
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A double-dysprosocenium single-molecule magnet bound together with neutral ligands†
Peter
Evans
 ,
Daniel
Reta
,
Conrad A. P.
Goodwin
,
Fabrizio
Ortu
,
Nicholas F.
Chilton
and
David P.
Mills
*
,
Daniel
Reta
,
Conrad A. P.
Goodwin
,
Fabrizio
Ortu
,
Nicholas F.
Chilton
and
David P.
Mills
*
Department of Chemistry, School of Natural Sciences, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.mills@manchester.ac.uk
First published on 16th April 2020
Abstract
A dinuclear dysprosocenium dication has been synthesised that is bound together by weak interactions between {Dy(Cp*)2}+ fragments and neutral NEt3AlMe3 molecules. The axiality of the Dy3+ crystal fields are perturbed by these equatorial interactions but a relatively large effective barrier to magnetisation reversal of 860(60) cm−1 and magnetic hysteresis up to 12 K are observed.
The potential use of single-molecule magnets (SMMs) in high-density data storage devices requires them to retain magnetisation on long timescales (i.e. months to years) at liquid nitrogen temperatures, but the best SMMs currently only have relaxation times of a few hours at liquid helium temperatures.1,2 The first lanthanide (Ln) SMM was reported in 20033 and this field has grown rapidly4 since design criteria to build Ln SMMs with large effective barriers to magnetisation reversal (Ueff) were popularised in 2011.5 For Dy3+ SMMs an axial ligand field is required to maximise Ueff,6 but a report of magnetic hysteresis at TH = 60 K in the isolated dysprosocenium cation [Dy(C5H2tBu3-1,2,4)2]+ in 2017 showed that a combination of charge-dense π-aromatic rings and an absence of equatorial anion interactions can provide high temperature magnetic remanence.2a This report also predicted that the removal of substituted cyclopentadienyl ring C–H bonds would provide even higher hysteresis temperatures, and this hypothesis has now been proved correct, with TH up to 80 K being reported in the last two years in peralkylated {Ln(CpR)2}-based SMMs.2b–d
Prior to the synthesis of [Dy(C5H2tBu3-1,2,4)2]+, much dysprosocene-based SMM research focused on the less bulky {DyCp*2} (Cp* = C5Me5) framework7 and the magnetic properties of a hypothetical [DyCp*2]+ cation were calculated.8 Two strategies were investigated in parallel to increase Ueff values for {DyCp*2} systems; in one approach the equatorial ligand field strength was progressively decreased,9 and a second tactic employed anionic radical ligands to promote magnetic exchange coupling between {DyCp*2} units.10 By utilising weakly coordinating equatorial anions Ueff = 662 cm−1 and TH < 1.8 K was found for [Dy(Cp*)2{μ-Fe(Cp)(CO)2}]2,9b and by blocking the equatorial sites with neutral Lewis base ligands a Ueff of 546 cm−1 and a TH of 5.2 K was found for [Dy(Cp*)2(NH3)2][BPh4].9c Given the improvement of the magnetic properties with weakening equatorial ligand fields, we set out to probe the magnetic behaviour of {Dy(Cp*)2}+ fragments with even weaker equatorial interactions; we report our results herein.
The dinuclear Ln3+ complexes [{Ln(Cp*)2(μ-Me3AlNEt3)}2][Al{OC(CF3)3}4]2 (1-Ln, Ln = Dy, Y) were prepared by the protonolysis reactions of parent [{Ln(Cp*)2(μ-Me4Al)}2]11 with two equivalents of [NEt3H][Al{OC(CF3)3}4]12 in benzene at room temperature; the elimination of two equivalents of methane provides an entropic driving force (Scheme 1). After stirring reaction mixtures overnight the solvent was removed in vacuo and crystals of 1-Ln were grown from chlorobenzene in good yields (1-Dy: 76%; 1-Y: 75%). A doped sample 1-Dy@Y containing a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95 ratio of Dy:Y (composition confirmed by ICP-OES) was synthesised by recrystallizing a mixture of 1-Dy and 1-Y. [{Ln(Cp*)2(μ-Me4Al)}2]11 and [NEt3H][Al{OC(CF3)3}4]12 were prepared by adapting literature procedures; see the ESI† for full details. We selected [NEt3H][Al{OC(CF3)3}4] as an abstraction agent as it was recently used to synthesise a bis-phospholyl dysprosium cation,12 and [B(C6F5)4]− anions have previously been shown to coordinate to a {Sc(Cp*)2}+ fragment.13 Elemental analysis results for 1-Ln·3C6H5Cl gave reproducibly low carbon values due to carbide formation (see ESI,† Section 1).
:95 ratio of Dy:Y (composition confirmed by ICP-OES) was synthesised by recrystallizing a mixture of 1-Dy and 1-Y. [{Ln(Cp*)2(μ-Me4Al)}2]11 and [NEt3H][Al{OC(CF3)3}4]12 were prepared by adapting literature procedures; see the ESI† for full details. We selected [NEt3H][Al{OC(CF3)3}4] as an abstraction agent as it was recently used to synthesise a bis-phospholyl dysprosium cation,12 and [B(C6F5)4]− anions have previously been shown to coordinate to a {Sc(Cp*)2}+ fragment.13 Elemental analysis results for 1-Ln·3C6H5Cl gave reproducibly low carbon values due to carbide formation (see ESI,† Section 1).
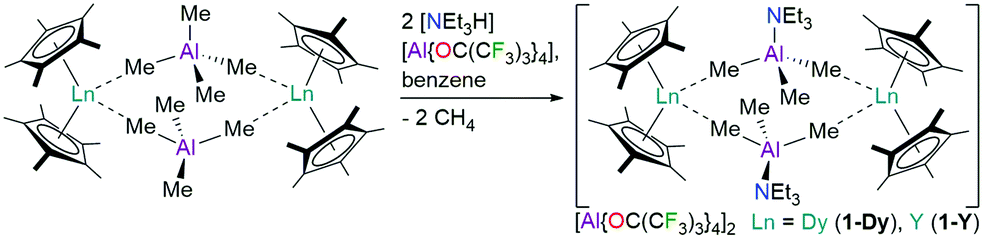 | ||
| Scheme 1 Synthesis of 1-Ln. | ||
Complexes 1-Ln are not soluble in non-polar solvents and decompose rapidly in CD2Cl2; as we avoided the use of coordinating solvents that could displace weakly-bound {(μ-Me3AlNEt3)} moieties our NMR characterisation of 1-Ln was limited to C6D5Cl solutions, though these also decomposed into unidentified species over several hours (see ESI,† Section 4). This precluded more detailed analysis of these equilibria by diffusion ordered 1H NMR spectroscopy and variable temperature (VT) experiments. However, a monomer–dimer equilibrium was previously established for C7D8 solutions of [{Y(Cp*)2(μ-Me4Al)}2] by VT 1H NMR spectroscopy,11a thus it is reasonable to assume that similar processes are in operation here for 1-Ln. The paramagnetism of 1-Dy thwarted its assignment by NMR spectroscopy apart from the presence of two signals in the 19F{1H} NMR spectrum at −81.40 and −80.28 ppm.
The solid state structures of 1-Ln were determined by single crystal XRD; 1-Dy·3C6H5Cl is depicted in Fig. 1 and isomorphous 1-Y·3C6H5Cl is shown in the ESI† together with [{Ln(Cp*)2(μ-Me4Al)}2] (Ln = Y, Dy). Each [{Dy(Cp*)2(μ-Me3AlNEt3)}2]2+ dication in 1-Dy is associated with two [Al{OC(CF3)3}4]− counterions and three co-crystallized chlorobenzene molecules in the lattice; the structural parameters of these latter components are unremarkable and there are no significant interactions with the dication. The dimeric structures in 1-Ln and [{Ln(Cp*)2(μ-Me4Al)}2] lie about inversion centres, with the two Ln3+ cations in each example exhibiting bent metallocene motifs, e.g.1-Dy: Cpcentroid⋯Dy⋯Cpcentroid = 138.03(13)°; Dy⋯Cpcentroid(av.) = 2.343 Å; Dy⋯Cring = 2.580(8)–2.665(7) Å (mean 2.634 Å). The Dy3+ coordination spheres are each completed by two Me groups of two separate {μ-Me3AlNEt3-κ1:κ1-C,C′} moieties (mean Dy⋯CMe: 2.794(8) Å) to give distorted pseudo-tetrahedral Dy3+ geometries (CMe⋯Dy⋯CMe: 82.8(2)°; range Cpcentroid⋯Dy⋯CMe: 103.6(2)–107.8(2)°) and a diamond-shaped Dy2Al2Me4 core, with two Dy and two Al vertices and Me groups on each face; the two {Dy(Cp*)2}+ units are orthogonal to the Dy2Al2 mean plane and the intramolecular Dy⋯Dy distance is 7.365(2) Å. As expected, the core Al–C bond lengths (mean 2.011(8) Å) are longer than the external Al–C distances (1.952(6) Å) as the latter do not interact with the Dy3+ centres.
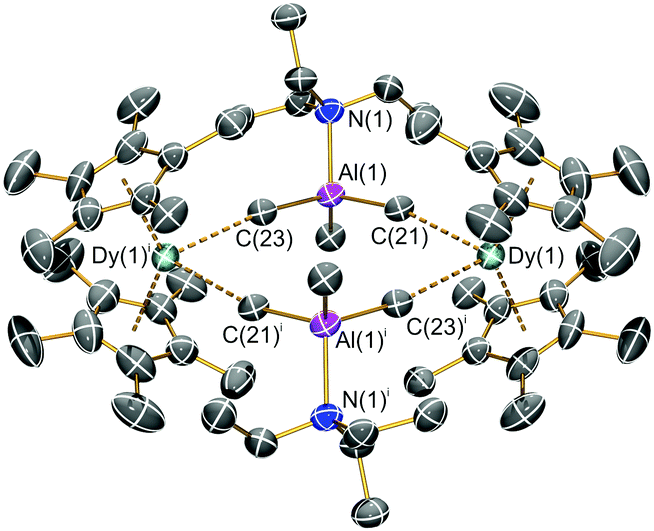 | ||
| Fig. 1 Molecular structure of the dication of 1-Dy·3C6H5Cl, shown at the 50% probability level; [Al{OC(CF3)3}4]− counterions and lattice solvent are omitted for clarity. Symmetry operation used to generate equivalent atoms: i = 2 − x, 1 − y, −z. Selected bond lengths (Å) and angles (°): Dy(1)⋯Cpcentroid1:2.388(3); Dy(1)⋯Cpcentroid2:2.348(3); Dy(1)⋯C(21): 2.800(6); Dy(1)⋯C(23)i: 2.788(6); range Dy(1)–Cring: 2.580(8)–2.665(7); Cpcentroid1⋯Dy(1)⋯Cpcentroid2:138.03(13); Cpcentroid1⋯Dy(1)⋯C(21): 103.74(14); Cpcentroid1⋯Dy(1)⋯C(23)i: 107.77(15); Cpcentroid2⋯Dy(1)⋯C(21): 107.29(14); Cpcentroid2⋯Dy(1)⋯C(23)i: 103.56(15); C(21)⋯Dy(1)⋯C(23)i: 82.80(17); Dy(1)⋯C(21)–Al(1): 169.9(3). | ||
The equatorial interactions in 1-Dy likely arise from the electrostatic attraction of the Dy3+ centres with Cδ−–Alδ+ partial dipoles. The two shortest equatorial Dy⋯H distances that result from these interactions are 2.407 and 2.482 Å, which are shorter than the two shortest equatorial Dy⋯H distances in [Dy(C5H2tBu3-1,2,4)2][B(C6F5)4] (mean 2.499 Å).2a Each {μ-Me3AlNEt3} unit in 1-Dy also exhibits ca. 10 C⋯C distances < 5 Å between its Me and Et groups and the Me groups of the Cp* ligands; it is postulated that the combined strength of these inter-ligand London dipole interactions is also crucial for binding the dication together as such forces are of particular importance in low-coordinate metal complexes.14 Five-coordinate C atoms have been invoked in related Ln complexes,15 but we do not comment on this aspect here. The bulk features of 1-Dy are most reminiscent of the dinuclear Dy3+ complex [Dy(Cp*)2{μ-Fe(Cp)(CO)2}]2 (mean Dy⋯Cpcentroid: 2.342(10) Å; Cpcentroid⋯Dy⋯Cpcentroid: 141.5(3)°; Dy⋯Dy: 7.14(2) Å),9b but this contains bridging {μ-Fe(Cp)(CO)2-κ1:κ1-O,O′}− anions rather than the neutral {μ-Me3AlNEt3} units in 1-Dy. The metrical parameters about the Dy3+ centres of 1-Dy are similar to those seen for [Dy(Cp*)2(NH3)2][BPh4] (mean Dy⋯Cpcentroid: 2.350(3) Å; Cpcentroid⋯Dy⋯Cpcentroid: 140.25(6)°),9c which also contains neutral donors in the equatorial plane, though the dative Dy–N distances in the literature complex (mean 2.47(1) Å)9c are shorter than the two equatorial Dy⋯CMe distances in 1-Dy.
Although the Dy3+ centres of 1-Dy have less axial geometries than several decamethyldysprosocene-based complexes in the literature,9,10 we envisioned that the combination of peralkylated Cp ligands and neutral equatorial ligands bound only through electrostatic interactions could endow favourable SMM properties for 1-Dy. The magnetic susceptibility temperature product (χMT) of a microcrystalline sample of 1-Dy suspended in eicosane is 27.72 cm3 K mol−1 at 300 K (Fig. S22, ESI†), as determined by SQUID magnetometry. This high-temperature value is in excellent agreement with twice the Dy3+ free-ion Curie value (14.17 cm3 K mol−1)16 and the predicted value from ab initio calculations (13.76 cm3 K mol−1 at 300 K, see below). As expected, the experimental χMT value decreases monotonically down to 24.16 cm3 K mol−1 at 10 K, as excited CF states are thermally depopulated (ab initio predicted 12.34 cm3 K mol−1), followed by a sharp decrease below 10 K due to the onset of slow dynamics; this is below the temperature at which we observe bifurcation of the field-cooled/zero-field-cooled (FC/ZFC) data (22 K, Fig. S34, ESI†), but is very close to the local ZFC maximum (8 K, Fig. S31, ESI†).
The dynamic magnetic behaviour of 1-Dy was probed by low temperature ac susceptibility measurements in zero-field. These data show slow relaxation of magnetisation, with clear in-phase and out-of-phase maxima between 25 and 68 K (Fig. S39, ESI†). We have used CC-FIT217 to fit the data to a generalised Debye model, where we can obtain estimated standard deviations for the relaxation times (Fig. 2). Below 25 K, the maxima in the ac data fall out of the range of accessible frequencies and we turn to magnetisation decay measurements to access the lowest temperature regime. The DC magnetisation decay data were fitted with stretched exponentials (Table S4, ESI†), where we have used a similar model to that employed by CC-FIT2 to obtain estimated standard deviations for the relaxation times.12
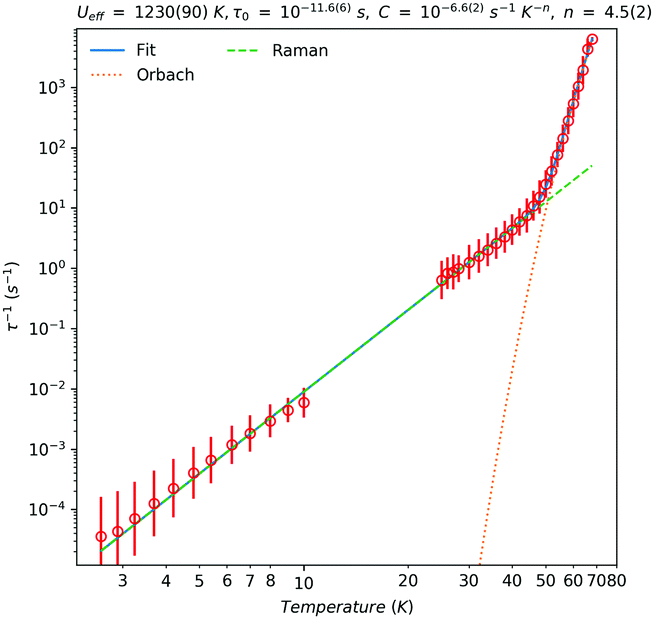 | ||
| Fig. 2 Temperature dependence of the magnetic relaxation rate of 1-Dy. Red circles are the relaxation rates extracted from AC susceptibility data (high temperature) and DC magnetisation decay data (low temperature); solid red lines are estimated standard deviations. The solid green line is given by τ−1 = τ0−1 exp[−Ueff/kT] + CTn, the dashed green line is given by τ−1 = τ0−1 exp[−Ueff/kT] and the dotted orange line is given by τ−1 = CTn with Ueff = 860 cm−1 (1230 K), τ0 = 2.51 × 10−12 s, C = 2.51 × 10−7 s−1 K−n and n = 4.5. | ||
In the high-temperature region (T > 46 K), an Orbach process dominates (τ−1 = τ0−1 exp[−Ueff/kT]) with an associated effective barrier of Ueff = 860(60) cm−1 (1230(90) K) and τ0 = 10−11.6(6) s (ca. 2.5 × 10−12 s); cf. structurally similar [Dy(Cp*)2{μ-Fe(Cp)(CO)2}]2 which has Ueff = 662 cm−1 (952 K),9b though the current record Ueff value is 1541 cm−1 (2217 K) for [Dy(C5iPr5)(Cp*)][B(C6F5)4].2c Below 46 K the relaxation rate is governed by a Raman process (τ−1 = CTn) with C = 10−6.6(2) s−1 K−n (ca. 2.5 × 10−7 s−1 K−n) and n = 4.5(2). The value of n is higher than typical values extracted for isolated Ln metallocenium cations (e.g. [Dy(Cpttt)2]+, n = 2.15;2a [Dy(C5iPr5)(Cp*)]+, n = 3.02c), likely due to the influence of the equatorial ligand interactions in 1-Dy.18 From these data we can directly measure a 100 s blocking temperature of 10.5 K.
Open magnetic hysteresis loops were observed for 1-Dy between 2 and 12 K at a sweep rate of ca. 20 Oe s−1 around the zero-field region (Fig. 3 and Fig. S27, ESI†); the waist-restricted “butterfly” shapes of these loops are typical for Ln3+ SMMs and arise due to strongly field-dependent relaxation near zero field.4 The highest temperature at which we observe an open loop is TH = 12 K; cf. 6.5 K at ca. 20 Oe s−1 for [{Dy(Cp*)2}2(μ-2,2′-bipyrimidine˙)][BPh4]10a and 5.2 K at ca. 20 Oe s−1 for [Dy(Cp*)2(NH3)2][BPh4].9c The current record value of TH = 80 K at ca. 25 Oe s−1 is seen for [Dy(C5iPr5)(Cp*)][B(C6F5)4],2c thus it is evident that even the very weak neutral equatorial contacts in 1-Dy promote faster magnetic relaxation than is observed for isolated [Dy(CpR)2]+ cations.2
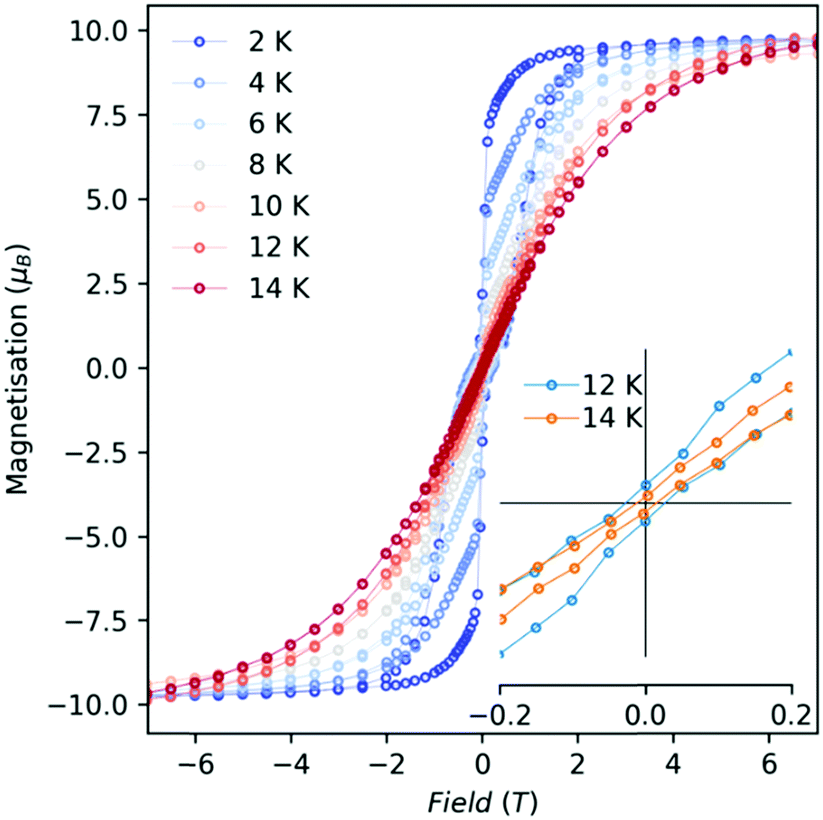 | ||
| Fig. 3 Magnetic hysteresis of solid 1-Dy, measured with a mean field sweep rate of 21(9) Oe s−1 for |H| < 10 kOe, 49(12) Oe s−1 for 10 < |H| < 20 kOe, and 88(17) Oe s−1 for 20 < |H| < 70 kOe. Hysteresis loops recorded from 2–14 K in 2 K steps; inset shows zoomed zero-field region. | ||
To confirm our implicit assumption that stronger equatorial interactions due to the anionic bridges in the precursor complex [{Dy(Cp*)2(μ-Me4Al)}2] result in a much poorer SMM, we have collected hysteresis and AC relaxation data for this compound. Hysteresis is only open up to 2 K (Fig. S24 and S28, ESI†) and relaxation rates are approximately an order of magnitude faster than for 1-Dy (Fig. S41 and S42, ESI†). To probe the effect of intramolecular exchange interactions on magnetic relaxation in 1-Dy, we have examined two doped samples prepared by different routes: 1-Dy@Y (scrambling of 1-Dy and 1-Y in a 5:95 ratio in chlorobenzene) and 1-Dy@Y_2 (reaction of a mixture of [{Ln(Cp*)2(μ-Me4Al)}2] (Ln = 5Dy:95Y) with [NEt3H] [Al{OC(CF3)3}4]). Both samples give practically identical data to one-another (Fig. S28, S29, S32, S33, S35 and S36, ESI†), ruling out any substantial contributions from pure {Dy2} dimers in either case. The doped samples show similar, yet slightly faster, relaxation rates in zero-field compared to 1-Dy (Fig. S43 (ESI†); note that we cannot collect AC susceptibility data on these doped samples due to low magnetic moments at high temperatures),19 and also show open hysteresis up to at least 8 K (Fig. S28, ESI;† we cannot reliably measure the low-moments of these samples near zero-field at higher temperatures). The doped samples also show slightly sharper steps near zero-field in the 2 K hysteresis loops compared to 1-Dy (Fig. S30, ESI†), where the drop occurs much closer to zero-field; this, along with faster zero-field relaxation rates, suggests that the intramolecular interactions in 1-Dy provide a small exchange-bias that reduces relaxation in zero-field.
An immobilised powdered sample of 1-Dy was found to be EPR silent at X-band at 5 K, thus we cannot measure the Dy⋯Dy interaction directly here.20 However, given the core-like nature of 4f electrons and the large separation of the two ions, we suggest that this interaction is purely dipolar. Indeed, simple dipolar calculations based on g-values obtained from complete active space self-consistent field spin–orbit (CASSCF-SO) calculations (see below) suggest a zero-field exchange splitting of ca. 0.42 cm−1 in 1-Dy, which would require a field of ca. 0.09 T to bring the states into resonance. This is in good agreement with the significant loss of magnetisation for 1-Dy between 0.05 and 0.1 T (ESI,† Section S7).17 Using a purely electrostatic model, equatorial ligands in Ln metallocene frameworks tend to increase Dy⋯Cpcentroid distances and decrease Cpcentroid⋯Ln⋯Cpcentroid angles, which disrupts the axiality of the ligand field, lowering the Ueff value and the purity of the mJ = ±15/2 ground state. The effect of molecular geometry and ligand vibrational modes on TH is a more complex relationship to fully disentangle. To probe the electronic structure of 1-Dy we performed CASSCF-SO calculations on a model of 1-Dy, where one of the Dy3+ centres was replaced by Lu3+ for computational efficiency (Fig. S42; see ESI† for details). The predicted Ueff for this model was 842 cm−1 (1211 K), in excellent agreement with experimentally obtained values, thus we confidently assign the Orbach relaxation pathway as proceeding via the fifth excited doublet.
To conclude, the double-decamethyldysprosocenium dication [{Dy(Cp*)2(μ-Me3AlNEt3)}2]2+ is held together by multiple weak dipolar interactions between the cationic {Dy(Cp*)2}+ fragments and neutral {μ-Me3AlNEt3} moieties. This dication fulfils the design criteria for high-performance dysprosocenium SMMs of an axial ligand field, rigid charge-dense π-aromatic rings, an absence of ring C–H bonds and a lack of equatorial anion interactions.2a However, magnetic relaxation promoted by the equatorial electrostatic contacts of the {μ-NEt3AlMe3} adducts have limited both the effective barrier to magnetisation reversal and maximum hysteresis temperature compared to dysprosocenium cations that lack such interactions. An alternative synthetic route is required to prepare an isolated decamethyldysprosocenium cation, which could represent the pinnacle of isolated [Dy(CpR)2]+ SMMs as it contains the smallest peralkylated CpR rings. Although the [Dy(Cp*)2]+ cation would exhibit short Dy⋯Cpcentroid distances, the Cpcentroid⋯Dy⋯Cpcentroid angle is more difficult to predict and is also crucial for determining its magnetic parameters.8
This work was supported by the EPSRC (EP/R002605X/1 for P. E., EP/P002560/1 for D. R. and F. O., Doctoral Prize Fellowship to C. A. P. G.), and the University of Manchester. N. F. C. thanks the University of Manchester for a Presidential Fellowship and The Royal Society for a University Research Fellowship. We thank B. L. L. Réant for providing Cp*H, Prof. R. E. P. Winpenny for stimulating discussions, the EPSRC UK National Electron Paramagnetic Resonance Service for access to the EPR facility and the SQUID magnetometer, and the University of Manchester for access to the Computational Shared Facility. Research data files supporting this publication are available from Mendeley Data at DOI: http://10.17632/vzwmzthy3z.2.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, 2006 Search PubMed.
- (a) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439 CrossRef CAS PubMed; (b) K. R. McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, Chem. Sci., 2018, 9, 8492 RSC; (c) F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400 CrossRef CAS PubMed; (d) C. A. Gould, K. R. McClain, J. Yu, T. J. Groshens, F. Furche, J. R. Long and B. G. Harvey, J. Am. Chem. Soc., 2019, 141, 12967 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS PubMed.
- R. A. Layfield and M. Murugesu, Lanthanides and Actinides in Molecular Magnetism, Wiley-VCH, 2015 Search PubMed.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC.
- See, for example: Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071 CrossRef CAS PubMed.
- B. M. Day, F.-S. Guo and R. A. Layfield, Acc. Chem. Res., 2018, 51, 1880 CrossRef CAS PubMed.
- Y.-S. Meng, Y.-Q. Zhang, Z.-M. Wang, B.-W. Wang and S. Gao, Chem. – Eur. J., 2016, 22, 12724 CrossRef CAS PubMed.
- (a) S. Demir, J. M. Zadrozny and J. R. Long, Chem. – Eur. J., 2014, 20, 9524 CrossRef CAS PubMed; (b) T. Pugh, N. F. Chilton and R. A. Layfield, Angew. Chem., Int. Ed., 2016, 55, 11082 CrossRef CAS PubMed; (c) S. Demir, M. D. Boshart, J. F. Corbey, D. H. Woen, M. I. Gonzalez, J. W. Ziller, K. R. Meihaus, J. R. Long and W. J. Evans, Inorg. Chem., 2017, 56, 15049 CrossRef CAS PubMed; (d) R. Collins, M. J. Heras Ojea, A. Mansikkamäki, J. Tang and R. A. Layfield, Inorg. Chem., 2020, 59, 642 CrossRef CAS PubMed.
- (a) S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546 CrossRef CAS PubMed; (b) S. Demir, M. Nippe, M. I. Gonzalez and J. R. Long, Chem. Sci., 2014, 5, 4701 RSC; (c) F.-S. Guo and R. A. Layfield, Chem. Commun., 2017, 53, 3130 RSC; (d) C. A. Gould, L. E. Darago, M. I. Gonzalez, S. Demir and J. R. Long, Angew. Chem., Int. Ed., 2017, 56, 10103 CrossRef CAS PubMed.
- (a) M. A. Busch, R. Harlow and P. L. Watson, Inorg. Chim. Acta, 1987, 140, 15 CrossRef CAS; (b) P. L. Watson and T. Herskovitz, Am. Chem. Soc. Symp. Ser., 1983, 212, 459 CrossRef CAS; (c) P. L. Watson, J. Am. Chem. Soc., 1983, 105, 6491 CrossRef CAS.
- P. Evans, D. Reta, G. F. S. Whitehead, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2019, 141, 19935 CrossRef CAS PubMed.
- A. Berkefeld, W. E. Piers, M. Parvez, L. Castro, L. Maron and O. Eisenstein, J. Am. Chem. Soc., 2014, 134, 10843 CrossRef PubMed.
- D. J. Liptrot and P. P. Power, Nat. Rev. Chem., 2017, 1, 0004 CrossRef.
- See, for example: (a) C. J. Burns and R. A. Andersen, J. Am. Chem. Soc., 1987, 109, 5853 CrossRef CAS; (b) W. T. Klooster, R. S. Lu, R. Anwander, W. J. Evans, T. F. Koetzle and R. Bau, Angew. Chem., Int. Ed., 1998, 37, 1268 CrossRef CAS , and references cited therein.
- D. A. Atwood, The Rare Earth Elements: Fundamentals and Applications, John Wiley and Sons Ltd., 2012 Search PubMed.
- D. Reta and N. F. Chilton, Phys. Chem. Chem. Phys., 2019, 21, 23567 RSC.
- (a) C. A. P. Goodwin, D. Reta, F. Ortu, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2017, 139, 18714 CrossRef CAS PubMed; (b) C. A. P. Goodwin, D. Reta, F. Ortu, J. Liu, N. F. Chilton and D. P. Mills, Chem. Commun., 2018, 54, 9182 RSC.
- (a) F. Habib, P.-H. Lin, J. Long, I. Korobkov, W. Wernsdorfer and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 8830 CrossRef CAS PubMed; (b) Y.-N. Guo, G.-F. Xu, W. Wernsdorfer, L. Ungur, Y. Guo, J. Tang, H.-J. Zhang, L. F. Chibotaru and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 11948 CrossRef CAS PubMed; (c) J. Xiong, H.-Y. Ding, Y.-S. Meng, C. Gao, X.-J. Zhang, Z.-S. Meng, Y.-Q. Zhang, W. Shi, B.-W. Wang and S. Gao, Chem. Sci., 2017, 8, 1288 RSC; (d) E. Moreno Pineda, G. Taran, W. Wernsdorfer and M. Ruben, Chem. Sci., 2019, 10, 5138 RSC.
- (a) E. Moreno Pineda, N. F. Chilton, R. Marx, M. Dörfel, D. O. Sells, P. Neugebauer, S.-D. Jiang, D. Collison, J. van Slageren, E. J. L. McInnes and R. E. P. Winpenny, Nat. Commun., 2014, 5, 5243 CrossRef CAS PubMed; (b) M. J. Giansiracusa, E. Moreno-Pineda, R. Hussain, R. Marx, M. Martínez Prada, P. Neugebauer, S. Al-Badran, D. Collison, F. Tuna, J. van Slageren, S. Carretta, T. Guidi, E. J. L. McInnes, R. E. P. Winpenny and N. F. Chilton, J. Am. Chem. Soc., 2018, 140, 2504 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic details, crystallography, NMR and FTIR spectra, magnetism and ab initio calculations. CCDC 1964114–1964119. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08945d |
| This journal is © The Royal Society of Chemistry 2020 |