
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of highly stable multinuclear AgnSn clusters in zinc fingers disrupts their structure and function†
Katarzyna
Kluska
a,
Manuel D.
Peris-Díaz
a,
Dawid
Płonka
b,
Alexander
Moysa
 b,
Michał
Dadlez
b,
Aurélien
Deniaud
c,
Wojciech
Bal
*b and
Artur
Krężel
*a
b,
Michał
Dadlez
b,
Aurélien
Deniaud
c,
Wojciech
Bal
*b and
Artur
Krężel
*a
aDepartment of Chemical Biology, Faculty of Biotechnology, University of Wrocław, Joliot-Curie 14a, 50-383 Wrocław, Poland. E-mail: artur.krezel@uwr.edu.pl
bInstitute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawińskiego 5a, 02-106 Warsaw, Poland. E-mail: wbal@ibb.waw.pl
cUniv. Grenoble Alpes, CNRS, CEA, IRIG, Laboratoire de Chimie et Biologie des Métaux, F-38000 Grenoble, France
First published on 8th January 2020
Abstract
Silver (Ag(I)) binding to consensus zinc fingers (ZFs) causes Zn(II) release inducing a gradual disruption of the hydrophobic core, followed by an overall conformational change and formation of highly stable AgnSn clusters. A compact eight-membered Ag4S4 structure formed by a CCCC ZF is the first cluster example reported for a single biological molecule. Ag(I)-induced conformational changes of ZFs can, as a consequence, affect transcriptional regulation and other cellular processes.
Zinc finger (ZF) domains are one of the most abundant structural motifs found in proteins, with a wide range of physiological roles including transcriptional regulation, signal transduction, DNA repair, cell migration, etc.1–5 Although ZFs contain Zn(II) in their native structures, they provide an impressive range of binding properties for monovalent (Cu(I) and Au(I)), divalent (Cd(II), Pb(II), Hg(II), Pt(II), Fe(II), Co(II), and Ni(II)) and trivalent (Sb(III) and As(III)) metal ions. These cations can compete with Zn(II), impacting the native domain structure (Fig. S1a, ESI†) and as a consequence influencing the biological functions of ZFs due to their various coordination geometries, ionic radii and ligand specificities.4–8 Moreover, they can promote multinuclear species formation, cysteinyl sulfur oxidation or incomplete coordination.9–11 All of these outcomes are important in the case of toxic metal ions, which can be absorbed into the body from diet, drinking water, by inhalation, dermal exposure or medical intervention, eventually finding their way to cells to exert toxicity by abolishing the ZF functions.
Extensive research has been devoted to the effects of heavy metals, like Pb(II), Hg(II) or Cd(II), on ZF domains, but much less is known about the impact of Ag(I) ions. Various Ag(I) complexes have been vigorously tested for antimicrobial,12,13 anti-inflammatory14,15 or anticancer16,17 properties. The use of elemental silver nanoparticles (AgNPs) is high and rising rapidly (30% of the products declared to contain NPs are AgNPs). They can be found in multiple daily life applications including food and textile industries, electronics and personal care. Moreover, the AgNP coatings are becoming popular in medical devices and bone implants as antibacterial additives.13,18,19 This rapid expansion of AgNPs in various fields elevates the population exposure to metallic nanoparticulates and ionic silver due to the high reactivity of Ag in aerobiosis.20
Animal studies have shown that exposure to Ag in any form leads to its accumulation mostly in the liver and spleen,21 while occupational health studies in humans reported deposition of insoluble silver sulfide or selenide precipitates in the dermis and cornea, causing argyria and argyrosis, respectively.22–24 Besides, a very slow clearance of Ag has been measured in the brain and testis, two crucial organs.21 Cellular experiments using hepatocytes, fibroblasts, and other human cell lines showed that endocytosed AgNPs undergo lysosome-assisted dissolution to Ag(I),25,26 which is the actual intracellular chemically reactive form of silver. This process leads to the subsequent formation of intracellular Ag(I)-thiolate complexes,20,25,27,28 strongly suggesting that Ag(I) ions target cysteine rich proteins and peptides including metallothioneins and glutathione that are abundant. Other proteins with solvent-exposed cysteines such as ZFs could also bind to Ag(I), with toxic consequences. This issue has not been properly explored, however.
In order to shed more light on how Ag(I) ions affect ZFs, we examined their interactions with consensus peptide 1 (CP1) ZF model peptides containing two (CCHH), three (CCCH) and four (CCCC) Cys residues using UV-Vis and CD spectroscopies, mass spectrometry (MS)–electrospray ionization (ESI) and ion-mobility (IM), quantum mechanics/molecular mechanics (QM/MM), and molecular dynamics (MD) and well-tempered metadynamics (WT-MetaD) simulations. CP1 ZF models have been extensively studied in terms of the coordination chemistry and thermodynamics.29,30 We used the recently updated CP1 sequence which represents natural ZF features even better (Fig. S1b, ESI†).31 The CD spectra were used to determine whether the Ag(I) binding can induce conformational changes of the ZF peptides. First, 0 to 4 equivalents of Ag(I) were titrated into each ZF. In the CCCC ZF the Ag4L species (L denotes a given ZF) was formed predominantly, as evidenced by the presence of isodichroic points and the overall stoichiometry (Fig. 1a). Its CD spectrum features two partially overlapping intensive negative bands at 210 and 225 nm indicating the presence of an α-helix with some contribution from a β-sheet. Ag(I) binding also induced a new band at 265 nm, assigned to a S → Ag(I) charge transfer (CT) band.32–34 Ag(I) titrations of other peptides (Fig. S2a, ESI†) yielded various negative and positive bands in the 200–225 nm range indicating the formation of multiple Ag(I) complexes. The comparison with Zn(II) induced CD spectra clearly demonstrates the fundamental alteration of the ZF conformation by Ag(I) coordination in all cases, but the far-UV features observed for CCHH and CCCH ZFs were significantly weaker, indicating a mostly random coil character of the formed Ag(I) complexes.35 The absorption spectra of the same titrations confirmed the formation of S → Ag(I) CT bands at 240–260 nm. Their molar absorption coefficients increase with the number of Cys residues (Fig. S2b (ESI†) and Fig. 1b).32,33 Interestingly when plotted against the Ag(I)-to-ZF molar ratio, the CT intensity linearly corresponds to the number of Cys residues in ZFs, confirming that predominant/final complexes formed for CCHH, CCCH and CCCC ZF peptides are Ag2L, Ag3L and Ag4L, respectively (see below). The course of titrations shows, however, that lower stoichiometry complexes were also formed. Very similar UV-Vis spectral patterns and complex stoichiometries observed in Ag(I) titrations of CCAA and CCCA ZFs (His residues replaced with alanines) indicate the absence of imidazole coordination to Ag(I) ions in CCHH and CCCH (Fig. S3, ESI†). The above observations, together with QM/MM MD simulations (Fig. S4–S6, ESI†), strongly support the formation of Ag2Cys2, Ag3Cys3 and Ag4Cys4 clusters in Ag2L, Ag3L and Ag4L species, respectively, with sulfur donors bringing two Ag(I) ions. The simulations indicated the absence of secondary structures in Ag2L and Ag3L, but the highly regular octagonal Ag4Cys4 cluster in Ag4L maintained the α-helix and a distorted β-sheet, in excellent agreement with the CD results. The AgnSn rings with n = 3–14 are known for low molecular weight thiolates,34 and recently a similar Ag4Cys4 cluster was found in the Cu(I) site of dimeric Atox1 copper chaperone (PDB: 5F0W), but the cluster formed by a CCCC ZF is the first one reported for a single biological molecule.
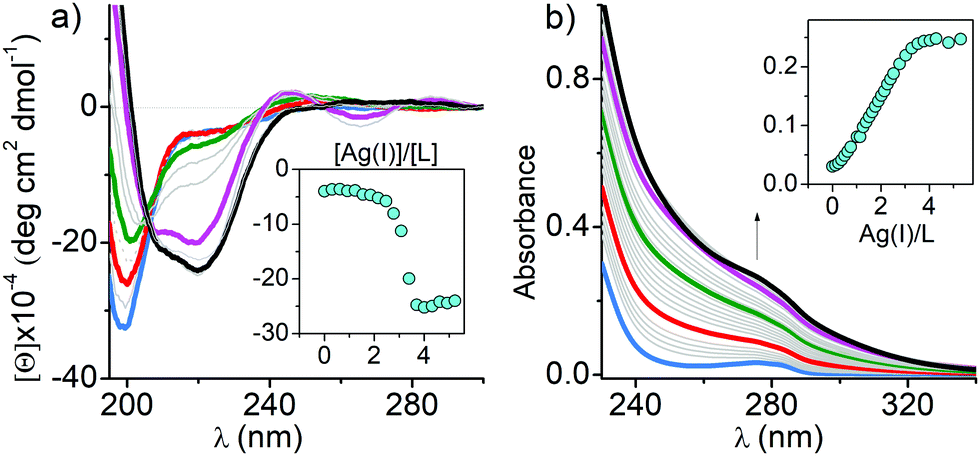 | ||
| Fig. 1 CD (a) and UV-Vis (b) titrations of 25 μM CCCC ZF in 20 mM TES buffer (pH 7.0) with Ag(I). Blue, red, green, magenta, and black indicate 0, 1, 2, 3, and 4 Ag(I) eq., respectively. The insets show the changes in molar ellipticity and absorption at 220 nm and 279 nm over the range of 0–4 eq. of Ag(I), respectively. | ||
The above results were corroborated by positive-ion ESI-MS-monitored Ag(I) titrations of ZFs performed in pH neutral solutions (Fig. S7, ESI†). The observed 3+ ions of metal free ZFs turn to mono- and then to multinuclear Ag(I) complexes in a stepwise manner, with Ag2L, Ag3L and Ag4L complexes being the most saturated ones for CCHH, CCCH and CCCC ZFs, respectively, confirming the spectroscopic results (Table S1, ESI†). These gas phase data, however, have only a qualitative character.36
The tight structure of the Ag4L complex of the CCCC ZF was additionally confirmed by MS/MS experiments, in which no molecular fragment could be obtained for this species, as opposed to other studied complexes (Tables S2–S6, ESI†).37 The [Ag4L]3+ molecular ion (Fig. S8 (ESI†) for the experimental and simulated isotopic patterns) was subjected to the IM-MS experiment, along with the [ZnL]3+ and L3+ ions. The shorter drift time (DT) and the narrower peak of [Ag4L]3+, compared to [ZnL]3+, perfectly agree with its simulated compacted structure (Fig. 2 and Fig. S8, ESI†). To further investigate the nature of these conformational changes, the average collision cross-sections (CCSs, Ω) of the ZF complexes in the 3+ charge state were calculated from the DT data (Fig. 2). The Ag4L complex was more compact (Ω = 439 Å2) than the ZnL complex (Ω = 461 Å2) and the apo-peptide L (Ω = 512 Å2) (Fig. 2). The CCS values calculated with IMPACT software from MD simulations are comparable to those obtained experimentally (Fig. S9, ESI†). To examine if Ag(I) ions could affect stable Zn(II)–ZF complexes, Zn(II) saturated ZF peptides were titrated with Ag(I) ions as monitored by CD and UV-Vis (Fig. S10, ESI†). In all three peptides Zn(II) ions were easily substituted by Ag(I) ions. The comparison of Ag(I) titrations of free and Zn(II)-complexed ZFs shows some differences in the course of signal changes, suggestive of low amounts of mixed metal intermediates, but at stoichiometric Ag(I) concentrations the Ag2L, Ag3L and Ag4L species were formed almost exclusively for peptides with two, three and four cysteinyl residues, respectively. The spectra recorded at those ratios and at a slight Ag(I) excess are identical to those recorded for free peptide titrations. This indicated that the Ag(I) affinity to the respective complexes is at least about two orders of magnitude higher than that of Zn(II). In order to correlate Zn(II) release with Ag(I) binding, the chromophoric Zn(II) chelating probe 4-(2-pyridylazo)resorcinol (PAR) was added to the Zn(II) saturated ZFs, followed by stepwise addition of single mol equiv. of Ag(I) ions, up to four per ZF for consistency.38 Firstly, to show that Ag(I) binding to PAR does not induce absorption at 492 nm, the control experiments (without peptides) were performed (Fig. S11, ESI†). After that, the concomitant Zn(II) release steps were determined in a time-dependent mode at 492 nm for Zn(PAR)2 (Fig. 3 and Fig. S12, ESI†). The Zn(II) release was rapid in all cases, with the apparent rate positively correlated with the number of Cys residues. A similar higher reactivity of ZnCys4 cores has been observed previously for a human/chicken estrogen receptor, E. coli N-Ada, HIV-1 nucleocapsid and FOG-1 protein.4,39–41 The addition of 1 mol equiv. of Ag(I) to the CCHH ZF caused the release of ca. 60% of Zn(II) from this peptide (Fig. S12, ESI†). The 1st Ag(I) effect was less pronounced for CCCH and CCCC ZFs, at ca. 40% and 30%, respectively (Fig. 3 and Fig. S12, ESI†). This trend reflects the increased cooperativity of the formation of Ag3Cys3 and Ag4Cys4 clusters. Furthermore, as showed by us previously, a Cys substitution to other noncoordinating residues weakens the Zn(II) binding more strongly than a His substitution.42,43 Individual Cys residues in ZnCys4 have comparable reactivities and energetic contributions to the Zn(II) complex.4,44
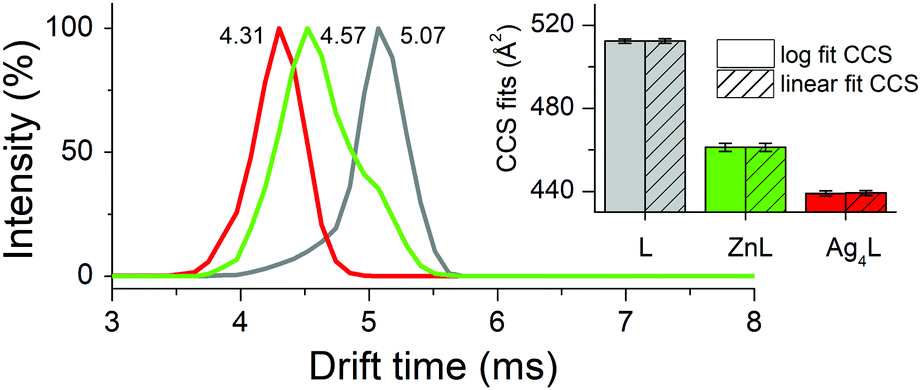 | ||
| Fig. 2 The arrival time distributions (ATDs) of the CCCC ZF species in the 3+ charge state derived from the IM-MS experiment. The inset shows average collision cross-sections (CCSs) of the CCCC ZF species in the 3+ charge state. | ||
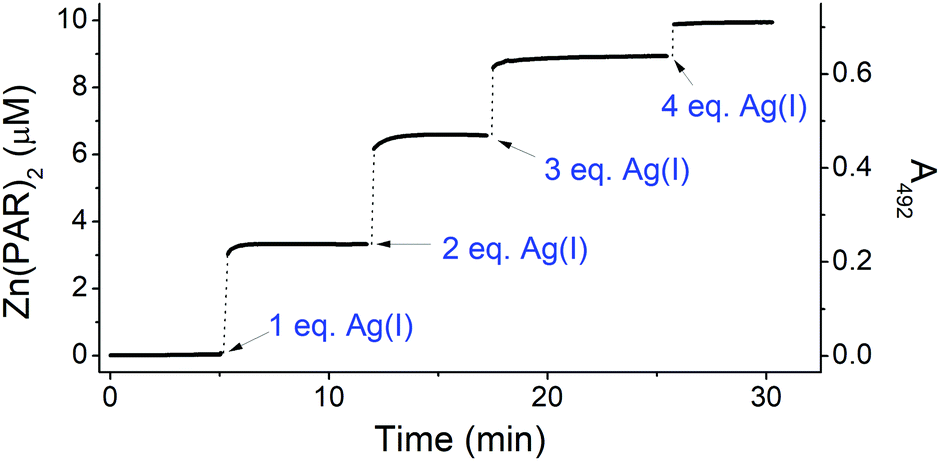 | ||
| Fig. 3 Zn(II) transfer from 10 μM CCCC ZF to 100 μM PAR during titration with 0–4 eq. of Ag(I). The measurement was performed in 20 mM TES buffer (pH 7.0). | ||
The Ag(I) complexes are too strong for the direct determination of affinity constants; therefore, we performed CD-monitored Ag(I) titration experiments in the presence of 150 μM cyanides (CN–), the strongest known simple Ag(I) ligand, used as a competitor.45 The binding isotherms were significantly shifted towards higher Ag(I) concentrations (Table S7 and Fig. S13, ESI†). This effect was used to estimate the constant for the CCCC ZF, for which the Ag4L species predominates at all concentrations of added Ag(I). Using KAg(CN)2− = 20.9 we obtained log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KAg4L = 54.9 corresponding to logK = 13.7 per one Ag(I) for pH 7.0 (see the ESI† for details of calculations).45 This value is indeed more than 2 log units higher than that for the Zn(II)–CCCC complex (logKZnL = 11.26 at pH 7.0) (Fig. S14 and Table S8, ESI†).46 Due to the significant overlap of intermediate and final complexes for CCHH and CCCH ZFs, their Ag(I) affinities could not be determined precisely, but the ratio of ca. 100 of Ag(I)/Zn(II) affinities appears to remain valid (Fig. S14 and Table S8, ESI†). This means that Zn(II) replacement in ZFs by Ag(I) can occur even at low silver exposure, and at higher Ag(I) concentrations the cooperative cluster formation will make this process essentially thermodynamically irreversible. Taking into account that exposure of AgNPs may load a cell with millions of silver atoms,47 our results indicate that ZFs may indeed be targeted intracellularly.
KAg4L = 54.9 corresponding to logK = 13.7 per one Ag(I) for pH 7.0 (see the ESI† for details of calculations).45 This value is indeed more than 2 log units higher than that for the Zn(II)–CCCC complex (logKZnL = 11.26 at pH 7.0) (Fig. S14 and Table S8, ESI†).46 Due to the significant overlap of intermediate and final complexes for CCHH and CCCH ZFs, their Ag(I) affinities could not be determined precisely, but the ratio of ca. 100 of Ag(I)/Zn(II) affinities appears to remain valid (Fig. S14 and Table S8, ESI†). This means that Zn(II) replacement in ZFs by Ag(I) can occur even at low silver exposure, and at higher Ag(I) concentrations the cooperative cluster formation will make this process essentially thermodynamically irreversible. Taking into account that exposure of AgNPs may load a cell with millions of silver atoms,47 our results indicate that ZFs may indeed be targeted intracellularly.
To illustrate the Ag(I)-coupled folding process of the CCCC ZF, a 1.3 μs long WT-MetaD simulation was performed along with two collective variables (CVs) capturing an α-helix and hydrophobic core formation (Tyr1, Phe10 and Phe18).48 To fully characterize how the Ag(I) cluster influences the folding, unbiased probability distributions were obtained for a set of CVs through a reweighting procedure (details in the ESI,† Fig. S15).49 The binding of the first Ag(I) ion (two Ag–S bonds) induces structures with the α-helix either folded or unfolded but the hydrophobic core always unpacked (Fig. 4a, Fig. S16a, b and S17, ESI†). The modulated formation of the α-helix depends on whether Ag(I) binds to Cys3 and Cys6 or Cys19 and Cys23, the latter located at the α-helix C-terminus (intermediate I2, Fig. S17a and b, ESI†). In agreement, the analysis of Ag(I) binding order demonstrated that either Cys6–Ag(I)–Cys3 or Cys23–Ag(I)–Cys19 could be formed first as shown in Fig. S18 (ESI†). The binding of the second Ag(I) (four Ag–S bonds) occurs through intermediate I4 forming the Cys3–Ag(I)–Cys19 bonds. This partially folds the α-helix and stabilizes the hydrophobic core (Fig. 4b, Fig. S16a–c, S17 and S18, ESI†). The Ag3L complex (six Ag–S bonds) is formed by the Cys19–Ag(I)–Cys23 coordination (Fig. 4c and Fig. S16a–c, ESI†). A basin for this intermediate I5 was not found, suggesting a transient character of this species (Fig. S16b and S17b, ESI†). The fourth Ag(I) ion binds via Cys6–Ag(I)–Cys23 and stabilizes the hydrophobic core by packing the Tyr1 residue (native state N, Fig. 4d, Fig. S16a–c, S17 and S18, ESI†). In the metal-coupled folding mechanism, proposed elsewhere,50 partial α-helix formation was observed for the CCHH ZF in the absence of a Zn(II) ion. In contrast, in the Ag(I)-coupled folding mechanism, proposed herein for the CCCC ZF, there is no α-helix formation in the absence of Ag(I). Therefore, stabilization of the α-helix and packing of the hydrophobic core are modulated by the number of Ag(I)–S bonds formed.4,50 Such a cooperative stabilization might point towards compact Ag4Cys4 cluster formation and indicate that breaking up any of these components will destabilize the CCCC ZF.
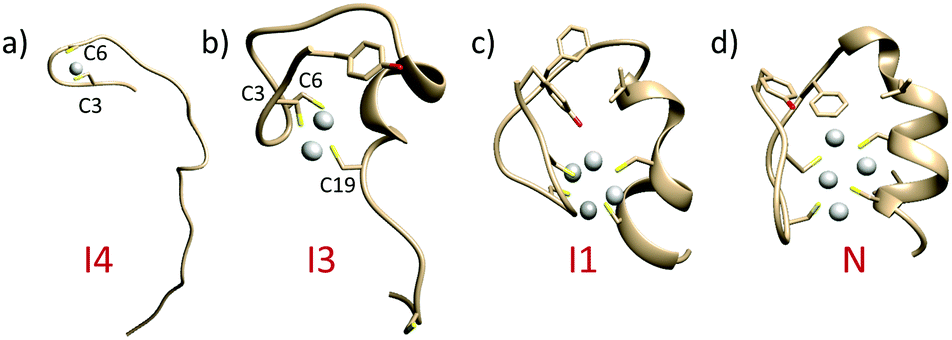 | ||
| Fig. 4 Average structures for the intermediate and native basins extracted from FES obtained by WT-MetaD. (a) Intermediate 4 (I4); (b) intermediate I3; (c) intermediate I1; and (d) native state (N) (see Fig. S16, ESI,† for more details). | ||
Our study demonstrates that Ag(I) ions can directly replace Zn(II) in ZFs of all kinds, forming complexes of various stoichiometries and inducing extensive conformational changes in the native ZF structures. They include the loss of secondary structure elements in CCHH and CCCH ZFs and formation of an alternative secondary structure in the CCCC ZF. The maximum number of Ag(I) ions bound per ZF is equal to the number of Cys residues, with the highly ordered eight-membered Ag4Cys4 structure formed in the CCCC ZF. The Ag(I)-induced conformational changes of biologically active ZF proteins might enhance or inhibit their binding to cognate DNA sequences. Very recently the ability of low dose AgNP exposure to inhibit the activity of hepatocyte nuclear receptors (NR) was demonstrated.47 NR are CCCC ZF transcription factors and thus our study provides the molecular basis for the understanding of this and other manifestations of silver toxicity. Our results warrant further research on the chemistry and biology of silver reactivity towards ZF proteins and other cellular thiols in the context of widespread AgNP exposure.
The study was sponsored by the National Science Centre of Poland, grant no. 2016/21/B/NZ1/02847. The authors thank Wrocław Centre for Networking and Supercomputing from Wrocław University of Technology for access to computational resources. The equipment used was sponsored in part by the Centre for Preclinical Research and Technology (CePT), a project co-sponsored by European Regional Development Fund and Innovative Economy, The National Cohesion Strategy of Poland.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. H. Laity, B. M. Lee and P. E. Wright, Curr. Opin. Struct. Biol., 2001, 11, 39–46 CrossRef CAS PubMed.
- J. M. Berg, J. Biol. Chem., 1990, 265, 6513–6516 CAS.
- J. H. Laity and G. K. Andrews, Arch. Biochem. Biophys., 2007, 463, 201–210 CrossRef CAS PubMed.
- K. Kluska, J. Adamczyk and A. Krężel, Coord. Chem. Rev., 2018, 367, 18–64 CrossRef CAS.
- M. Padjasek, A. Kocyła, K. Kluska, O. Kerber, J. B. Tran and A. Krężel, J. Inorg. Biochem., 2020, 204, 110955 CrossRef PubMed.
- Y. Shi, R. D. Beger and J. M. Berg, Biophys. J., 1993, 64, 749–753 CrossRef CAS PubMed.
- A. Hartwig, M. Asmuss, I. Ehleben, U. Herzer, D. Kostelac, A. Pelzer, T. Schwerdtle and A. Bürkle, Environ. Health Perspect., 2002, 110, 797–799 CrossRef CAS PubMed.
- M. Padjasek, M. Maciejczyk, M. Nowakowski, O. Kerber, M. Pyrka, W. Koźmiński and A. Kreżel, Chemistry, 2020 DOI:10.1002/chem.201904942.
- S. M. Quintal, Q. A. dePaula and N. P. Farrell, Metallomics, 2011, 3, 121–139 RSC.
- A. Witkiewicz-Kucharczyk and W. Bal, Toxicol. Lett., 2006, 162, 29–42 CrossRef CAS PubMed.
- W. Bal, T. Schwerdtle and A. Hartwig, Chem. Res. Toxicol., 2003, 16, 242–248 Search PubMed.
- S. M. Modak, L. Sampath and C. L. Fox Jr, J. Burn Care Rehabil., 1988, 9, 359–363 CrossRef CAS.
- S. Eckhardt, P. S. Brunetto, J. Gagnon, M. Priebe, B. Giese and K. M. Fromm, Chem. Rev., 2013, 113, 4708–4754 CrossRef CAS.
- P. L. Nadworny, J. Wang, E. E. Tredget and R. E. Burnell, Nanomedicine, 2008, 4, 241–251 CrossRef CAS PubMed.
- K. C. Bhol and P. J. Schechter, Dig. Dis. Sci., 2007, 52, 2732–2742 CrossRef CAS.
- S. J. Allison, M. Sadiq, E. Baronou, P. A. Cooper, C. Dunnill, N. T. Georgopoulos, A. Latif, S. Shepherd, S. D. Shnyder, I. J. Stratford, R. T. Wheelhouse, C. E. Willans and R. M. Phillips, Cancer Lett., 2017, 403, 98–107 CrossRef CAS PubMed.
- D. F. Shen, S. S. Wu, R. R. Wang, Q. Zhang, Z. J. Ren, H. Liu, H. D. Guo and G. G. Gao, Small, 2016, 12, 6153–6159 CrossRef CAS.
- K. Chaloupka, Y. Malam and A. M. Seifalian, Trends Biotechnol., 2010, 28, 580–588 CrossRef CAS.
- R. A. Bapat, T. V. Chaubal, C. P. Joshi, P. R. Bapat, H. Choudhury, M. Pandey, B. Gorain and P. Kesharwani, Mater. Sci. Eng., C, 2018, 91, 881–898 CrossRef CAS PubMed.
- M. Marchioni, P. H. Jouneau, M. Chevallet, I. Michaud-Soret and A. Deniaud, Coord. Chem. Rev., 2018, 364, 118–136 CrossRef CAS.
- M. van der Zande, R. J. Vandebriel, E. Van Doren, E. Kramer, Z. Herrera Rivera, C. S. Serrano-Rojero, E. R. Gremmer, J. Mast, R. J. Peters, P. C. Hollman, P. J. Hendriksen, H. J. Marvin, A. A. Peijnenburg and H. Bouwmeester, ACS Nano, 2012, 6, 7427–7442 CrossRef CAS PubMed.
- K. D. Rosenman, A. Moss and S. Kon, J. Occup. Med., 1979, 21, 430–435 CAS.
- G. D. DiVincenzo, C. J. Giordano and L. S. Schriever, Int. Arch. Occup. Environ. Health, 1985, 56, 207–215 CrossRef CAS PubMed.
- P. L. Drake and K. J. Hazelwood, Ann. Occup. Hyg., 2005, 49, 575–585 CAS.
- G. Veronesi, A. Deniaud, T. Gallon, P. H. Jouneau, J. Villanova, P. Delangle, M. Carriere, I. Kieffer, P. Charbonnier, E. Mintz and I. Michaud-Soret, Nanoscale, 2016, 8, 17012–17021 RSC.
- V. De Matteis, M. A. Malvindi, A. Galeone, V. Brunetti, E. De Luca, S. Kote, P. Kshirsagar, S. Sabella, G. Bardi and P. P. Pompa, Nanomedicine, 2015, 11, 731–739 CrossRef CAS PubMed.
- X. Jiang, T. Miclăuş, L. Wang, R. Foldbjerg, D. S. Sutherland, H. Autrup, C. Chen and C. Beer, Nanotoxicology, 2014, 9, 181–189 CrossRef PubMed.
- G. Veronesi, C. Aude-Garcia, I. Kieffer, T. Gallon, P. Delangle, N. Herlin-Boime, T. Rabilloud and M. Carriere, Nanoscale, 2015, 7, 7323–7330 RSC.
- B. A. Krizek, B. T. Amann, V. J. Kilfoil, D. L. Merkle and J. M. Berg, J. Am. Chem. Soc., 1991, 113, 4518–4523 CrossRef CAS.
- B. A. Krizek, D. L. Merkle and J. M. Berg, Inorg. Chem., 1993, 32, 937–940 CrossRef CAS.
- A. N. Besold, L. R. Widger, F. Namuswe, J. L. Michalek, S. L. Michael and D. P. Goldberg, Mol. BioSyst., 2016, 12, 1183–1193 RSC.
- A. J. Żelazowski, Z. Gasyna and M. J. Stillman, J. Biol. Chem., 1989, 264, 17091–17099 Search PubMed.
- A. J. Żelazowski and M. J. Stillman, Inorg. Chem., 1992, 31, 3363–3370 CrossRef.
- B. O. Leung, F. Jalilehvand, V. Mah, M. Parvez and Q. Wu, Inorg. Chem., 2013, 52, 4593–4602 CrossRef CAS PubMed.
- G. Park, Z. N. Amaris, M. K. Eiken, K. V. Baumgartner, K. A. Johnston, M. A. Williams, J. G. Marckwordt, J. E. Millstone, K. E. Splanc and K. E. Wheeler, Environ. Sci.: Nano, 2019, 6, 2367–2378 RSC.
- H. Mattapalli, W. B. Monteith, C. S. Burns and A. S. Danell, J. Am. Soc. Mass Spectrom., 2009, 20, 2199–2205 CrossRef CAS PubMed.
- Z. Du, R. E. F. de Paiva, K. Nelson and N. P. Farrell, Angew. Chem., Int. Ed., 2017, 56, 4464–4467 CrossRef CAS PubMed.
- A. Kocyła, A. Pomorski and A. Krężel, J. Inorg. Biochem., 2015, 152, 82–92 CrossRef PubMed.
- Y. M. Lee and C. Lim, J. Am. Chem. Soc., 2011, 133, 8691–8703 CrossRef CAS PubMed.
- C. Abbehausen, Metallomics, 2019, 11, 15–28 RSC.
- L. H. Wang, X. Y. Yang, X. Zhang, K. Mihalic, Y. X. Fan, W. Xiao, O. M. Howard, E. Appella, A. T. Maynard and W. L. Farrar, Nat. Med., 2004, 10, 40–47 CrossRef CAS PubMed.
- K. Kluska, J. Adamczyk and A. Krężel, Metallomics, 2018, 10, 248–263 RSC.
- A. Miłoch and A. Krężel, Metallomics, 2014, 6, 2015–2024 RSC.
- T. Kochańczyk, M. Nowakowski, D. Wojewska, A. Kocyła, A. Ejchart and W. Koźmiński, Sci. Rep., 2016, 6, 36346 CrossRef PubMed.
- M. Beck, Pure Appl. Chem., 1987, 59, 1703–1720 CAS.
- O. Sénèque and J. M. Latour, J. Am. Chem. Soc., 2010, 132, 17760–17774 CrossRef PubMed.
- V. Tardillo-Suarez, E. Karepina, B. Gallet, C. Cottet-Rousselle, M. Chevallet, P. Charbonnier, C. Moriscot, I. Michaud-Soret, W. Bal, A. Fuchs, R. Tucoulou, P.-H. Jouneau, G. Veronesi and A. Deniaud, BioRxiv, 2019 DOI:10.1101/825919.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- P. Tiwary and M. Parrinello, J. Phys. Chem. B, 2015, 119, 736–742 CrossRef CAS PubMed.
- W. Li, J. Zhang, J. Wang and W. Wang, J. Am. Chem. Soc., 2008, 130, 892–900 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc09418k |
| This journal is © The Royal Society of Chemistry 2020 |