
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon-11 carboxylation of trialkoxysilane and trimethylsilane derivatives using [11C]CO2†
Salvatore
Bongarzone
 *,
Nicola
Raucci
,
Igor Camargo
Fontana
,
Federico
Luzi
and
Antony D.
Gee
*
*,
Nicola
Raucci
,
Igor Camargo
Fontana
,
Federico
Luzi
and
Antony D.
Gee
*
School of Biomedical Engineering & Imaging Sciences, King's College London, King's Health Partners, St Thomas’ Hospital, London SE1 7EH, UK. E-mail: salvatore.bongarzone@kcl.ac.uk; antony.gee@kcl.ac.uk
First published on 25th March 2020
Abstract
A novel carboxylation radiosynthesis methodology is described starting from cyclotron-produced [11C]CO2 and fluoride-activated silane derivatives. Six carbon-11 labelled carboxylic acids were obtained from their corresponding trimethylsilyl and trialkoxysilyl precursors in a one-pot labelling methodology. The radiochemical yields ranged from 19% to 93% within 12 minutes post [11C]CO2 delivery with a trapping efficiency of 21–89%.
Carbon-11 (11C) is a short-lived radionuclide (t1/2 = 20.4 min) commonly applied in positron emission tomography (PET) imaging.1 The isotopic substitution of carbon-12 for a carbon-11 atom in bioactive molecules maintains the chemical and biological properties of the non-radioactive autologue, allowing the study of the pharmacokinetics and biodistribution of a wide range of biologically active molecules in living subjects.1
11C is cyclotron-produced in the form of carbon dioxide ([11C]CO2) which can be directly incorporated into various biologically relevant molecules, such as [carbonyl-11C]carboxylic acids.2 Traditionally, aromatic carbon-11 labelled carboxylic acids have been labelled directly from [11C]CO2 using either (i) Grignard reagents3 or (ii) aromatic boronic esters as supporting reagents (Scheme 1).4
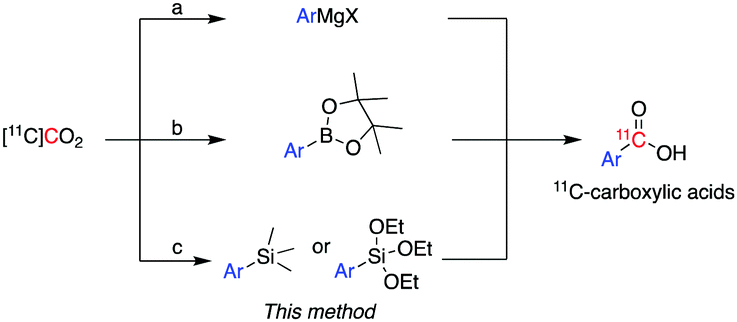 | ||
| Scheme 1 Current methods for the preparation of aromatic carbon-11 labelled carboxylic acids from [11C]CO2 using: (a) Grignard reagents, (b) boronic esters and (c) trialkoxysilane and trimethylsilane derivatives – the latter is used in this work. | ||
However, these methodologies present some challenges which limit their wider application. For instance, the high reactivity of Grignard reagents is not well tolerated by many functional groups, limiting their utility to labelling functionally simple substrates.3 In addition, Grignard reagents are very sensitive to moisture or reaction with atmospheric CO2, even if great care is used in the storage and use of these reagents, leading to isotopic dilution of [11C]CO2 and concomitant low molar activity (Am) of 11C-labelled products.
Compared to Grignard reagents, boronic esters have greater stability to atmospheric CO2 and moisture which broadens their use for radiolabelling aromatic and heteroaromatic compounds.4 However, the radiolabelling of the latter class of compounds (e.g. pyridyl, pyrazyl and thienyl boronic ester derivatives) is inconsistent and gives low-moderate radiochemical yields (RCYs: 3–69%).4a Recently, a dynamic carbon isotope exchange (isotopic enrichment) of carboxylates using [13C]CO2 and [14C]CO2 has been reported; however, the applicability of this methodology to carbon-11 chemistry would require an extensive study on the range of molar activity that could be obtained.5
Based on a search of the traditional synthetic chemistry literature, improved methods for the 11C-carboxylation of aryl and heteroaryl groups might be achieved by the use of trialkoxysilyl and trimethylsilyl derivatives via a so-called copper-catalysed desilylative carboxylation reaction.6 Arylsilanes reacted readily with a fluoride anion source, such as cesium fluoride (CsF), potassium fluoride (KF), and tetramethylammonium fluoride (Me4NF), to form a pentavalent silicate.6,7 The pentavalent silicate was then converted in the presence of a copper catalyst to an aryl copper intermediate which reacted with non-radioactive CO2 in moderate to excellent yields (27–99%).6,7 Varying the substitution patterns of the aromatic ring with electron-withdrawing or electron donating groups did not alter the efficiency of substrate carboxylation.6a–c Excellent results were also reported for the carboxylation of heteroaromatic compounds, such as thiophenyl, pyridyl and furanyl silane derivatives, and their derivatization to ester products (89–93%).6b,c
Compared to the traditional 11C-carboxylation methodologies, the use of silyl derivatives would provide greater air and moisture stability and therefore easier handling and storage. Moreover, trimethylsilyl and trialkoxysilyl precursors are readily obtained via a plethora of synthetic reagents: Grignard or organolithium reagents8 or functionalization of arylamides,9 aryl acyl fluorides,10 aryl esters,11 and aryl cyanides12via transition metals (nickel, copper, and ruthenium).
With the aim of developing more robust and versatile 11C-carboxylation methodologies, we herein present the development of a novel 11C-carboxylation protocol involving the use of arylsilyl derivatives. The carbon-11 labelled carboxylic acids were obtained in short synthesis times, with high molar activities and with broad applicability to a range of trimethylsilane and trialkoxysilane derivatives.
2-(Thienyl)trimethylsilane (1a, Scheme 2) was initially chosen as a model substrate for cyclotron-produced [11C]CO2 carboxylation reactions. Liu et al. reported that the combination of CsF and 18-crown-6 in the presence of CO2 (1 atm) allowed the carboxylation of trimethylsilane derivatives in high yields.13
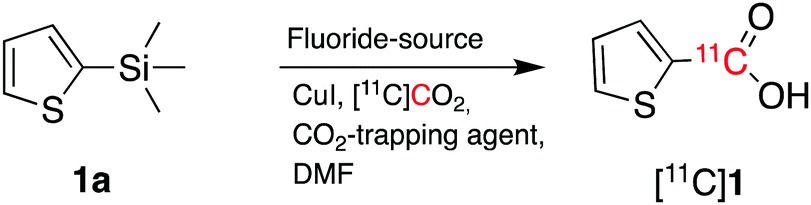 | ||
| Scheme 2 Radiosynthetic approach to radiolabelled carbon-11 labelled carboxylic acids from cyclotron-produced [11C]CO2. | ||
As a starting point, we applied the same approach of using CsF and 18-crown-6 (CsF–crown) in the presence of [11C]CO2 to carboxylate 1a. However, when 1a (100 μmol, 1 equiv.) was reacted with [11C]CO2 for 5 minutes at 100 °C in dimethylformamide (DMF), no [11C]1 was formed and the resulting [11C]CO2 trapping efficiency (TE, see footnote ‡) was poor (entry 1, Table 1).
| Entrya | Fluoride source (eq.) | Additive (eq.) | DBU (eq.) | CuI (%) | TE (%) | RCY [11C]1 (%) |
|---|---|---|---|---|---|---|
Reaction conditions: [11C]CO2 was bubbled in a solution of 1a (100 μmol, 1 equiv.), DBU (0.6–0.9 equiv.), CuI (10%), fluoride source CsF or KF (3–0.25 equiv.) and additive 18-crown-6 or K2.2.2 (3–0.25 equiv.) in DMF (500 μL) at 0 °C. Then, the reaction mixture was heated (100 °C) for 5 minutes and then the system was flushed with helium (60 mL min−1) for 20 seconds. Subsequently, the temperature was reduced to 0 °C and the reaction was quenched with a solution of 0.5% trifluoroacetic acid (TFA) in water and acetonitrile (H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN, 1:1, 1 mL).a n = 3.b n = 1.c 140 °C.d 70 °C.e THF. :MeCN, 1:1, 1 mL).a n = 3.b n = 1.c 140 °C.d 70 °C.e THF. |
||||||
| 1b | CsF (3) | 18-Crown-6 (3) | — | — | 6 | 0 |
| 2b | CsF (3) | 18-Crown-6 (3) | — | 10 | 99 | 0 |
| 3b | CsF (3) | 18-Crown-6 (3) | 0.6 | 10 | 77 | 0 |
| 4b | KF (3) | K2.2.2 (3) | 0.6 | 10 | 96 | 21 |
| 5 | KF (0.5) | K2.2.2 (0.5) | 0.6 | 10 | 63 ± 14 | 41 ± 9 |
| 6 | KF (0.25) | K2.2.2 (0.25) | 0.6 | 10 | 52 ± 5 | 53 ± 23 |
| 7a,c | KF (0.5) | K2.2.2 (0.5) | 0.6 | 10 | 77 | 40 |
| 8c | KF (0.25) | K2.2.2 (0.25) | 0.6 | 10 | 67 ± 13 | 55 ± 7 |
| 9b,d | KF (0.25) | K2.2.2 (0.25) | 0.6 | 10 | 37 | 0 |
| 10 | KF (0.25) | K2.2.2 (0.25) | 0.9 | 10 | 86 ± 5 | 28 ± 11 |
| 11 | KF (0.25) | K2.2.2 (0.25) | 0.6 | 20 | 61 ± 31 | 44 ± 26 |
| 12b,c,e | KF (0.25) | K2.2.2 (0.25) | 0.6 | 10 | 7 | 15 |
This might be due to the poor reactivity of the pentavalent silicate intermediate and/or the absence of any [11C]CO2 trapping agent. The transmetallation of hypervalent silicates with copper catalysts (10%), however, has been shown to form aryl copper intermediates that readily react with non-radioactive CO2.6a Despite this finding, in our hands, the addition of 10% CuI to the reaction mixture did not promote the formation of [11C]1 (entry 2, Table 1). Moreover, the addition of a [11C]CO2 trapping agent (1,8-diazabicyclo[5.4.0]undec-7-ene, DBU, 0.6 equiv.) did not favour the formation of [11C]1 either, although the TE increased from 6% to 77% (entry 1 versus 3).
We subsequently focused on selecting alternative fluoride sources as CsF is highly hygroscopic and poorly soluble in organic solvents – even in the presence of 18-crown-6, which might have hampered the formation of [11C]1. KF was investigated as a fluoride source as it has previously been used for the carboxylation of aryltrimethylsilanes; however, due to the low reactivity of KF in organic solvents the corresponding carboxylic acid derivative was only obtained with a low to moderate yield (17–74%).6c,14 To increase the reactivity of KF in organic solvents, we opted to explore the use of the polyether kryptofix (K2.2.2) to form a K+–cryptand complex.
Interestingly, replacing CsF–crown with KF–K2.2.2 improved the formation of [11C]1 (100 °C, 5 minutes) giving radiochemical yields (RCY, see footnote ‡) of 21% and high TE (96%, entry 4).15
In order to further increase the RCY of [11C]1, an optimization process was subsequently performed by modifying: (i) the amount of fluoride source, (ii) the reaction temperature, (iii) the amount of trapping reagent, (iv) the amount of copper catalyst and (v) the solvent.
The effect of the equivalents of fluoride source was initially investigated. Lowering the equivalents of the KF–K2.2.2 complex from 3 to 0.5 and 0.25 equivalents, and keeping the temperature at 100 °C, enhanced the RCY of [11C]1 (21% with 3 equiv., 41% with 0.5 equiv., and 53% with 0.25 equiv., entries 4–6). A similar trend was obtained at 140 °C (40% with 0.5 equiv. and 55% with 0.25 equiv., entries 7 and 8).
Additionally, we observed that higher temperatures favoured the formation of [11C]1 – either when 0.5 equivalents (41% at 100 °C versus 40% at 140 °C, entries 5 and 7) or 0.25 equivalents (53% at 100 °C versus 55% at 140 °C, entries 6 and 8) of the KF–K2.2.2 complex were used. Conversely, when lowering the temperature to 70 °C, [11C]1 was not obtained (entry 9) as high temperature is needed for the activation of the desilylation reaction. Similarly, the carboxylation of arylsilane derivatives with CO2 is promoted by high temperature.6a
Increasing the amount of the trapping agent (DBU) from 0.6 to 0.9 equivalents halved the RCY of [11C]1 (53% versus 28%, entries 6 and 10, respectively). Similarly, increasing the content of CuI from 10% to 20% did not markedly affect the RCY of [11C]1 (53% at 10% versus 44% at 20%, entries 6 and 11).
The use of a different solvent was investigated. Using tetrahydrofuran (THF) instead of DMF had a negative effect on reactivity, with the RCY of [11C]1 dropping to 15% (entry 12).
Optimal conditions were obtained when 1a (100 μmol, 1 equiv.) was reacted with the cyclotron-produced [11C]CO2 at 140 °C in the presence of 0.25 equiv. of KF–K2.2.2, 10% of CuI and DMF (entry 8, Table 1).
Aiming to further increase the RCY of [11C]1, DBU was substituted with BEMP as a CO2 trapping agent. Although no significant difference in RCY was observed at 100 °C (33 ± 15% with BEMP, entry 1, Table 2versus 53 ± 23% with DBU, entry 6, Table 1), higher yields of [11C]1 were obtained when the temperature was increased to 140 °C (93 ± 6% with BEMP, entry 2, Table 2versus 55 ± 7% with DBU, entry 8, Table 1). Using BEMP over DBU, a significant increase in TE was observed at 100 °C (84% with BEMP, entry 2, Table 2versus 52% with DBU, entry 6, Table 1) and at 140 °C (89% with BEMP, entry 2, Table 2versus 67% with DBU, entry 8, Table 1).
| Entrya | KF (eq.) | K2.2.2 (eq.) | BEMP (eq.) | CuI (%) | TE (%) | RCY [11C]1 (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: [11C]CO2 was bubbled in a solution of 1a (100 μmol, 1 equiv.), BEMP (0.6 equiv.), CuI (10%), KF (0.125–0.25 equiv.), and K2.2.2 (0.125–0.25 equiv.) in DMF (500 μL) at 0 °C. Then, the reaction mixture was heated (140 °C) for 5 minutes and then the system was flushed with helium (60 mL min−1) for 20 seconds. Subsequently, the temperature was reduced to 0 °C and the reaction was quenched with a solution of 0.5% TFA in H2O:MeCN (1:1, 1 mL).a n = 3.b 100 °C.c 2.5 minutes.d n = 2.e n = 1.f 1a (50 μmol).g THF.h MeCN. |
||||||
| 1b | 0.25 | 0.25 | 0.6 | 10 | 84 ± 3 | 33 ± 15 |
| 2 | 0.25 | 0.25 | 0.6 | 10 | 89 ± 8 | 93 ± 6 |
| 3c | 0.25 | 0.25 | 0.6 | 10 | 76 ± 12 | 58 ± 9 |
| 4 | 0.25 | 0.25 | — | 10 | 6 ± 2 | 95 ± 1 |
| 5 | 0.25 | 0.25 | 0.6 | — | 76 ± 22 | 24 ± 18 |
| 6d | — | 0.25 | 0.6 | 10 | 40, 30 | 0 |
| 7e | 0.25 | — | 0.6 | 10 | 48 | 0 |
| 8f | 0.125 | 0.125 | 0.6 | 10 | 55 ± 15 | 47 ± 9 |
| 9g | 0.25 | 0.25 | 0.6 | 10 | 12 ± 5 | 20 ± 7 |
| 10e,h | 0.25 | 0.25 | 0.6 | 10 | 50 | 0 |
Encouraged by these results, BEMP was used as a trapping agent for the following experiments which initially focused on the effect of shorter reaction times. Halving the reaction time from 5 to 2.5 minutes resulted in reducing the RCY of [11C]1 (58% at 2.5 min versus 93% at 5 min, entries 2 and 3, Table 2).
To understand the role of each reagent in the reaction mechanism, experiments were conducted with the omission of key reagents (KF, K2.2.2, BEMP, or CuI) from the reaction mixture. Removing BEMP yielded [11C]1 with high RCY but with a significantly lower TE (6% without BEMP and 89% with BEMP, entries 2 and 4). Removing CuI yielded [11C]1 with significantly lower RCY (93% with CuI and 24% without CuI, entries 2 and 5) but the TE was not clearly affected. Notably, [11C]1 was not formed at all when KF or K2.2.2 was eliminated from the reaction mixture (entries 6 and 7, respectively). Similarly, when the amount of 1a and KF–K2.2.2 was halved, the RCY of [11C]1 was reduced 2-fold (47%, entry 8). These results highlight the primary role of the concentration of 1a and fluoride source to promote the formation of a highly nucleophilic intermediate, which is stabilized by the copper catalyst. The effect of the solvent was also investigated during the optimisation of the reaction conditions. The use of THF and acetonitrile (MeCN) gave low or zero RCYs of [11C]1 (20% in THF and 0% in MeCN, entries 9 and 10, Table 2).
The results presented in Tables 1 and 2 show that the RCY of [11C]1 is maximized when 100 μmol of 1a is reacted with 0.6 equiv. of BEMP, 0.25 equiv. of KF–K2.2.2 and 0.1 equiv. of CuI in DMF for 5 minutes at 140 °C (entry 2, Table 2). Following this protocol, for [11C]1 the isolated (by semipreparative HPLC) decay-corrected-RCY of 17 ± 5% and an Am of 3.1 ± 0.4 Gbq μmol−1 at the end of bombardment (EOB) were obtained starting from 2.30 ± 0.3 GBq of [11C]CO2.§
The reaction conditions were subsequently kept constant while studying the substrate scope of additional trialkoxysilyl and trimethylsilyl compounds. Initially, the effect of silyl substituents other than the trimethyl silyl moiety on the thienyl ring was explored using a triethoxysilyl substituent (triethoxy-2-thienylsilane, 1b, Table 3). Both precursors 1a and 1b yielded the corresponding [11C]1 with high RCY (93% and 90%, respectively). However, the use of 1b resulted in lower TE (57%, entry 1, Table 3) compared with 1a (89%, entry 2, Table 2).
| Entrya | Reagent | R | Product | Temp. (°C) | TE (%) | RCY (%) | |
|---|---|---|---|---|---|---|---|
| Reaction conditions: [11C]CO2 was bubbled in a solution of 1b, 2a–b, 3a–b, 4a–6a (100 μmol, 1 equiv.), BEMP (0.6 equiv.), CuI (10%), KF (0.25 equiv.), and K2.2.2 (0.25 equiv.) in DMF (500 μL) at 0 °C. Then, the reaction mixture was heated (30–140 °C) for 5 minutes and then the system was flushed with helium (60 mL min−1) for 20 seconds. Subsequently, the temperature was reduced to 0 °C and the reaction was quenched with a solution of 0.5% TFA in H2O:MeCN (1:1, 1 mL).a n = 3. |
|||||||
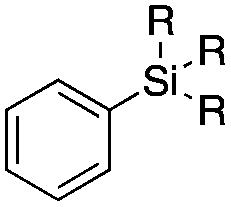
| 1 |
|
1b | OEt |
|
140 | 57 ± 18 | 90 ± 4 |
| 2 |
|
2a | Me |
|
140 | 13 ± 8 | 0 |
| 3 | 2b | OEt | 140 | 76 ± 8 | 84 ± 2 | ||
| 4 |
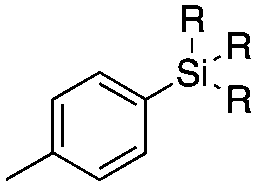
|
3a | Me |
|
140 | 15 ± 8 | 0 |
| 5 | 3b | OEt | 140 | 81 ± 2 | 78 ± 2 | ||
| 6 |
|
4a | Me |
|
140 | 23 ± 15 | 18 ± 7 |
| 7 |
|
5a | Me |
|
140 | 40 ± 1 | 87 ± 6 |
| 8 |
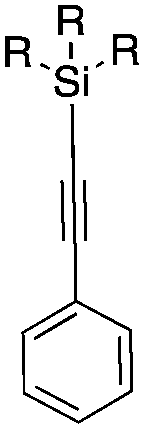
|
6a | Me |
|
140 | 5 ± 4 | 0 |
| 9 | 100 | 3 ± 2 | 9 ± 8 | ||||
| 10 | 30 | 21 ± 12 | 19 ± 15 | ||||
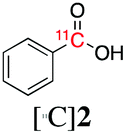
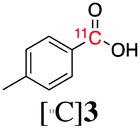
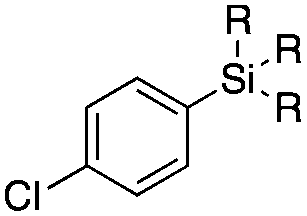
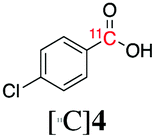
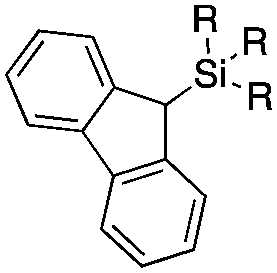
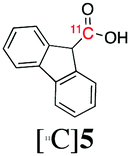
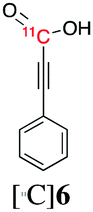
Next, we directed our attention on radiolabelling other 11C-labelled aromatic carboxylic acids such as [11C]benzoic acid ([11C]2, entries 2 and 3) and [11C]p-toluic acid ([11C]3, entries 4 and 5) using trimethyl silyl (2a and 3a) and the triethoxysilyl (2b and 3b) precursors. In contrast to that observed with [11C]1, the trimethyl silyl derivatives showed a different reactivity to triethoxysilyl analogues. 2b and 3b produced the corresponding carbon-11 labelled carboxylic acids with high RCYs (RCY of [11C]2 = 84%, entry 3; RCY of [11C]3 = 78%, entry 5), whereas the trimethylsilyl derivatives, 2a and 3a, did not form the desired products (entries 2 and 4). As expected, the low reactivity of benzyl-trimethylsilyl substrates was also observed using 1-chloro-4-(trimethylsilyl)benzene (4a), yielding only small amounts of [11C]4 (18%, entry 6). Further studies focused on non-aromatic silane precursors such as fluorene and alkyne derivatives (entries 7–10). The radiolabelling of a fluorene moiety (5a) was effective, producing [11C]fluorene-9-carboxylic acid ([11C]5) with high RCY (87%, entry 7). The radiolabelling of prop-1-yn-1-ylbenzene (6a) to [11C]3-phenylpropiolic acid ([11C]6), instead, was ineffective at 140 °C (entry 8) and 100 °C (entry 9). However, lowering the temperature to 30 °C yielded [11C]6, although with low RCY (19%, entry 10).
To demonstrate that the arylcopper intermediates were obtained by the KF–K2.2.2-mediated desilylation of trimethylsilyl derivatives, we replaced [11C]CO2 with [11C]CH3I. [11C]7 was obtained by direct aromatic 11C-methylation of 1a (Scheme 3), with a RCY of 16 ± 4% (n = 3). Although this method has not been optimised here, we note a potential application of this strategy as an alternative route to produce 11C-methylaromatic radiopharmaceuticals such as (15R)-[11C]TIC, [11C]MNQP, [11C]M-MTEB, [11C]celecoxib, [11C]cibbi-772, and [11C]UCB-J by direct aromatic 11C-methylation.2
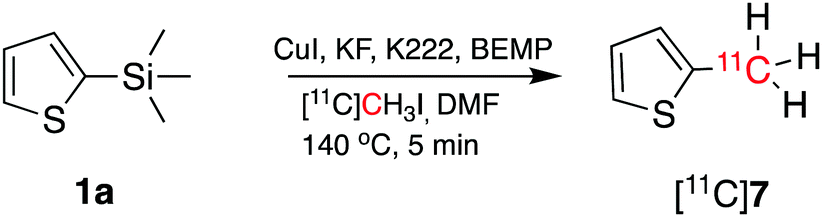 | ||
| Scheme 3 Aromatic 11C-methylation of 1a using [11C]CH3I to obtain [11C]7. | ||
In summary, we have developed a novel carbon-11 reaction using cyclotron-produced [11C]CO2 and aryltrimethylsilanes and aryltrialkoxysilanes to obtain 11C-carboxylic acid derivatives. Aryltrimethylsilanes and aryltrialkoxysilanes are activated by a fluoride source (KF–K2.2.2) and a copper catalyst which readily react with cyclotron-produced [11C]CO2. We have also expanded the use of activated aryltrimethylsilanes as nucleophilic compounds for aromatic 11C-methylation using [11C]CH3I. This one-pot methodology, similar to other one-pot reactions using 11C-syntons,16 has the compatibility to be fully automated using a commercial radiochemistry synthesis module. The application of silane-mediated 11C-carboxylation reactions has the potential to be an alternative route to produce a plethora of radiopharmaceuticals bearing an aryl carboxylic acid such as [11C]bexarotene, [11C]eprosartan, and [11C]Am80, or an aryl carboxylic acid that is subsequently converted into an 11C-amide by an amide-coupling reaction such as [11C]raclopride, [11C]olaparib, [11C]JNJ-31020028, [11C]FIMX, [11C]tubastatin A, and [11C]AZ11136118.2
This work was supported by Medical Research Council (MRC, MR/K022733/1) and European Commission, FP7-PEOPLE-2012-ITN (316882, RADIOMI). The authors acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) Biomedical Research Centre at Guy's & St Thomas’ NHS Foundation Trust and King's College London and the Centre of Excellence in Medical Engineering funded by the Wellcome Trust and EPSRC under grant number WT 203148/Z/16/Z. Igor Camargo Fontana acknowledges financial support from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, project number: 88887.185806/2018-00).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. H. Rotstein, S. H. Liang, J. P. Holland, T. L. Collier, J. M. Hooker, A. A. Wilson and N. Vasdev, Chem. Commun., 2013, 49, 5621–5629 RSC; (b) G. Boscutti, M. Huiban and J. Passchier, Drug Discovery Today: Technol., 2017, 25, 3–10 CrossRef PubMed.
- (a) X. Deng, J. Rong, L. Wang, N. Vasdev, L. Zhang, L. Josephson and S. H. Liang, Angew. Chem., Int. Ed., 2019, 58, 2580–2605 CrossRef CAS PubMed; (b) P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- (a) A. Pekošak, U. Filp, L. Rotteveel, A. J. Poot and A. D. Windhorst, J. Labelled Compd. Radiopharm., 2015, 58, 342–348 CrossRef PubMed; (b) D. Y. Tang, A. Lipman, G.-J. Meyer, C.-N. Wan and A. P. Wolf, J. Labelled Compd. Radiopharm., 1979, 16, 435–440 CrossRef CAS.
- (a) P. J. Riss, S. Lu, S. Telu, F. I. Aigbirhio and V. W. Pike, Angew. Chem., Int. Ed., 2012, 51, 2698–2702 CrossRef CAS PubMed; (b) B. H. Rotstein, J. M. Hooker, J. Woo, T. L. Collier, T. J. Brady, S. H. Liang and N. Vasdev, ACS Med. Chem. Lett., 2014, 5, 668–672 CrossRef CAS PubMed.
- (a) G. Destro, O. Loreau, E. Marcon, F. Taran, T. Cantat and D. Audisio, J. Am. Chem. Soc., 2019, 141, 780–784 CrossRef CAS PubMed; (b) C. Kingston, M. A. Wallace, A. J. Allentoff, J. N. de Gruyter, J. S. Chen, S. X. Gong, S. Bonacorsi and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 774–779 CrossRef CAS PubMed.
- (a) T. V. Q. Nguyen, W.-J. Yoo and S. Kobayashi, Asian J. Org. Chem., 2018, 7, 116–118 CrossRef CAS; (b) X. Frogneux, N. von Wolff, P. Thuery, G. Lefevre and T. Cantat, Chemistry, 2016, 22, 2930–2934 CrossRef CAS PubMed; (c) M. Yonemoto-Kobayashi, K. Inamoto and Y. Kondo, Chem. Lett., 2014, 43, 477–479 CrossRef CAS; (d) T. Mita, H. Tanaka, K. Michigami and Y. Sato, Synlett, 2014, 1291–1294 CrossRef.
- L. A. Babadzhanova, N. V. Kirij and Y. L. Yagupolskii, J. Fluorine Chem., 2004, 125, 1095–1098 CrossRef CAS.
- A. S. Manoso, C. Ahn, A. Soheili, C. J. Handy, R. Correia, W. M. Seganish and P. Deshong, J. Org. Chem., 2004, 69, 8305–8314 CrossRef CAS PubMed.
- S.-C. Lee, L. Guo, H. Yue, H.-H. Liao and M. Rueping, Synlett, 2017, 2594–2598 CAS.
- X. Wang, Z. Wang and Y. Nishihara, Chem. Commun., 2019, 55, 10507–10510 RSC.
- L. Guo, A. Chatupheeraphat and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 11810–11813 CrossRef CAS PubMed.
- M. Tobisu, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2006, 128, 8152–8153 CrossRef CAS PubMed.
- B. Yu, P. Yang, X. Gao, Z. Z. Yang, Y. F. Zhao, H. Y. Zhang and Z. M. Liu, New J. Chem., 2017, 41, 9250–9255 RSC.
- F. Effenberger and W. Spiegler, Chem. Ber., 1985, 118, 3900–3914 CrossRef CAS.
- H. H. Coenen, A. D. Gee, M. Adam, G. Antoni, C. S. Cutler, Y. Fujibayashi, J. M. Jeong, R. H. Mach, T. L. Mindt, V. W. Pike and A. D. Windhorst, Nucl. Med. Biol., 2017, 55, v–xi CrossRef CAS PubMed.
- (a) P. Buccino, E. Savio and W. Porcal, EJNMMI Radiopharm. Chem., 2019, 4, 14 CrossRef PubMed; (b) V. Pichler, M. Ozenil, K. Bamminger, C. Vraka, M. Hacker, O. Langer and W. Wadsak, EJNMMI Radiopharm. Chem., 2019, 4, 31 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00449a |
| ‡ Radiochemical yield (RCY) of the crude product has been determined by analytical radio-HPLC (non-isolated). The trapping efficiency (TE) has been calculated as a ratio of the decay corrected radioactivity in the vial and the total radioactivity produced by the cyclotron. |
| § This work describes a method development study using short, low current, cyclotron irradiations where obtaining high molar activities (Am) was not the main focus. Assuming that the stable 12C carrier content would be in the same range for a standard clinical [11C]CO2 production (30 GBq), it is estimated that a molar activity of ∼45 GBq μmol−1 would be obtained. |
| This journal is © The Royal Society of Chemistry 2020 |