
Copper-mediated tandem ring-opening/cyclization reactions of cyclopropanols with aryldiazonium salts: synthesis of N-arylpyrazoles†
Jidan
Liu
 *,
Erjie
Xu
,
Jinyuan
Jiang
,
Zeng
Huang
,
Liyao
Zheng
and
Zhao-Qing
Liu
*,
Erjie
Xu
,
Jinyuan
Jiang
,
Zeng
Huang
,
Liyao
Zheng
and
Zhao-Qing
Liu
School of Chemistry and Chemical Engineering/Institute of Clean Energy and Materials/Guangzhou Key Laboratory for Clean Energy and Materials, Guangzhou University, Guangzhou, 510006, P. R. China. E-mail: jdliu@gzhu.edu.cn
First published on 17th January 2020
Abstract
A general method for the synthesis of structurally diverse N-arylpyrazoles from readily available cyclopropanols and aryldiazonium salts is disclosed. The reaction was conducted at room temperature within minutes with a broad substrate scope and excellent regioselectivity.
Pyrazole, a five-membered heterocycle with two adjacent nitrogen atoms used extensively in agrochemical and pharmaceutical applications, displays a broad spectrum of biological activities, such as anti-inflammatory, antimicrobial, antiobesity, antidepressant, antidiabetic, analgesic, and antiviral activities,1 as exemplified by the presence of this core structure in several commercial drugs, including rimonabant, lonazolac and celecoxib, as well as the insecticide fipronil (Fig. 1).2 In addition to their medicinal value, N-arylpyrazoles have also been applied as ligands in transition-metal-catalyzed cross-coupling reactions,3 N-heterocyclic carbene (NHC) precursors4 and directing groups for C–H bond functionalizations.5
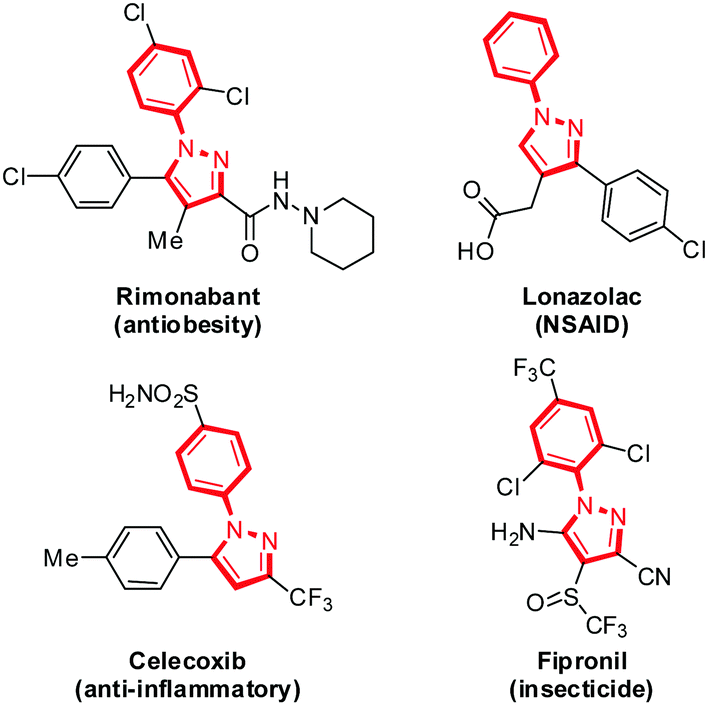 | ||
| Fig. 1 Representative commercialized bioactive N-arylpyrazoles. | ||
Due to the important applications of N-arylpyrazole scaffolds, the construction of these useful compounds has attracted considerable attention during the past few decades.6 Conventional approaches for the preparation of N-arylpyrazoles mainly focus on the condensation of arylhydrazines with 1,3-dicarbonyl or α,β-unsaturated carbonyl compounds.7 Alternatively, the 1,3-dipolar cycloaddition of diazo compounds or their surrogates with olefins or alkynes8 followed by transition-metal-catalyzed N-arylation represents another popular approach.9 Although these strategies can provide efficient routes to prepare N-arylpyrazoles, some drawbacks, such as harsh reaction conditions, narrow substrate scope, and poor regioselectivity, limit their synthetic application to some extent. In order to overcome these problems, several alternative strategies involving transition metal catalyzed or mediated intermolecular coupling reactions have been developed.10 Albeit great achievements have been achieved, further exploration of convenient, efficient, and mild protocols to afford structurally diverse N-arylpyrazoles from readily available starting materials is still highly desirable.
Cyclopropanols are important and useful synthetic intermediates in organic synthesis and can be readily prepared via Kulinkovich cyclopropanation of the corresponding esters in one step or Simmons–Smith cyclopropanation of silyl-enol ethers.11 The tandem radical ring-opening couplings of cyclopropanols that proceed via single-electron oxidation can be easily realized owing to the intrinsic ring strain associated with the three-membered ring, and many efforts have been devoted to synthesizing β-functionalized carbonyl compounds with different single-electron oxidants.12 In these transformations, the β-keto alkyl radical intermediates generated from the oxidative ring-opening of cyclopropanols were involved. However, despite progress in this area, the tandem radical ring-opening/coupling strategy for the construction of nitrogen-containing heterocycles via the cyclization with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of β-functionalized carbonyl compounds is quite rare. In 2009, Chiba and co-workers reported an efficient Mn(III)-mediated ring-opening coupling reaction of cyclopropanols with vinyl azides for the synthesis of substituted pyridines (Scheme 1a).13 Mechanistic studies revealed that this transformation was initiated with the generation of β-keto alkyl radical intermediates via the Mn(acac)3-mediated oxidative radical ring-opening of cyclopropanols, followed by radical addition to vinyl azides and annulations. In light of our continuous interest in the chemistry of cyclopropanols and the synthesis of heterocycles,14 we envisioned that the β-keto alkyl radicals derived from cyclopropanols may be trapped by aryldiazonium salts followed by the cyclization with intramolecular carbonyl groups to afford important nitrogen-containing heterocycles. Herein, we described a copper-mediated tandem ring-opening/cyclization reaction of cyclopropanols with aryldiazonium salts for the construction of diversely substituted N-arylpyrazoles (Scheme 1b). This procedure can be conducted at room temperature with high efficiency, excellent regioselectivity and broad substrate scope.
O bond of β-functionalized carbonyl compounds is quite rare. In 2009, Chiba and co-workers reported an efficient Mn(III)-mediated ring-opening coupling reaction of cyclopropanols with vinyl azides for the synthesis of substituted pyridines (Scheme 1a).13 Mechanistic studies revealed that this transformation was initiated with the generation of β-keto alkyl radical intermediates via the Mn(acac)3-mediated oxidative radical ring-opening of cyclopropanols, followed by radical addition to vinyl azides and annulations. In light of our continuous interest in the chemistry of cyclopropanols and the synthesis of heterocycles,14 we envisioned that the β-keto alkyl radicals derived from cyclopropanols may be trapped by aryldiazonium salts followed by the cyclization with intramolecular carbonyl groups to afford important nitrogen-containing heterocycles. Herein, we described a copper-mediated tandem ring-opening/cyclization reaction of cyclopropanols with aryldiazonium salts for the construction of diversely substituted N-arylpyrazoles (Scheme 1b). This procedure can be conducted at room temperature with high efficiency, excellent regioselectivity and broad substrate scope.
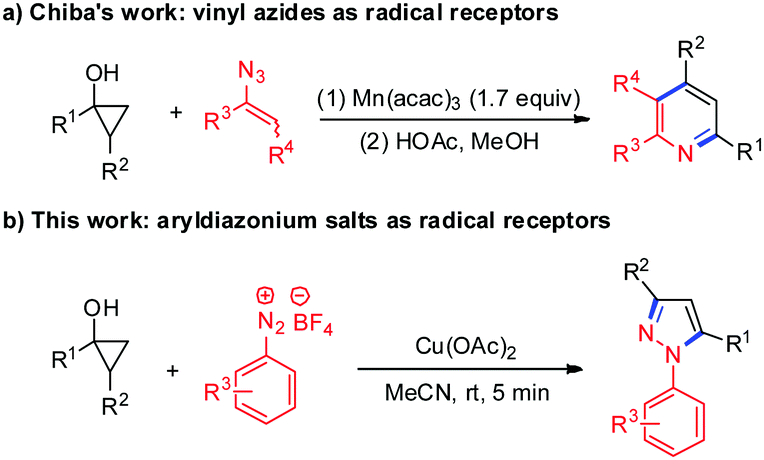 | ||
| Scheme 1 Ring-opening coupling of cyclopropanols to construct N-heterocycles. | ||
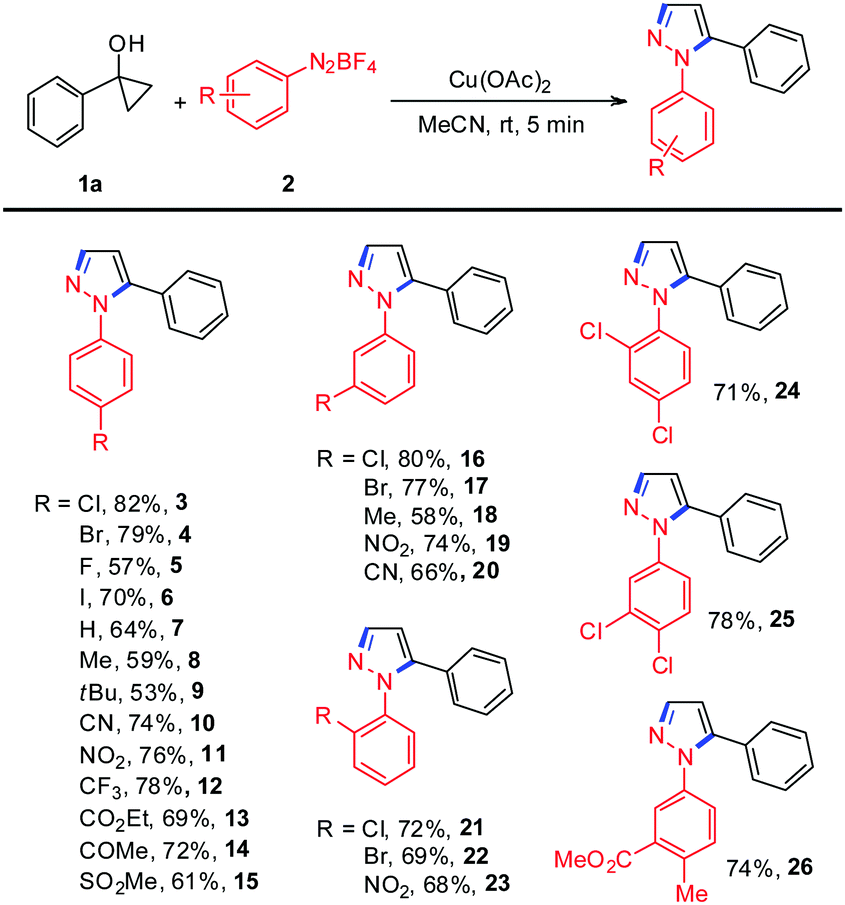
We began our investigations with the use of 1-phenylcyclopropanol 1a and aryldiazonium tetrafluoroborate 2a as model substrates to optimize the reaction conditions and these results are summarized in Table S1 (see the ESI†). Encouragingly, treating 1-phenylcyclopropanol 1a with aryldiazonium tetrafluoroborate 2a in the presence of 20 mol% Cu(OAc)2 in MeCN at room temperature under N2 gave the expected N-arylpyrazole 3 in 18% yield (Table S1, ESI,† entry 1). The yield of the desired product was dramatically improved (82%) by increasing the loading of Cu(OAc)2 to 1.0 equiv. (Table S1, ESI,† entry 4). Extensive solvent screening revealed that MeCN was important since reactions conducted in other solvents, such as DMSO, DMF, toluene, DCE, and 1,4-dioxane, generated the desired product in less than 5% yield (Table S1, ESI,† entries 6–10). Among various copper salts tested, Cu(OAc)2 gave the highest yield, whereas the others were far less effective (Table S1, ESI,† entries 11–17), indicating that the counter ions of copper salts have a remarkable impact on the reaction yield. Subsequently, various other metal salts were also examined, and it was observed that only the acetate-type metal salts, such as Fe(OAc)2, Mn(OAc)2, and Mn(OAc)3, could efficiently promote the reaction to deliver the desired product in moderate yields (Table S1, ESI,† entries 18–26).
With the optimized reaction conditions in hand, we set out to investigate the scope and generality of the reaction between various aryldiazonium tetrafluoroborates and 1-phenylcyclopropanol 1a. As shown in Table 1, aryldiazonium salts bearing either electron-withdrawing groups or electron-donating groups at the para-position of the benzene rings produced the corresponding N-arylpyrazoles in moderate to good yields (Table 1, 3–15). It should be mentioned that the halogens in N-arylpyrazoles 3–6 remained intact, offering new possibilities for cross-coupling-type manipulations. Similar yields could be achieved with ortho- and meta-substituted aryldiazonium salts, indicating that this transformation is not sensitive to steric effects (Table 1, 16–23). Disubstituted aryldiazonium salts were also compatible to generate the desired N-arylpyrazole derivatives in yields ranging from 71% to 78% (Table 1, 24–26). Unfortunately, diazonium salts possessing other aromatic motifs such as 3-thienyl and 3-pyridinyl were not suitable substrates for the reaction.
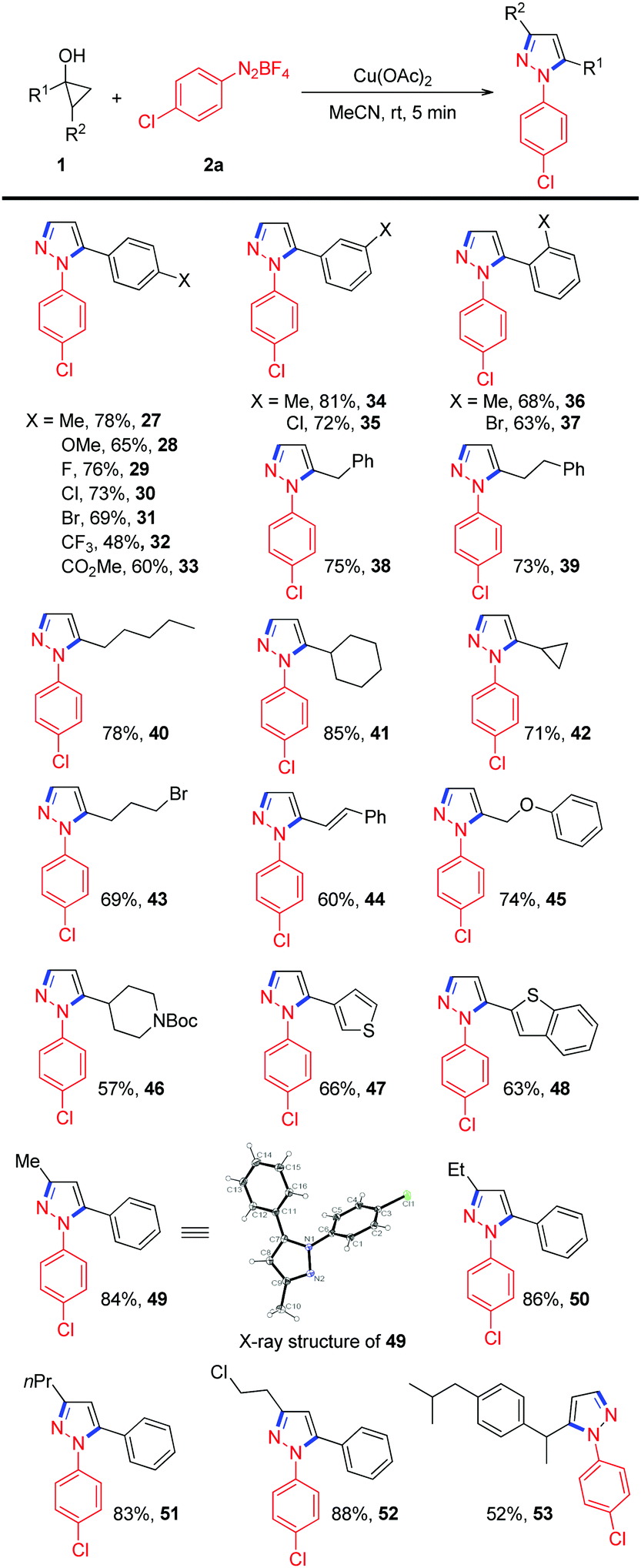
Next, the substrate scope with respect to cyclopropanol was also tested, and the results are summarized in Table 2. Phenylcyclopropanols with different substituents on the phenyl rings, such as p-Me, p-OMe, p-F, p-Cl, p-Br, p-CO2Me, m-Me, m-Cl, o-Me, and o-Br, reacted smoothly with 2a to give the desired N-arylpyrazoles in moderate to good yields. A slightly decreased yield was observed for cyclopropanol with a strong electron-withdrawing CF3 group (Table 2, 32). Alkyl substituted cyclopropanols were also viable for this reaction, generating the corresponding products 38–42 in 71% to 85% yields. Functional groups, such as alkyl bromide, alkene, aryl ether and alkylsulfonamide, were all well tolerated (Table 2, 43–46). In addition, thiophene- and benzothiophene-containing cyclopropanols were also compatible to deliver the expected N-arylpyrazoles in 66% and 63% yields, respectively. It is worth mentioning that when 1,2-disubstituted cyclopropanols were employed, the desired 1,3,5-trisubstituted pyrazoles 49–52 could be obtained in better yields than those of 1-substituted cyclopropanols, which could be attributed to the formation of thermodynamically more stable secondary alkyl radicals via the oxidative radical ring-opening of 1,2-disubstituted cyclopropanols. The regioselectivity of the coupling was further confirmed by single-crystal X-ray analysis of 49. Finally, cyclopropanol derived from an anti-inflammatory drug, ibuprofen, participated smoothly in this transformation to provide the desired product 53 in a synthetically useful yield.
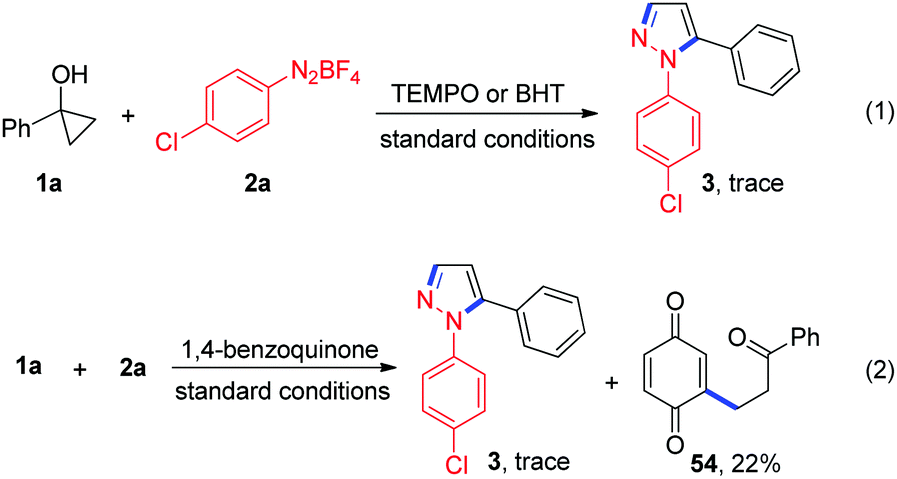
To gain more insights into the reaction mechanism, several control experiments were conducted. First, the reaction of 1a with 2a in the presence of 3.0 equiv. of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT) afforded only a trace amount of the desired product 3, thus indicating that a radical pathway might be involved in this transformation (Scheme 2, eqn (1)). To trap the possibly formed β-keto alkyl radical in this reaction, 1,4-benzoquinone (3 equiv.) was subjected to the reaction and the expected coupling product 54 was observed (Scheme 2, eqn (2)). This result supports the generation of the β-keto alkyl radical during the reaction.
 | ||
| Scheme 2 Control experiments. | ||
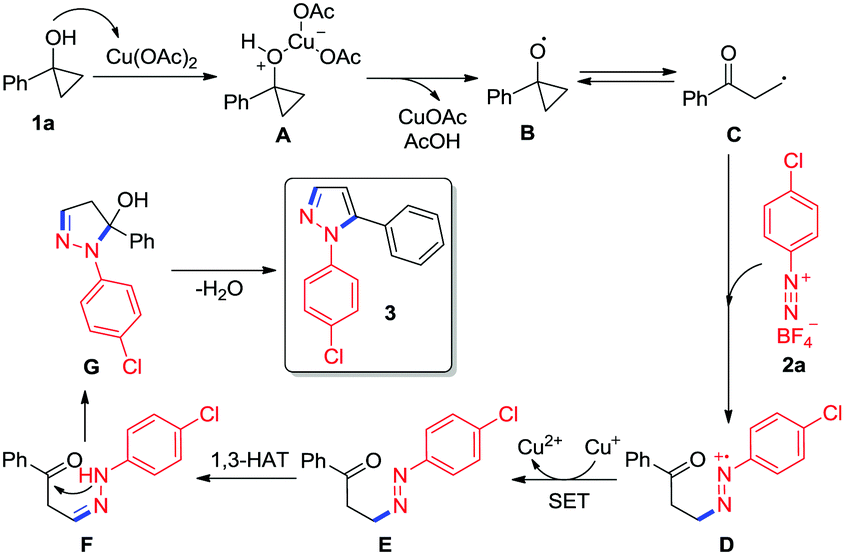
Based on the above control experiments and literature precedents,13,15 a plausible mechanism for copper-mediated tandem ring-opening/cyclization of cyclopropanols with aryldiazonium salts is proposed in Scheme 3. Initially, the coordination of the hydroxyl group of cyclopropanol 1a to Cu(OAc)2 leads to the formation of a Lewis base-acid complex A, which undergoes an inner-sphere electron-transfer to produce the cyclopropoxy radical B with the elimination of CuOAc and AcOH.15a Subsequently, the oxygen-centered radical B undergoes ring-opening to generate a β-keto alkyl radical C. The in situ generated β-keto alkyl radical C can be readily trapped by aryldiazonium salt 2a to afford a radical cation D.15b,c Next, the radical cation D is reduced to intermediate Evia single-electron transfer,12j,15d,16 which is followed by a formal 1,3-hydrogen atom transfer (1,3-HAT) process to give a phenylhydrazone F. Finally, the intarmoleculer nucleophilic attack of the amino group on the ketone in intermediate F followed by the elimination of water from intermediate G yields the desired N-arylpyrazole 3.
 | ||
| Scheme 3 Plausible mechanism. | ||
In conclusion, we have developed a general method for the synthesis of various N-arylpyrazoles from readily available cyclopropanols and aryldiazonium salts through a copper-mediated tandem ring-opening/cyclization reaction. This procedure can be conducted at room temperature with high efficiency, excellent regioselectivity and broad substrate scope with respect to cyclopropanol and the aryldiazonium salt substrate. Due to the importance of this structural motif as well as the easy availability of starting materials and the simplicity of the reaction conditions, we expect that the protocol will find broad applications in synthetic and medicinal chemistry.
We thank the National Natural Science Foundation of China (Grant No. 21875048 and 21576056), the Guangdong Natural Science Foundation (Grant No. 2017A030310620), and the Science and Technology Research Project of Guangzhou (Grant No. 201707010483) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. Szabó, J. Fischer, Á. Kis-Varga and K. Gyires, J. Med. Chem., 2008, 51, 142–147 CrossRef PubMed; (b) J. B. Thomas, A. M. Giddings, R. W. Wiethe, S. Olepu, K. R. Warner, P. Sarret, L. Gendron, J.-M. Longpre, Y. Zhang, S. P. Runyon and B. P. Gilmour, J. Med. Chem., 2014, 57, 5318–5332 CrossRef CAS PubMed; (c) J. V. Faria, P. F. Vegi, A. G. C. Miguita, M. S. dos Santos, N. Boechat and A. M. R. Bernardino, Bioorg. Med. Chem., 2017, 25, 5891–5903 CrossRef CAS PubMed.
- Ş. G. Küçükgüzel and S. Şenkardeş, Eur. J. Med. Chem., 2015, 97, 786–815 CrossRef PubMed.
- (a) R. A. Singer, S. Caron, R. E. McDermott, P. Arpin and N. M. Do, Synthesis, 2003, 1727–1731 CrossRef CAS; (b) R. A. Singer, M. Doré, J. E. Sieser and M. A. Berliner, Tetrahedron Lett., 2006, 47, 3727–3731 CrossRef CAS.
- A. Schmidt and A. Dreger, Curr. Org. Chem., 2011, 15, 2897–2970 CrossRef CAS.
- (a) T. Asaumi, N. Chatani, T. Matsuo, F. Kakiuchi and S. Murai, J. Org. Chem., 2003, 68, 7538–7540 CrossRef CAS PubMed; (b) N. Umeda, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 7094–7099 CrossRef CAS PubMed; (c) V. S. Thirunavukkarasu, K. Raghuvanshi and L. Ackermann, Org. Lett., 2013, 15, 3286–3289 CrossRef CAS PubMed; (d) S.-S. Zhang, C.-Y. Jiang, J.-Q. Wu, X.-G. Liu, Q. Li, Z.-S. Huang, D. Li and H. Wang, Chem. Commun., 2015, 51, 10240–10243 RSC; (e) T.-J. Gong, M.-Y. Xu, S.-H. Yu, C.-G. Yu, W. Su, X. Lu, B. Xiao and Y. Fu, Org. Lett., 2018, 20, 570–573 CrossRef CAS PubMed.
- S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed.
- For selected examples, see: (a) B. C. Bishop, K. M. J. Brands, A. D. Gibb and D. J. Kennedy, Synthesis, 2004, 43–52 CAS; (b) S. Peruncheralathan, T. A. Khan, H. Ila and H. Junjappa, J. Org. Chem., 2005, 70, 10030–10035 CrossRef CAS PubMed; (c) S. T. Heller and S. R. Natarajan, Org. Lett., 2006, 8, 2675–2678 CrossRef CAS PubMed; (d) B. S. Gerstenberger, M. R. Rauckhorst and J. T. Starr, Org. Lett., 2009, 11, 2097–2100 CrossRef CAS PubMed; (e) A. DeAngelis, D.-H. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 3434–3437 CrossRef CAS PubMed; (f) Y. Tu, Z. Zhang, T. Wang, J. Ke and J. Zhao, Org. Lett., 2017, 19, 3466–3469 CrossRef CAS PubMed; (g) R. S. Thombal and Y. R. Lee, Org. Lett., 2018, 20, 4681–4685 CrossRef CAS.
- For selected recent reviews, see: (a) T. V. Baiju and I. N. N. Namboothiri, Chem. Rec., 2017, 17, 939–955 CrossRef CAS PubMed; (b) Y. Xia and J. Wang, Chem. Soc. Rev., 2017, 46, 2306–2362 RSC ; For selected examples, see: ; (c) X. Qi and J. M. Ready, Angew. Chem., Int. Ed., 2007, 46, 3242–3244 CrossRef CAS PubMed; (d) M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2013, 52, 7219–7223 CrossRef PubMed; (e) G. Zhang, H. Ni, W. Chen, J. Shao, H. Liu, B. Chen and Y. Yu, Org. Lett., 2013, 15, 5967–5969 CrossRef CAS PubMed; (f) F.-G. Zhang, Y. Wei, Y.-P. Yi, J. Nie and J.-A. Ma, Org. Lett., 2014, 16, 3122–3125 CrossRef CAS; (g) Z. Chen, Y. Zheng and J.-A. Ma, Angew. Chem., Int. Ed., 2017, 56, 4569–4574 CrossRef CAS PubMed; (h) Y. Shao, H. Zheng, J. Qian and X. Wan, Org. Lett., 2018, 20, 2412–2415 CrossRef CAS PubMed; (i) W. Wu, M. Li, J. Zheng, W. Hu, C. Li and H. Jiang, Chem. Commun., 2018, 54, 6855–6858 RSC; (j) X. Peng, X. Zhang, S. Li, Y. Lu, L. Lan and C. Yang, Org. Chem. Front., 2019, 6, 1775–1779 RSC.
- (a) J. C. Antilla, J. M. Baskin, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2004, 69, 5578–5587 CrossRef CAS PubMed; (b) H. Kaddouri, V. Vicente, A. Ouali, F. Ouazzani and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 333–336 CrossRef CAS PubMed; (c) S. Onodera, T. Kochi and F. Kakiuchi, J. Org. Chem., 2019, 84, 6508–6515 CrossRef CAS PubMed.
- (a) X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636–3646 CrossRef CAS; (b) X. Tang, L. Huang, J. Yang, Y. Xu, W. Wu and H. Jiang, Chem. Commun., 2014, 50, 14793–14796 RSC; (c) D. Li, X. Mao, H. Chen, G. Chen and P. Liu, Org. Lett., 2014, 16, 3476–3479 CrossRef CAS PubMed; (d) H. Guo, D. Zhang, C. Zhu, J. Li, G. Xu and J. Sun, Org. Lett., 2014, 16, 3110–3113 CrossRef CAS PubMed; (e) J. J. Neumann, M. Suri and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7790–7794 CrossRef CAS PubMed.
- For selected reviews, see: (a) O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597–2632 CrossRef CAS PubMed; (b) A. Nikolaev and A. Orellana, Synthesis, 2016, 1741–1768 CAS; (c) X. Cai, W. Liang and M. Dai, Tetrahedron, 2019, 75, 193–208 CrossRef CAS; (d) X. Wu and C. Zhu, Chem. Commun., 2019, 55, 9747–9756 RSC.
- For selected examples, see: (a) N. Iwasawa, S. Hayakawa, M. Funahashi, K. Isobe and K. Narasaka, Bull. Chem. Soc. Jpn., 1993, 66, 819–827 CrossRef CAS; (b) S. Chiba, Z. Cao, S. A. A. E. Bialy and K. Narasaka, Chem. Lett., 2006, 35, 18–19 CrossRef CAS; (c) A. Ilangovan, S. Saravanakumar and S. Malayappasamy, Org. Lett., 2013, 15, 4968–4971 CrossRef CAS PubMed; (d) D. G. Kananovich, Y. A. Konik, D. M. Zubrytski, I. Järving and M. Lopp, Chem. Commun., 2015, 51, 8349–8352 RSC; (e) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490–3493 CrossRef CAS PubMed; (f) S. Wang, L.-N. Guo, H. Wang and X.-H. Duan, Org. Lett., 2015, 17, 4798–4801 CrossRef CAS PubMed; (g) X. Fan, H. Zhao, J. Yu, X. Bao and C. Zhu, Org. Chem. Front., 2016, 3, 227–232 RSC; (h) K. Jia, F. Zhang, H. Huang and Y. Chen, J. Am. Chem. Soc., 2016, 138, 1514–1517 CrossRef CAS PubMed; (i) Z. Ye, X. Cai, J. Li and M. Dai, ACS Catal., 2018, 8, 5907–5914 CrossRef CAS; (j) Q. Tan, Z. Yang, D. Jiang, Y. Cheng, J. Yang, S. Xi and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 6420–6424 CrossRef CAS PubMed.
- (a) Y.-F. Wang and S. Chiba, J. Am. Chem. Soc., 2009, 131, 12570–12572 CrossRef CAS PubMed; (b) Y.-F. Wang, K. K. Toh, E. P. J. Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 6411–6421 CrossRef CAS PubMed.
- (a) J. Sheng, J. Liu, L. Chen, L. Zhang, L. Zheng and X. Wei, Org. Chem. Front., 2019, 6, 1471–1475 RSC; (b) J. Liu, J. Zou, J. Yao and G. Chen, Adv. Synth. Catal., 2018, 360, 659–663 CrossRef CAS; (c) J. Sheng, J. Liu, H. Zhao, L. Zheng and X. Wei, Org. Biomol. Chem., 2018, 16, 5570–5574 RSC; (d) J. Liu, Z. Xue, Z. Zeng, Y. Chen and G. Chen, Adv. Synth. Catal., 2016, 358, 3694–3699 CrossRef CAS.
- (a) E. Hasegawa, M. Tateyama, R. Nagumo, E. Tayama and H. Iwamoto, Beilstein J. Org. Chem., 2013, 9, 1397–1406 CrossRef PubMed; (b) M. R. Heinrich, O. Blank and S. Wölfel, Org. Lett., 2006, 8, 3323–3325 CrossRef CAS PubMed; (c) F. Liu, J.-Y. Wang, P. Zhou, G. Li, W.-J. Hao, S.-J. Tu and B. Jiang, Angew. Chem., Int. Ed., 2017, 56, 15570–15574 CrossRef CAS PubMed; (d) S. E. Schaafsma, R. Jorritsma, H. Steinberg and T. J. de Boer, Tetrahedron Lett., 1973, 14, 827–830 CrossRef.
- The need for a stoichiometric amount of Cu(OAc)2 in our reaction suggests that the Cu(II) which was re-generated from Cu(I) and intermediate D may not react again from A to B.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1959009. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc09657d |
| This journal is © The Royal Society of Chemistry 2020 |