Zhang, C. Loh, J. Chen and N. Mainolfi, Drug Discovery Today: Technol., 2019, 31, 53–60 CrossRef PubMed.
- S. Röth, L. J. Fulcher and G. P. Sapkota, Cell. Mol. Life Sci., 2019, 76, 2761–2777 CrossRef PubMed.
- D. P. Bondeson, A. Mares, I. E. D. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. Den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed.
- M. Pettersson and C. M. Crews, Drug Discovery Today: Technol., 2019, 31, 15–27 CrossRef PubMed.
- S. Tomoshige, S. Nomura, K. Ohgane, Y. Hashimoto and M. Ishikawa, Angew. Chem., Int. Ed., 2017, 56, 11530–11533 CrossRef CAS PubMed.
- P. Ottis and C. M. Crews, ACS Chem. Biol., 2017, 12, 892–898 CrossRef CAS PubMed.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef PubMed.
- Q. Delacour, C. Li, M. A. Plamont, E. Billon-Denis, I. Aujard, T. Le Saux, L. Jullien and A. Gautier, ACS Chem. Biol., 2015, 10, 1643–1647 CrossRef CAS PubMed.
- P. Pfaff, K. T. G. Samarasinghe, C. M. Crews and E. M. Carreira, ACS Cent. Sci., 2019, 5(10), 1682–1690 CrossRef CAS PubMed.
- M. Reynders, B. S. Matsuura, M. Bérouti, D. Simoneschi, A. Marzio, M. Pagano and D. Trauner, Sci. Adv. DOI:10.1126/sciadv.aay5064.
- G. Xue, K. Wang, D. Zhou, H. Zhong and Z. Pan, J. Am. Chem. Soc., 2019, 141, 18370–18374 CrossRef CAS PubMed.
- Y. Naro, K. Darrah and A. Deiters, J. Am. Chem. Soc., 2020, 142, 2193–2197 CrossRef CAS PubMed.
- J. Liu, H. Chen, L. Ma, Z. He, D. Wang, Y. Liu, Q. Lin, T. Zhang, N. Gray, H. Ü. Kaniskan, J. Jin and W. Wei, Sci. Adv., 2020, 6, 1–12 Search PubMed.
- D. L. Buckley, I. Van Molle, P. C. Gareiss, H. S. Tae, J. Michel, D. J. Noblin, W. L. Jorgensen, A. Ciulli and C. M. Crews, J. Am. Chem. Soc., 2012, 134, 4465–4468 CrossRef CAS PubMed.
- M. Zengerle, K. H. Chan and A. Ciulli, ACS Chem. Biol., 2015, 10, 1770–1777 CrossRef CAS PubMed.
- M. M. Savitski, N. Zinn, M. Faelth-Savitski, D. Poeckel, S. Gade, I. Becher, M. Muelbaier, A. J. Wagner, K. Strohmer, T. Werner, S. Melchert, M. Petretich, A. Rutkowska, J. Vappiani, H. Franken, M. Steidel, G. M. Sweetman, O. Gilan, E. Y. N. Lam, M. A. Dawson, R. K. Prinjha, P. Grandi, G. Bergamini and M. Bantscheff, Cell, 2018, 173, 260–274 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- D. M. Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–88 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00523a |
| This journal is © The Royal Society of Chemistry 2020 |