
A pre-synthetic strategy to construct single ion conductive covalent organic frameworks†
Juan
Li
 *a,
Fu-Qiang
Zhang
b,
Falian
Li
a,
Zhenzhen
Wu
a,
Canliang
Ma
ac,
Qinchao
Xu
d,
Pengfei
Wang
d and
Xian-Ming
Zhang
*abe
*a,
Fu-Qiang
Zhang
b,
Falian
Li
a,
Zhenzhen
Wu
a,
Canliang
Ma
ac,
Qinchao
Xu
d,
Pengfei
Wang
d and
Xian-Ming
Zhang
*abe
aInstitute of Crystalline Materials, Shanxi University, Wucheng Rd, No. 92, Taiyuan 030006, China. E-mail: lj0511@sxu.edu.cn; xmzhang@sxu.edu.cn
bKey Laboratory of Magnetic Molecules & Magnetic Information Materials (Ministry of Education), Shanxi Normal University, Linfen, 041004, China
cInstitute of Molecular Science, Shanxi University, 030006, Taiyuan, China
dInstitute of Coal Chemistry, Chinese Academy of Sciences, 030001 Taiyuan, China
eInstitute of Chemistry and Culture, School of Chemistry & Material Science, Shanxi Normal University, Linfen 041004, China
First published on 29th January 2020
Abstract
A pre-synthetic strategy was developed for the construction of single ion conductive covalent organic frameworks (COFs). A high Li+ conductivity of 1.6 × 10−3 S cm−1 at 273 K was achieved, and single Na+ and K+ COFs were also obtained by using Na+ and K+ salts as monomers according to this synthetic method.
Crystalline porous materials, such as covalent organic frameworks (COFs), metal–organic frameworks (MOFs), zeolitic imidazolate frameworks (ZIFs) and cages, have been reported as an advanced platform to design fast ion conductors over bulk ion conduction compounds.1–13 Their highly ordered nanochannels enable facile electrolyte penetration and fast ion transportation through them. In addition, the X-ray diffraction technique combined with theoretical simulation methods can provide strong evidence to establish their structure–performance relationships.2,9,14–24 Their electronically insulating framework, well-defined pore structure, and tunable surface polarities also offer them infinite possibilities for their application as candidate ion conductors in solid state electrolytes.2,18–23
Accumulation of anions at the anode is one of the primary culprits for decreasing lithium ion battery performance over time. In single ion solid conductors, the anions are fixed to the underlying matrix, which prevents the sedimentation of anions and eliminates the polarization effects present in dual ion electrolytes.25–28 Different matrixes such as polymers and silicas have been adopted to construct single lithium ions.29–33 But the uncontrollability and great difficulties in synthesis limited the development of single Li+ conductors.
As a novel class of porous crystalline solids, covalent organic frameworks (COFs) are excellent platforms for exploring single-ion conductors, which benefit from their chemical stability and flexibility. Their ordered open channels would favor fast ion migration in principle. Very recently, post-synthesized loading, modification and organometallic methods have been used to construct single lithium MOFs, and good performance like 7.4 × 10−3 S cm−1 has been achieved in those single-ion conductive MOFs and COFs,10–13,34–42 which nearly reached the practical application standard. However, we found that most of the crystalline porous single lithium conductors were synthesized by using a post-synthetic method, but pre-synthetic methods for single Li+ COFs have rarely been reported.
In this study, we propose a pre-synthetic strategy to synthesize single Li+ COFs by using lithium salt (lithium 2,5-diaminobenzenesulfonate, PaSO3Li) as the monomer. A very small amount of acetic acid was used as the catalyst to avoid the possible H+/Li+ exchange problem during the synthesis process (Scheme 1). We first synthesized the lithium salt PaSO3Li through the acid–base reaction of 2,5-diaminobenzenesulfonic acid (PaSO3H) with lithium hydroxide (LiOH). The complete conversion to the lithium salt was verified by the 1H-NMR spectrum. Then PaSO3Li and triformylphloroglucinol (Tp) were condensed to form a lithium salt-based COF (Tp-PaSO3Li-COF) through keto–enamine covalent bonds in a solvothermal environment. The chemical structure of Tp-PaSO3Li-COF was characterized by solid-state 13C NMR spectrometry, FT-IR spectroscopy, and powder X-ray diffraction (PXRD) analysis. In the 13C NMR spectrum in Fig. 1G, a sharp signal at δ = 184.38 ppm (a) corresponds to the carbonyl carbon atom of the β-ketoenamine linker. The absence of N–H stretching peaks (ν = 3367 and 3293 cm−1) due to the free diamine –NH2 group and of a peak due to the aldehyde group –CHO (ν = 1645 cm−1) of Tp indicated complete consumption of the reactants. The appearance of new peaks at ν = 1583 and 1234 cm−1 could be ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N stretching bands (Fig. 1F).
C and C–N stretching bands (Fig. 1F).
 | ||
| Scheme 1 Illustration of the pre-synthetic strategy for single-ion (Li+, Na+, K+) conductive COFs. | ||
 | ||
| Fig. 1 Synthesis of Tp-PaSO3Li-COF (A). Unit cell (B), top (C) and side (D) views of the AA stacking mode of Tp-PaSO3Li-COF (red, oxygen; blue, nitrogen; grey, carbon; purple, lithium). PXRD profiles of Tp-PaSO3Li-COF (E): experimentally observed (black), Pawley refined (red), and their difference. FTIR spectra (F) of Tp-PaSO3Li-COF (cyan), PaSO3Li (blue), PaSO3H (red) and Tp (black). Solid-state 13C-NMR spectrum of Tp-PaSO3Li-COF (G). N2 adsorption isotherm of Tp-PaSO3Li-COF (H). | ||
Thermogravimetric analysis (TGA) revealed that Tp-PaSO3Li-COF showed no decomposition up to 400 °C under a nitrogen atmosphere (Fig. S1, ESI†). The permanent porosity of Tp-PaSO3Li-COF was evaluated from the N2 adsorption isotherm at 77 K. Activated Tp-PaSO3Li-COF showed a regular type II adsorption isotherm (Fig. 1H), and its surface area was evaluated according to the Brunauer–Emmett–Teller (BET) model as 488 m2 g−1. Its pore volume was evaluated as 0.639 cm3 g−1 at P/P0 = 0.99. Its pore diameter was evaluated as 1.2 nm, consistent with the BET analysis (Fig. S2, ESI†).
The crystalline structure and unit cell parameters of Tp-PaSO3Li-COF were determined by powder X-ray diffraction (PXRD) together with structural simulations using density functional theory (DFT) tight-binding and Pawley refinement. Tp-PaSO3Li-COF exhibited a PXRD pattern with intense peaks at 4.69°, 8.01°, and 26.96°, which were assigned to the (1 0 0), (1 1 0), and (0 0 1) planes, respectively. It adopts the space group P3, with unit cell parameters a = b = 22.58 Å, c = 5.34 Å, α = β = 90° and γ = 120° (Fig. 1C). Pawley refinement yielded a PXRD pattern (Fig. 1E, red line) that was consistent with the experimentally observed profile, as evidenced by negligible difference (Fig. 1E, black plot), with RWP and RP values of 2.88% and 2.24%, respectively. Table S1 (ESI†) shows the unit cell parameters and atomistic coordinates of Tp-PaSO3Li-COF. We speculate that the large interlayer distance of 5.34 Å is the net result of competing coulomb and π–π stacking forces.
We used X-ray photoelectron spectroscopy (XPS) and 7Li-NMR spectroscopy to study the chemical nature and Li+ dynamic motion of Tp-PaSO3Li-COF. Compared with monomer PaSO3Li, the binding energy of Li 1s (Fig. 2B) in Tp-PaSO3Li-COF shifted from 55.05 eV to 55.48 eV, indicating strong interaction between Li+ and the framework of Tp-PaSO3Li-COF. The O1s spectra of monomer PaSO3Li and Tp-PaSO3Li-COF are compared and deconvoluted in Fig. 2C. The peaks at 531.3 eV of both PaSO3Li and Tp-PaSO3Li-COF are assigned to the SO3− and the additional peak at 532.7 eV in Tp-PaSO3Li-COF is attributed to the oxygen atom in the β-ketoenamine linkers. As can be seen from the solid-state 7Li-NMR spectrum in Fig. 2D, after the temperature increases from 293 K to 353 K, the chemical shift of lithium ions migrates to a higher field and the peak shape reflects this. This may be because the high temperature accelerates the migration of lithium ions and weakens the interaction between anions and lithium ions, resulting in a weakened shielding effect around lithium ions.
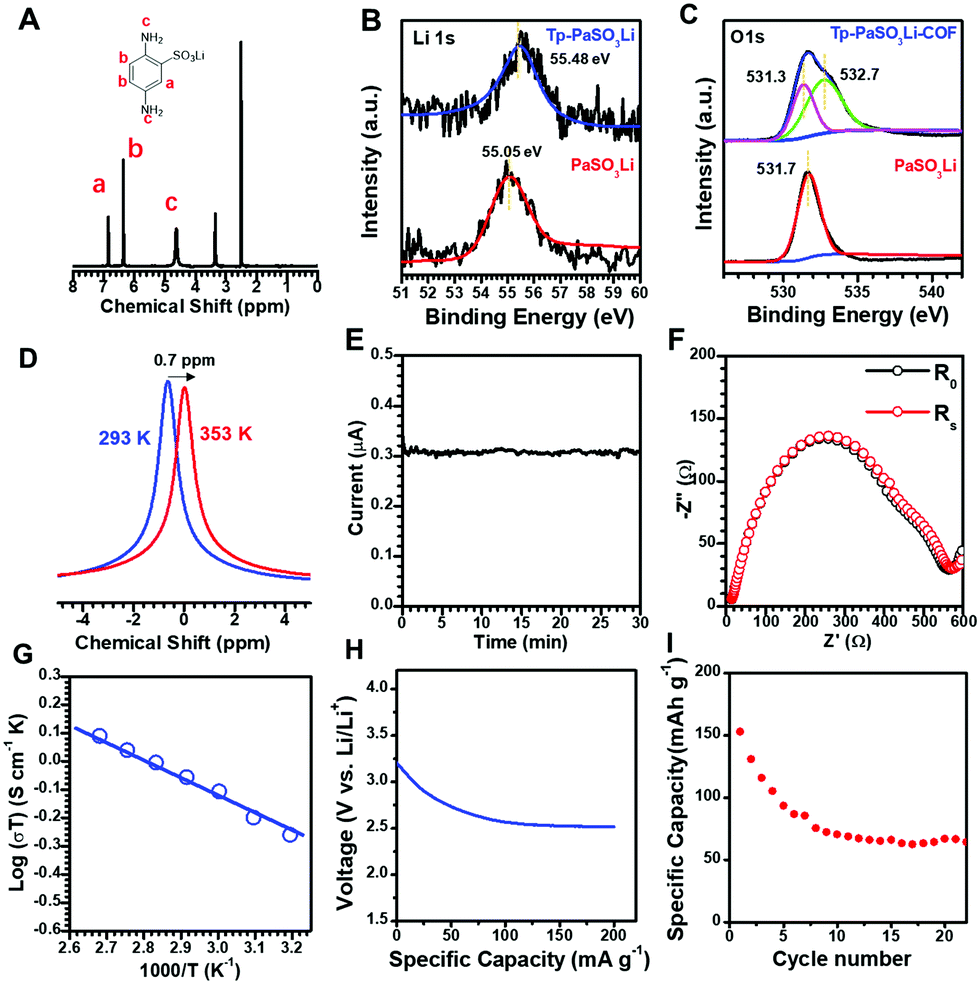 | ||
| Fig. 2 1H-NMR spectrum of PaSO3Li (A). High-resolution XPS spectra: Li 1s (B) and O 1s (C) of Tp-PaSO3Li-COF and PaSO3Li. Solid-state 7Li-NMR spectra of Tp-PaSO3Li-COF at 293 K and 353 K (D). I–t curve (E) and Nyquist plots (F) of Tp-PaSO3Li-COF before and after an EIS test. Arrhenius plot (G), discharge curve (H) and capacity retention (I) for the typical prototype Li/Tp-PaSO3Li-COF/LiFePO4 at a current density of 34 mA g−1. | ||
The lithium-ion conductivity was characterized by alternating current impedance spectroscopy using a Solartron 1260 instrument. Tp-PaSO3Li-COF powder was compressed into a robust pellet under 200 kN. The medium did not disrupt the robustness of the Tp-PaSO3Li-COF pellet. To estimate the activation energy (Ea) of the solid electrolyte and to understand the mechanism of the lithium-ion conduction behaviours, the conductivity was measured at various temperatures from 20 °C to 90 °C. In Nyquist plots (Fig. S8 and S9, ESI†), the intercepts on the Z′ axis yielded the resistance. The conductivity was calculated according to the equation:
| σ = l/SRs |
The conductivities were calculated as 1.6 × 10−3, 1.7 × 10−3, 1.9 × 10−3, 2.4 × 10−3, 2.6 × 10−3, 2.8 × 10−3, and 3.0 × 10−3 S cm−1 at 293, 313, 323, 333, 343, 353, and 363 K, respectively. It should be noted that about 10 μL EC/DMC (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent mixture was added to the pellet as the plasticizer to facilitate ion migration. We calculated the activation energy of Tp-PaSO3Li-COF as 0.13 eV according to the Arrhenius equation (Fig. 2G). This insignificantly small Ea indicates that Tp-PaSO3Li-COF can be classed as a superionic conductor.
:1) solvent mixture was added to the pellet as the plasticizer to facilitate ion migration. We calculated the activation energy of Tp-PaSO3Li-COF as 0.13 eV according to the Arrhenius equation (Fig. 2G). This insignificantly small Ea indicates that Tp-PaSO3Li-COF can be classed as a superionic conductor.
In order to detect the possible electronic conductivity of Tp-PaSO3Li-COF, we conducted an impedance test on the equivalent COF without the lithium sulfonate group, named Tp-Pa-COF, and the results showed that Tp-Pa-COF was an electrical insulator (Fig. S10, ESI†). At the same time, to exclude the proton conduction caused by partial sulfonic acid ions that may exist in the structure, we tested the conductivity of Tp-PaSO3H-COF, and the results showed that Tp-PaSO3H had no conductivity at room temperature or high temperature (Fig. S11 and S12, ESI†). A symmetric cell (Li/Tp-PaSO3Li-COF pellet/Li) was assembled by connecting Li-metal electrodes to both sides of a Tp-PaSO3Li-COF pellet. The Li+ transference number was calculated as 0.94 by using the Bruce–Vincent–Evans (BVE) technique.43 As shown in Table S7 (ESI†), the lithium conduction performance of Tp-PaSO3Li-COF is among the best performances of MOF- and COF-based single Li+ conductors.
To confirm the practical application of this single Li+ electrolyte, a battery prototype based on Tp-PaSO3Li-COF was assembled using a lithium-metal negative electrode (anode) and an aluminum-coated LiFePO4 as a model active material for the positive electrode. As the technical optimization of the electrode formulation is beyond our current ability and is not the main aim of this work, we just conducted the cycling tests with general technical parameters. The maximum specific capacity of 152 mA g−1 was observed in the first cycle, and due to the transport limitations of a composite electrode with 100 μm thickness (that make the average ion pathway longer and tortuous), the specific capacity decreased to a minimum value of 67 mA g−1. Although good capacity retention needs to be optimized in the future, it is the first time that a single Li+ COF based Li+ battery has been assembled to check the practical applications of single Li+ COFs.
Further, we performed theoretical elucidation by DFT calculations, and two migration pathways (axial and planar) were considered. As can be seen, the rate-determining steps were initial 1 to intermediate 2 (Fig. 3d) and intermediate 5 to 6 (Fig. 3h) in the axial and planar migration pathways, respectively, and the corresponding migration barriers (Em) are 1.0 kcal mol−1 and 2.8 kcal mol−1, lower than those of the post-synthesized sample. For the axial route in the rate determining step with a Li migration distance of 3.004 Å, the migrating Li ion is less chelated by two oxygen species from sulfonate with a Li–O distance of 2.413 Å in 1 but strongly coordinated by two chelating sulfonate and one keto oxygen species with a Li–O distance of 1.893–2.003 Å in 2. In contrast, for the planar route in the rate determining step with a Li migration distance of 4.475 Å, the migrating Li is singly coordinated by one sulfonate oxygen with a short Li–O distance of 1.614 Å in 5, while it is coordinated by sulfonate oxygen and one keto oxygen with Li–O distances of 1.831 Å and 2.005 Å in 6.
 | ||
| Fig. 3 Theoretically calculated Li-ion migration behaviour along the axial (left) and planar (right) directions, with the migrating Li ions shown as large orange balls in a [1 × 1 × 2] superlattice. Note: the migrating Li ion sites in structures 1, 4 and 7 are equivalent. | ||
A single Na+ COF (Tp-PaSO3Na-COF) and a single K+ COF (Tp-PaSO3K-COF) were then obtained by using sodium salt (PaSO3Na) and potassium salt (PaSO3K) as the monomers, and they were characterized by PXRD, solid-state 13C-NMR, FTIR spectroscopy, TGA and nitrogen sorption tests (Scheme 1 and Fig. S14–S23, ESI†). The PXRD patterns show that they are isostructural to Tp-PaSO3Li-COF and the decreased relative peak intensity might be influenced by the different cations. The BET surface areas of Tp-PaSO3Na-COF and Tp-PaSO3K-COF are 525.66 m2 g−1 and 369.08 m2 g−1, respectively. The significant crystallinity and porosity verified the universality of this pre-synthetic strategy.
In summary, we have presented a pre-synthetic strategy for the design and construction of single Li+ conductive COFs. The fast ion migration behaviour was verified by theoretical calculation and cycle testing results proved the practicability of the material. Further, single Na+ (Tp-PaSO3Na-COF) and single K+ (Tp-PaSO3K-COF) conductors were successfully obtained by using sodium and potassium salt monomers, which verifies the universality of this pre-synthetic strategy. This work provides a novel solution to construct advanced solid electrolytes in the future.
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21805173 & 21871167).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Jiang, X. Chen, K. Geng, R. Liu, K. T. Tan, Y. Gong, Z. Li, S. Tao and Q. Jiang, Angew. Chem., Int. Ed., 2019, 58, 2 CrossRef.
- L. Bai, B. Tu, Y. Qi, Q. Gao, D. Liu, Z. Liu, L. Zhao, Q. Li and Y. Zhao, Chem. Commun., 2016, 52, 3003 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC.
- T. Yamada, K. Otsubo, R. Makiura and H. Kitagawa, Chem. Soc. Rev., 2013, 42, 6655 RSC.
- C. Qian, Q.-Y. Qi, G.-F. Jiang, F.-Z. Cui, Y. Tian and X. Zhao, J. Am. Chem. Soc., 2017, 139, 6736 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, 1585 CrossRef PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su and J. Li, Science, 2018, 361, 48 CrossRef CAS PubMed.
- J. Bae, Y. Li, J. Zhang, X. Zhou, F. Zhao, Y. Shi, J. B. Goodenough and G. Yu, Angew. Chem., Int. Ed., 2018, 130, 2118 CrossRef.
- Z. Xie, B. Wang, Z. Yang, X. Yang, X. Yu, G. Xing, Y. Zhang and L. Chen, Angew. Chem., Int. Ed., 2019, 131, 15889 CrossRef.
- H. Chen, H. Tu, C. Hu, Y. Liu, D. Dong, Y. Sun, Y. Dai, S. Wang, H. Qian, Z. Lin and L. Chen, J. Am. Chem. Soc., 2018, 140, 896 CrossRef CAS PubMed.
- Q. Xu, S. Tao, Q. Jiang and D. Jiang, J. Am. Chem. Soc., 2018, 140, 7429 CrossRef CAS.
- N. Huang, P. Wang, M. A. Addicoat, T. Heine and D. Jiang, Angew. Chem., Int. Ed., 2017, 56, 4982 CrossRef CAS PubMed.
- S. Chandra, T. Kundu, K. Dey, M. Addicoat, T. Heine and R. Banerjee, Chem. Mater., 2016, 28, 1489 CrossRef CAS.
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- Z. Wang, R. Tan, H. Wang, L. Yang, J. Hu, H. Chen and F. Pan, Adv. Mater., 2018, 30, 1704436 CrossRef PubMed.
- J. Cepeda, S. Pérez-Yáñez, G. Beobide, O. Castillo, E. Goikolea, F. Aguesse, L. Garrido, A. Luque and P. A. Wright, Chem. Mater., 2016, 28, 2519 CrossRef CAS.
- K. Fujie, R. Ikeda, K. Otsubo, T. Yamada and H. Kitagawa, Chem. Mater., 2015, 27, 7355 CrossRef CAS.
- M. L. Aubrey, R. Ameloot, B. M. Wiers and J. R. Long, Energy Environ. Sci., 2014, 7, 667 RSC.
- M. L. Aubrey and J. R. Long, J. Am. Chem. Soc., 2015, 137, 13594 CrossRef CAS PubMed.
- Z. Wang, Z. Wang, L. Yang, H. Wang, Y. Song, L. Han, K. Yang, J. Hu, H. Chen and F. Pan, Nano Energy, 2018, 49, 580 CrossRef CAS.
- B. C. Patra, S. K. Das, A. Ghosh, A. Raj K, P. Moitra, M. Addicoat, S. Mitra, A. Bhaumik, S. Bhattacharya and A. Pradhan, J. Mater. Chem. A, 2018, 6, 16655 RSC.
- L. Fan, S. Wei, S. Li, Q. Li and Y. Lu, Adv. Energy Mater., 2018, 8, 1702657 CrossRef.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403 CrossRef CAS PubMed.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797 RSC.
- M. Miner Elise and M. Dincă, Philos. Trans. R. Soc., A, 2019, 377, 20180225 CrossRef CAS PubMed.
- Q. Ma, H. Zhang, C. Zhou, L. Zheng, P. Cheng, J. Nie, W. Feng, Y.-S. Hu, H. Li, X. Huang, L. Chen, M. Armand and Z. Zhou, Angew. Chem., Int. Ed., 2016, 55, 2521 CrossRef CAS PubMed.
- J. F. Van Humbeck, M. L. Aubrey, A. Alsbaiee, R. Ameloot, G. W. Coates, W. R. Dichtel and J. R. Long, Chem. Sci., 2015, 6, 5499 RSC.
- H. Oh, K. Xu, H. D. Yoo, D. S. Kim, C. Chanthad, G. Yang, J. Jin, I. A. Ayhan, S. M. Oh and Q. Wang, Chem. Mater., 2016, 28, 188 CrossRef CAS.
- H. Zhao, F. Asfour, Y. Fu, Z. Jia, W. Yuan, Y. Bai, M. Ling, H. Hu, G. Baker and G. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 19494 CrossRef CAS PubMed.
- Y. Fei, S. Liu, Y. Long, L. Lu, Y. He, X. Ma and Y. Deng, ChemElectroChem, 2019, 6, 2219 CrossRef CAS.
- L. Shen, H. B. Wu, F. Liu, J. L. Brosmer, G. Shen, X. Wang, J. I. Zink, Q. Xiao, M. Cai and G. Wang, Adv. Mater., 2018, 30, 1707476 CrossRef.
- S. Fischer, J. Roeser, T. C. Lin, R. H. DeBlock, J. Lau, B. S. Dunn, F. Hoffmann, M. Fröba, A. Thomas and S. H. Tolbert, Angew. Chem., Int. Ed., 2018, 130, 16925 CrossRef.
- X. Liu, X. Li, H. Li and H. B. Wu, Chem. – Eur. J., 2018, 24, 18293 CrossRef CAS PubMed.
- R. Zhao, Z. Liang, R. Zou and Q. Xu, Joule, 2018, 21, 2235 CrossRef.
- E. M. Miner, S. S. Park and M. Dinca, J. Am. Chem. Soc., 2019, 141, 4422 CrossRef CAS.
- S. S. Park, Y. Tulchinsky and M. Dinca, J. Am. Chem. Soc., 2017, 139, 13260 CrossRef CAS.
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S. H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1737 CrossRef CAS.
- Y. Hu, N. Dunlap, S. Wan, S. Lu, S. Huang, I. Sellinger, M. Ortiz, Y. Jin, S.-H. Lee and W. Zhang, J. Am. Chem. Soc., 2019, 141, 7518 CrossRef CAS.
- K. Jeong, S. Park, G. Y. Jung, S. H. Kim, Y.-H. Lee, S. K. Kwak and S.-Y. Lee, J. Am. Chem. Soc., 2019, 141, 5880 CrossRef CAS PubMed.
- J. Mindemark, B. Sun, E. Törmä and D. Brandell, J. Power Sources, 2015, 298, 166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00454e |
| This journal is © The Royal Society of Chemistry 2020 |