
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvidence for enzyme catalysed intramolecular [4+2] Diels–Alder cyclization during the biosynthesis of pyrichalasin H†
Verena
Hantke
*,
Elizabeth J.
Skellam
 and
Russell J.
Cox
*
and
Russell J.
Cox
*
Institute for Organic Chemistry and BMWZ, Leibniz University Hannover, Schneiderberg 38, Hannover 30167, Germany. E-mail: Verena.Hantke@oci.uni-hannover.de; russell.cox@oci.uni-hannover.de
First published on 10th February 2020
Abstract
Cytochalasans are highly complex fungal metabolites which exhibit diverse biological activities. Little is known of the chemical steps involved in the construction of the tricyclic core, which consists of an octahydro-isoindole skeleton fused to a macrocyclic ring. Here, using a directed gene knockout and complementation strategy, we show that PyiF is implicated as the proposed intramolecular [4+2] Diels–Alderase required for construction of the tricyclic core of pyrichalasin H 1.
Cytochalasans are a diverse group of fungal polyketide-non-ribosomal peptide secondary metabolites with varied biological properties including antibacterial, antiviral and antitumor potential.1,2 All cytochalasans contain a tricyclic core, which consists of a macrocyclic ring fused to an amino acid-derived octahydro-isoindole. Differences arise from variations in the size and the substitution pattern of the macrocycle and the incorporated amino acid (Fig. 1).3
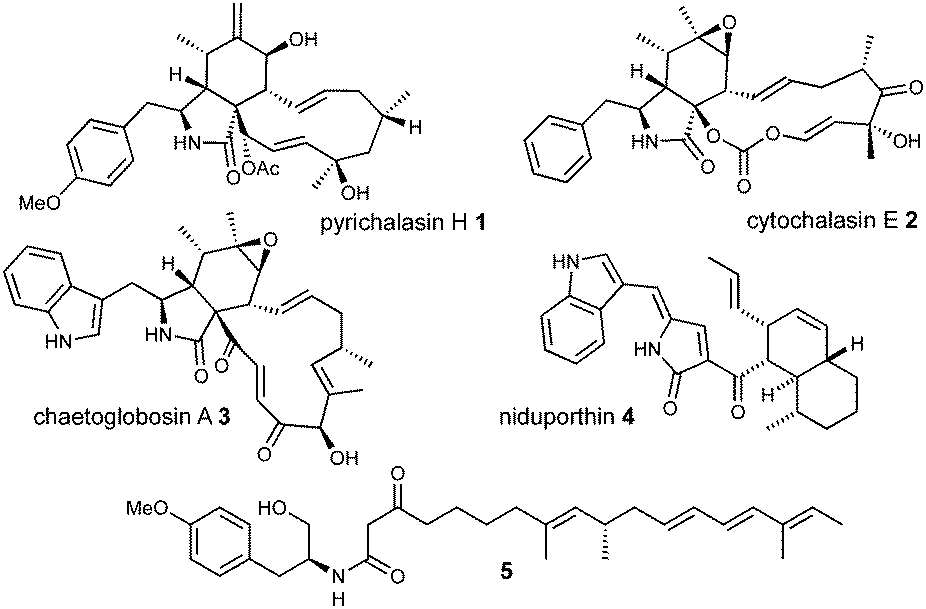 | ||
| Fig. 1 Chemical structures of selected cytochalasans and related metabolites. | ||
Several biosynthetic gene clusters (BGC) involved in cytochalasan biosynthesis are known, and their biosynthetic pathways have been partially elucidated, including those for pyrichalasin H 1,4 cytochalasin E 2,5 chaetoglobosin A 36 and compounds such as niduporthin 47 and the ACE1 shunt 58 which are genetically and chemically closely related. All cytochalasan BGC encode: a polyketide synthase/non-ribosomal peptide synthetase (PKS-NRPS); a trans-acting enoyl reductase (ER); an αβ-hydrolase (HYD); several tailoring enzymes such as cytochrome P450 monooxygenases (P450) and oxidoreductases (OXR); and transcriptional regulators (TF) and transporters. Recently, we identified the pyi BGC encoding the phytotoxin pyrichalasin H 1 in Magnaporthe grisea NI980 (Scheme 1) and elucidated the late-stage biosynthetic pathway of 1 using targeted gene inactivations.4
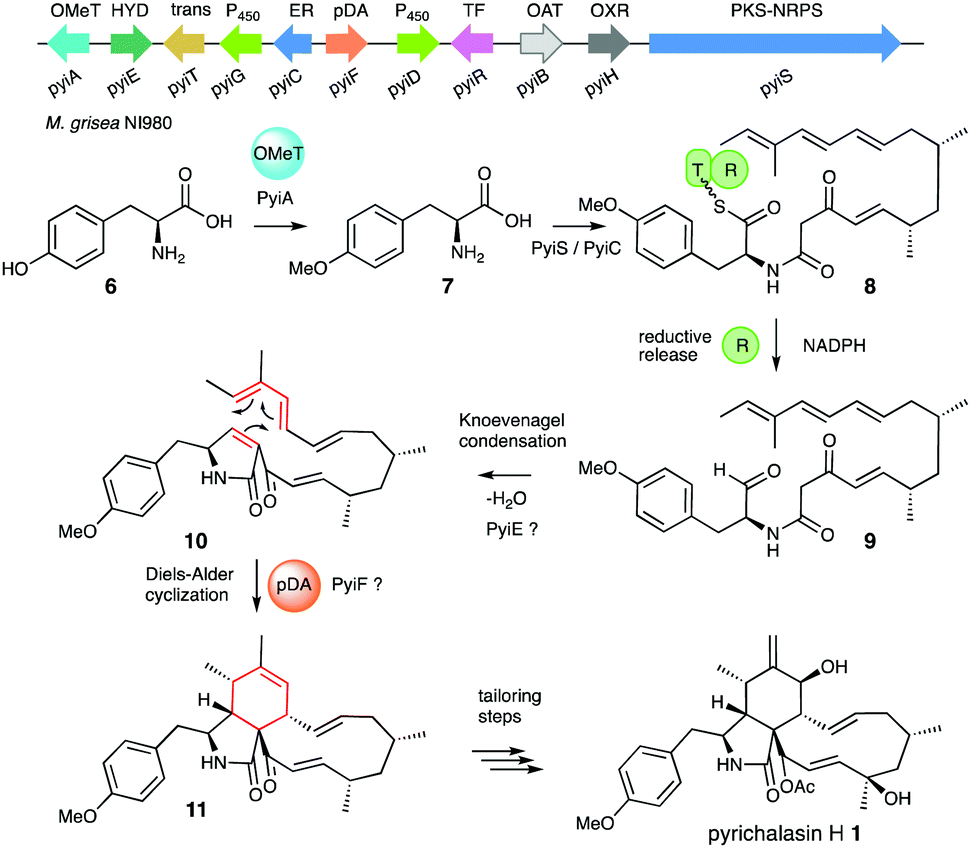 | ||
| Scheme 1 Proposed early biosynthetic steps of pyrichalasin H 1. | ||
Formation of the tricyclic core of 1 and other cytochalasans is thought to occur in two steps. First, it is proposed that the PKS-NRPS reductively releases an N-acyl α-amino aldehyde 9 which is cyclised to form a 1,5-dihydropyrrolone 10. This is then proposed to undergo an endo-selective intramolecular [4+2] Diels Alder (DA) reaction (Scheme 1).9 Intermediates 9 and 10 have not been observed for pyrichalasin H 1 or related cytochalasans, although similar α-aminoaldehyde and pyrrolone intermediates have been proposed during the biosynthesis of fusarin C, for example.3,10 In other related systems non-reductive release (i.e. Dieckmann cyclisation) occurs during the biosynthesis of, e.g., pretenellin A.11 The heterocyclic core seems to be formed by the PKS-NRPS and trans-ER alone in the cases of niduporthin 47 and cytochalasans from Parastagonospora nodorum.12
The assumption that the DA reaction is not spontaneous is supported by observations in the total synthesis of cytochalasans.13 Stork and Nakamura developed the first biomimetic macrocyclization strategy in 1982 while synthesizing cytochalasin B. To achieve the DA reaction a precursor analogous to 10 had to be heated to 180–190 °C in neat mesitylene for nearly a week to yield 35% product (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of endo:exo), strongly suggesting that the DA reaction is not favoured under physiological conditions.14
:1 mixture of endo:exo), strongly suggesting that the DA reaction is not favoured under physiological conditions.14
Enzymatic DA15 and hetero DA (hDA)16 catalysts have been discovered and validated in several biosynthetic pathways. However, while the action of a DA enzyme has been frequently proposed as the key element of cytochalasan pathways no evidence has been so-far gathered for the identity of this catalyst. In the case of pyrichalasin H 1 we have recently demonstrated the function of all the genes in its BGC except pyiE and pyiF.4 Homologs of these two genes are present in all known cytochalasan BGC and they may thus be involved in the formation of the isoindole. PyiE is a member of the αβ-hydrolase family,17 but PyiF and its homologs have no known function. Here we investigated the function of PyiF and homologs from other systems by knockout (KO) and complementation in M. grisea NI980.
Disruption of pyiF was achieved by replacing it with a hygromycin resistance cassette.18 Wild-type (WT) M. grisea NI980 produces 1 at around 60 mg L−1 (Fig. 2A), but inactivation of pyiF resulted in the abolition of 1 (Fig. 2B).
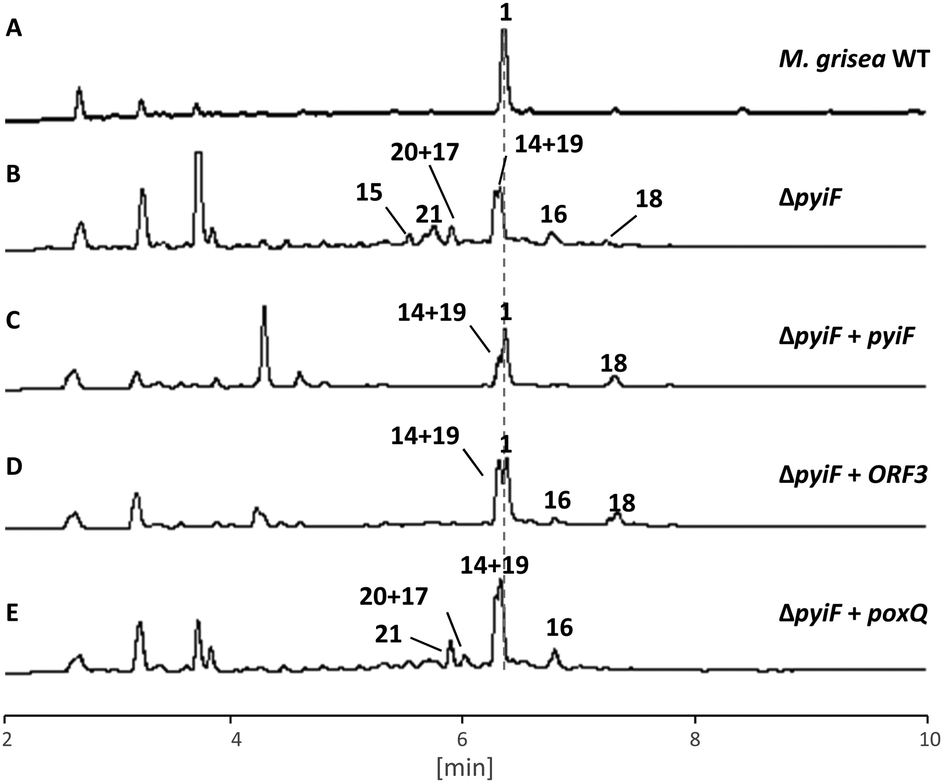 | ||
| Fig. 2 LCMS traces (diode array) for key experiments: (A) M. grisea wild type (WT); (B) M. grisea ΔpyiF; (C) as B, complemented with pyiF; (D) as B, complemented with ORF3; (E) as B, complemented with poxQ. Unnumbered peaks are unrelated. | ||
We expected knockout of pyiF to result in an accumulation of the pyrrolone 10 (m/z = 447 g mol−1) but this was not observed. Instead the organic extracts of ΔpyiF strains contained several new compounds which, based on their UV absorption spectra at 275 nm (see ESI†), derived from tyrosine 6 or O-methyl tyrosine 7 (Fig. 1B). Eight compounds (15, 0.4 mg; 21, 0.3 mg; 20 and 17 (mixture, 20:17ca. 2:1), 0.5 mg; 19 and 14 (mixture, 19:14ca. 1:1.2), 1.5 mg; 16, 0.5 mg; and 18, 1.0 mg) were purified by preparative LCMS from a 1 litre ΔpyiF M. grisea culture after 7 days.
The molecular formula for each compound was obtained (HRMS) and structure elucidation (NMR, see ESI† for full NMR and HRMS data) was then performed. In the case of the major compound 14, HRMS indicated a molecular formula of C29H43NO6 (obs. 546.3073, calc., 546.3067, ES−, [M + HCO2H–H]−) consistent with the skeleton of 1. HMBC correlations from the methoxy group (δH 3.7 ppm) to C-27, supported by HMBC from H-25 and H-26, confirmed the presence of the p-substituted aromatic ring and also located benzylic C-23 (δC 39.7 ppm). COSY correlation from H-23 via H-5, H-4 and H-3 led to methine H-6, and HMBC from H-4 and H-5 located C-2 (δC 177.2 ppm) and confirmed the presence of the γ-lactam. Further COSY correlations then located the 7/8 olefin, and methine H-9 and methylenes H-10. HMBC correlations to methyls 21 and 22 located carbons 8–12, and further COSY correlations then revealed the diene system to C-16. Finally, HMBC from methyls-19 and 20 revealed C-17 as a tertiary alcohol and C-18 as a secondary alcohol (Scheme 2B). The related compounds were identified by comparison and close analysis of HMBC signals, particularly around the γ-lactam.
 | ||
| Scheme 2 Putative Diels–Alder reaction catalysed by PyiF and isolated shunt products (14–21) purified from a ΔpyiF M. grisea strain, and related chemistry. | ||
All isolated compounds displayed an underlying linear structure of the polyketide moiety. The C-17/C-18 olefin was dihydroxylated in all compounds, probably via epoxide formation and hydrolysis. Genome analysis of M. grisea showed it contains the dihydropyriculol/dihydropyriculariol 22/23 BGC19 and dihydropyriculol 22 was purified and characterised from M. grisea NI980 producing strains (see ESI†). The same oxygenase responsible for the formation of the diols in 22/23 could give the diols observed in 14–21.
Compound 21 also features C-13/C-14 olefin dihydroxylation; compounds 14 and 15 are hydroxyled at C-4; and compound 20 is hydroxylated at C-11. Another deviation from the expected pyrrolone 10 is that all compounds carry a hydroxyl at C-6 instead of a carbonyl. Furthermore, the C-3/C-4 olefin of 10 (part of the pyrrolone ring) is missing in all compounds, except 16 and 17. Five compounds (14, 16, 18, 20, 21) derive from O-methyl tyrosine 7; the others from tyrosine 6. The observed compounds appeared to be shunt products of 10 (or its 27-O-desmethyl cogener) formed via oxidative and reductive steps.
A functional copy of pyiF was then introduced downstream of the inducible amyB promoter (PamyB) from Aspergillus oryzae and transformed into two ΔpyiF M. grisea strains.
As expected, in all strains carrying an ectopic functional copy of pyiF, the production of 1 was restored. However, the obtained yields of 1 were lower compared to the wild type (ca. 10%). In addition, some of the shunt intermediates (14–21) observed in the ΔpyiF M. grisea deletion strain were also detected in most of the transformants (mainly compound 14, Fig. 2C).
Heterologous complementation studies were carried out to investigate if the closely related homologous protein ORF3 (62% identity to PyiF, see ESI†) from the cryptic ACE1 BGC in P. oryzae Guy11,20 is able to restore production of 1. The pyi and ACE1 gene clusters are closely related, suggesting that ORF3 might be able to replace PyiF as a DAase. This was confirmed when ΔpyiF M. grisea strains carrying ORF3 driven by PamyB restored the production of 1, albeit in similar titres compared to the homologous complementation with pyiF (ca. 10%, Fig. 2D). No new compounds were observed.
An analogous complementation experiment was also carried out with the poxQ gene from Penicillium oxalicum. PoxQ is proposed to be a decalin-forming DAase (29% identical to PyiF) during the biosynthesis of oxaleimide.21 PoxQ and related enzymes22 are proposed to intercept an intermediate 24 in which the polyketide moiety is nearly identical to that proposed in the case of pyrichalasin H 1 (e.g.8, Scheme 2D). If the PoxQ protein recognised 8 then decalins such as 26 might be expected. However, all M. grisea ΔpyiF:PamyBPoxQ transformants demonstrated the same secondary metabolite profile as the M. grisea ΔpyiF control strain (Fig. 2E). Accordingly, the production of 1 could not be restored and no new decalin-containing compounds were produced.
Finally, in an attempt to demonstrate in vitro activity of PyiF very extensive efforts were made to obtain soluble PyiF protein via heterologous expression. However this proved impossible.
In conclusion, targeted gene disruption of pyiF in M. grisea resulted in complete abolition of 1, and the accumulation of 14–21 which appear to arise from a redox shunt pathway from 10 (Scheme 2). Compound 16 probably arises by reduction of the C-6 carbonyl and likely epoxidation and hydrolysis of the C-17/18 olefin. Conjugate addition of water at C-4/5 could then give the main compound 14. Further reduction of 16 leads to compound 18, and compounds 20 and 22 are oxidation products of 18. Dihydroxylation of the triene sidechain may be catalysed by enzymes from the pyriculol pathway, known to be present and active in M. grisea. Compounds 15, 17 and 19 lacking the O-methyl likely arise from use of tyrosine by the PyiS NRPS.18
All compounds isolated from the ΔpyiF mutant lack the cyclohexenyl moiety indicating that the DA reaction has not occurred. This, therefore, shows that PyiF is the likely DA enzyme required for the formation of the core cytochalasan isoindole. Complementation studies showed that the closely related ORF3 (62% identical) from the cryptic ACE1 pathway is also a DA enzyme, and since all known cytochalasan BGCs contain a homolog of pyiF/ORF3, the results suggest this is the likely function of these genes. The results of this experiment also add to the growing evidence that the cryptic ACE1 metabolite is a cytochalasan.18
It is notable that the titres of the DA products from the complementation experiments are lower than the WT titres of 1. This most likely arises because the A. oryzae PamyB promoter, used to drive the expression in these cases, is not strong when used in M. grisea.23 Lebrun and coworkers showed in a previous study that ORF3 is the most highly expressed gene in the ACE1 BGC, perhaps indicating a requirement for relatively high protein concentrations for rapid DA reaction.24
Heterologous complementation of the ΔpyiF mutant with poxQ from Penicillium oxalicum did not restore the production of 1, or produce a decalin metabolite. PoxQ is proposed to catalyse a decalin forming DA reaction of a near-identical polyketide intermediate (e.g.8) during the biosynthesis of 1 (Scheme 2D).19 This may indicate that PoxQ and related decalin-forming catalysts20 recognise more than just the polyketide intermediates, or that the triene of 8 was rapidly destroyed by olefin oxidation before PoxQ could act. Intermediates such as 8 are also known to undergo spontaneous decalin formation (albeit with different stereoselectivity)20 and rapid conversion of 8 to 14 in M. grisea could also explain the lack of spontaneous decalin formation.
Collectively, our results strongly suggest that PyiF and ORF3 catalyse the long-proposed intramolecular DA reaction forming the tricyclic core structure of cytochalasans. For the first time the involvement of a DA enzyme in the biosynthesis of cytochalasans has been experimentally confirmed. DA enzymes involved in the biosynthesis of cytochalasans belong to the stand-alone class of pericyclases with no additional function besides catalysing the [4+2] cyclisation. Whether this reaction requires conformational or chemical catalysis is unknown and will require in vitro studies of soluble protein which is not yet available. To gain more insights into the biosynthetic role of the hydrolase PyiE, in vitro studies are underway and results of these experiments will be reported in due course.25
This work was supported by DFG [(CO 1328/2-1, INST 187/621-1, INST 187/686-1)]. The authors also thank: Professor Marc-Henri Lebrun (UMR BIOGER INRA AgroParisTech) and Dr Didier Tharreau (CIRAD UMR BGPI) for the gift of M. grisea NI980 and useful discussions; and Chongqing Wang for assistance with identification of dihydropyriculol 22.
Conflicts of interest
There are no conflicts to declare.References
- K. Scherlach, D. Boettger, N. Remme and C. Hertweck, Nat. Prod. Rep., 2010, 27, 869–886 RSC.
- R. Kretz, L. Wendt, S. Wongkanoun, J. Luangsa-ard, F. Surup, S. Helaly, S. Noumeur, M. Stadler and T. Stradal, Biomolecules, 2019, 9, 73 CrossRef CAS PubMed.
- E. Skellam, Nat. Prod. Rep., 2017, 34, 1252–1263 RSC.
- C. Wang, V. Hantke, R. J. Cox and E. Skellam, Org. Lett., 2019, 21, 4163–4167 CrossRef CAS PubMed.
- K. Qiao, Y. H. Chooi and Y. Tang, Metab. Eng., 2011, 13, 723–732 CrossRef CAS PubMed.
- K. Ishiuchi, T. Nakazawa, F. Yagishita, T. Mino, H. Noguchi, K. Hotta and K. Watanabe, J. Am. Chem. Soc., 2013, 135, 7371–7377 CrossRef CAS PubMed.
- M. L. Nielsen, T. Isbrandt, L. M. Petersen, U. H. Mortensen, M. R. Andersen, J. B. Hoof and T. O. Larsen, PLoS One, 2016, 11, 1–18 Search PubMed.
- Z. Song, W. Bakeer, J. Marshall, A. Yakasai, R. Mohd-Khalid, J. Collemare, E. Skellam, D. Tharreau, M.-H. Lebrun, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Sci., 2015, 6, 4837–4845 RSC.
- R. Wyss and C. Tamm, Croat. Chem. Acta, 1985, 58, 537–546 CAS.
- Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS PubMed.
- K. L. Eley, L. M. Halo, Z. Song, H. Powles, R. J. Cox, A. M. Bailey, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2007, 8, 289–297 CrossRef CAS PubMed.
- H. Li, H. Wei, J. Hu, E. Lacey, A. Sobolev, K. Stubbs, P. Solomon and Y. Chooi, ACS Chem. Biol., 2019 DOI:10.1021/acschembio.9b00791.
- E. J. Thomas, Acc. Chem. Res., 1991, 24, 229–235 CrossRef CAS.
- G. Stork and E. Nakamura, J. Am. Chem. Soc., 1983, 105, 5510–5512 CrossRef CAS.
- C. Jamieson, M. Ohashi, F. Liu, Y. Tang and K. Houk, Nat. Prod. Rep., 2019, 36, 698–713 RSC.
- R. Schor, C. Schotte, D. Wibberg, J. Kalinowski and R. J. Cox, Nat. Commun., 2018, 9, 1963 CrossRef PubMed.
- Knockout of pyiE shows that it is not essential for the biosynthesis of 1.
- M. L. Nielsen, L. Albertsen, G. Lettier, J. B. Nielsen and U. H. Mortensen, Fungal Genet. Biol., 2006, 43, 54–64 CrossRef CAS PubMed.
- S. Jacob, T. Grötsch, A. J. Foster, A. Schüffler, P. H. Rieger, L. P. Sandjo, J. C. Liermann, T. Opatz and E. Thines, Microbiology, 2017, 163, 541–553 CrossRef CAS PubMed.
- C. Wang, K. Becker, S. Pfutze, E. Kuhnert, M. Stadler, R. J. Cox and E. J. Skellam, Org. Lett., 2019, 21, 8756–8760 CrossRef CAS PubMed.
- M. Sato, J. E. Dander, C. Sato, Y. Hung, S.-S. Gao, M.-C. Tang, L. Hang, J. M. Winter, N. K. Garg, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2017, 139, 5317–5320 CrossRef CAS PubMed.
- M. Sato, F. Yagishita, T. Mino, N. Uchiyama, A. Patel, Y. Chooi, Y. Goda, W. Xu, H. Noguchi, T. Yamamoto, K. Hotta, K. N. Houk, Y. Tang and K. Watanabe, ChemBioChem, 2015, 16, 2294–2298 CrossRef CAS PubMed.
- V. Hantke, C. Wang, E. J. Skellam and R. J. Cox, RSC Adv., 2019, 9, 35797–35802 RSC.
- J. Collemare, M. Pianfetti, A.-E. Houlle, D. Morin, L. Camborde, M.-J. Gagey, C. Barbisan, I. Fudal, M.-H. Lebrun and H. U. Böhnert, New Phytol., 2008, 179, 196–208 CrossRef CAS PubMed.
- Full experimental details are supplied in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc09590j |
| This journal is © The Royal Society of Chemistry 2020 |