
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent SAM analogues for methyltransferase based DNA labeling†
Vince
Goyvaerts
 a,
Sven
Van Snick
a,
Laurens
D'Huys
a,
Raffaele
Vitale
ab,
Milena
Helmer Lauer
a,
Su
Wang
ac,
Volker
Leen
a,
Wim
Dehaen
a and
Johan
Hofkens
*ad
a,
Sven
Van Snick
a,
Laurens
D'Huys
a,
Raffaele
Vitale
ab,
Milena
Helmer Lauer
a,
Su
Wang
ac,
Volker
Leen
a,
Wim
Dehaen
a and
Johan
Hofkens
*ad
aLaboratory of Molecular Imaging and Photonics, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: johan.hofkens@kuleuven.be
bLASIR CNRS, Université de Lille, 59655 Villeneuve d’Ascq, France
cLaboratory for Quantitative Biology of the Nucleus, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, SE-171 77 Stockholm, Sweden
dDepartment of Chemistry, Max Planck Institute for Polymer Research, 55128 Mainz, Germany
First published on 6th February 2020
Abstract
In this work, the preparation of new S-adenosyl-L-methionine (SAM) analogues for sequence specific DNA labeling is evaluated. These non-natural analogues, comprising cysteine rather than the natural homolog, were obtained in near quantitative conversions from readily available starting materials without relying on using an excess amount of labor intensive molecules. The synthetic strategy was used to generate fluorescent cofactors, with colours spanning the whole visible spectrum, and their applicability in methyltransferase based optical mapping is shown.
Site-specific DNA labeling holds the potential of unlocking promising advances in DNA-based applications.1 Through modifications of known positions on DNA, researchers are able to extract vital information about the genetic material, making site-specific DNA labeling an indispensable tool for species identification or medical diagnostics.2–4 In the last two decades, several approaches to such site-specific labeling have been developed, mainly relying on hairpin polyamides,5 triplex-helix-forming oligodeoxynucleotides6 or enzyme-directed DNA modifications.7–9
Perhaps one of the most exciting examples of site-specific labeling is DNA methyltransferases (MTases) based labeling. In nature, these enzymes play a key role in the DNA methylation process by catalyzing the transfer of a methyl group from an S-adenosyl-L-methionine (SAM) cofactor onto adenine (N6-position) or cytosine (N4- or C5-position).10 Generally, the catalytic transfer will only occur after the enzyme successfully binds its recognition sequence, ensuring site-specific labeling. While a methyl group is an inert moiety, pioneering work by Weinhold and coworkers shows that MTase enzymes tolerate non-natural cofactors to effectively transfer functionalized labels onto DNA, and such strategies have been developed to introduce (fluorescent) labels which allow for localization,11 capture,12 photolabile caging,13,14 photocrosslinking,15 selective scission,16 … and is a fundamental technology in the field of optical mapping.
In variants of this technology, replacing the methionine side chain of SAM with an aziridine ring resulted in functional cofactors with a pending aziridine moiety.17 MTase enzymes are tricked into catalyzing the nucleophilic ring opening, in line with nucleophilic attack by the nucleobase, thus covalently coupling the whole cofactor onto DNA. By introducing a functional group on the nucleobase, these molecules are able to label DNA. This research was later extended by Rajski and co-workers,18 who used reactive nitrogen mustards for in situ aziridine generation. However, during the enzymatic labeling, both research groups noticed a strong affinity of the labeled DNA for the enzymatic binding pocket, which inhibited further turnovers and thus forcing the use of stoichiometric amounts of enzyme to fully label DNA.
This downside prompted Weinhold et al. to develop more efficient cofactors, and a second class called the doubly activated cofactors was evaluated.9 Here, a double or triple bond is introduced at the β-position to the sulfonium center as part of the functionalized label which is transferred by the MTase enzymes. As only the label is transferred, this class is less affected by enzyme inhibition. The unsaturated bond stabilizes the transition state in the transfer reaction, and hence, no enzymatic transfer was observed for saturated labels.
Commonly, doubly activated cofactors are used to transfer a chemically reactive group, e.g. amine,19 azide20 or alkyne,21,22 followed by conjugation to a fluorescent dye.23–26 This class was rapidly adopted by the research community and extended with synthetic modifications introduced on the sulfonium center,27–29 the amino acid30,31 or the transferrable linker.32 While this method can be used to quickly introduce different labels, the conjugation of the dye often results in a lower yield. Hence, in pursuit of high degrees of fluorescent labeling, the research was continued to fluorescent SAM analogues which contain the fluorescent molecule and directly transfer this functionality to its substrate.11,33
Unfortunately, the described pathways towards these non-natural cofactors are time consuming and often result in low yields. This is especially true for the final step, where S-adenosyl-L-homocysteine (SAH) is coupled with a highly electrophilic alkyl triflate or allyl bromide and a high excess of this linker (50–200 equivalents) is needed to drive the reaction towards completion (Fig. 1). As the reagent used in large excess actually contains the functionality introduced to the substrate later-on, this imposes significant restraints on the flexibility of the approach. Furthermore, even with this large excess, reported yields do not exceed 25% for a single epimer after HPLC purification.9 Efforts to increase the formation of the correct epimer by a chemoenzymatic synthesis pathway also failed to give reasonable yields for larger systems.31,34
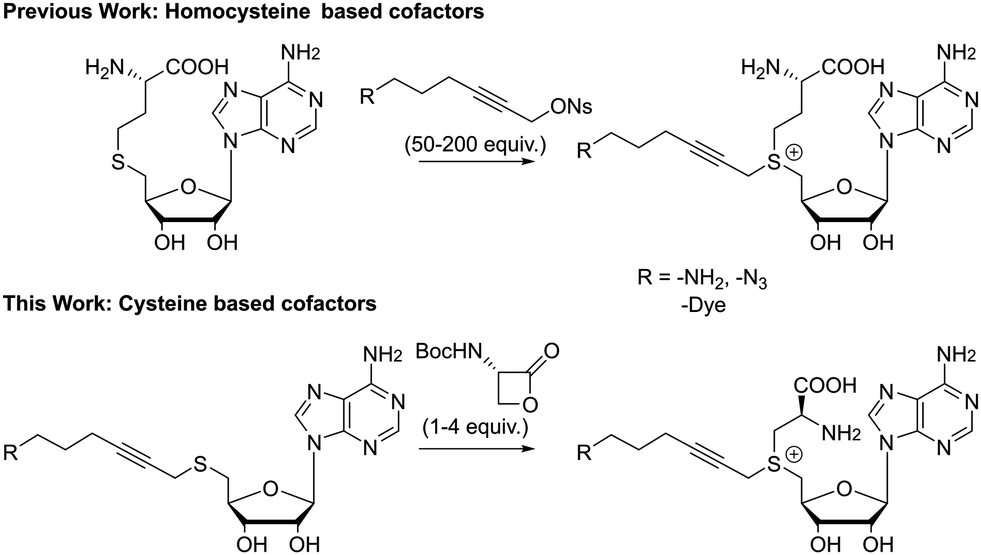 | ||
| Fig. 1 Summary of previous work and present work. | ||
In our pursuit of advanced applications of enzymatic DNA labeling, we were interested in the development of doubly activated cofactors, unhindered by the synthetic issues discussed above. To this end, we developed and evaluated new cysteine based SAM analogues. Our findings show that they are easily accessible and possess comparable transfer behavior as their homocysteine counterparts. The late stage introduction of the amino acid moiety allows to reach functional MTase cofactors without sacrificing labor intensive molecules.
Our first efforts were aimed at the synthesis of cofactors which can be used in a two-step labeling protocol. In such schemes, a reactive functionality, e.g. azide or amine, is appended to the transferable group, for modification at either the cofactor stage or after transfer to the substrate. Thus, we started with the coupling of 5′-thioadenosine35 and 6-azido-1-bromohex-2-yne. These starting materials are well described in literature and their synthesis can be carried out at gram scale. The substitution reaction furnished thioether 1a in good yield, which is readily reduced to the amino substituted product 1b under Staudinger conditions. Much to our delight, these molecules are converted into the desired cofactors 2 in acidic media in the presence of one to four equivalents of the β-lactone (Scheme 1). In line with the soft nucleophilic character of the sulfur, the reaction with the strained β-lactone favors α-carbon attack over nucleophilic acylation.36 For simple systems, conversion was quantitative without the formation of byproducts, and final removal of the protecting group proceeded smoothly after addition of a stronger acid. Though lactone salts (tosylate, tetrafluoroborate, trifluoroacetate) can provide the desired compounds directly upon reaction with the thioethers 1, reactions are sluggish, and the deprotection route is preferred. Due to the good conversion and low amount of byproducts formed, these cofactors can be used directly or with minimal purification (trituration or reverse phase silica gel filtration). While this is an effective and scalable method for cofactor synthesis, it should be noted that the formed cofactors are diastereomeric in nature, and the sulfonium centers are approximately a 50/50 mixture of R and S isomers. Diastereomerically pure cofactors can be obtained from HPLC purification, though the inactive isomer does not hinder further applications. The cofactors can be stored for several months at low temperatures and low pH.
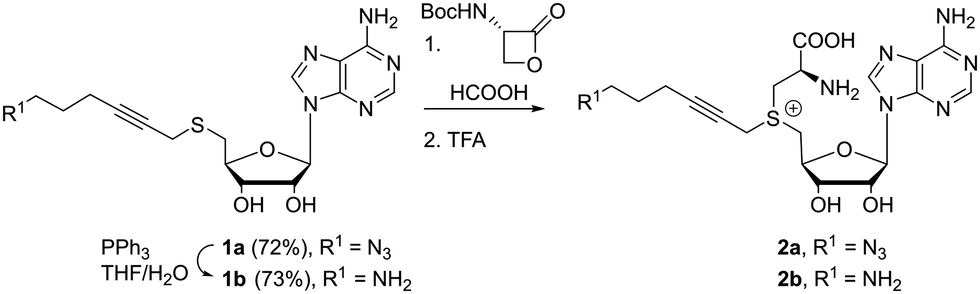 | ||
| Scheme 1 Formation of cofactors 2a and 2b. | ||
Shifting from homocysteine appended cofactors to cysteine containing systems will have an effect on the position of the cofactor within the enzymatic pocket. Fortunately, gel based DNA restriction analysis indicated that MTase enzymes tolerate cysteine based cofactors, though at increased concentration (ESI,† Fig. S1–S3), proving that this pathway provides efficient access to azide and amine substituted functional cofactors.
Next, these cofactors were converted to fluorescent versions through coupling of their reactive handles with dyes. However, these reactions were always accompanied by severe side product formation, attributed to high pH (amide coupling) or incompatible reagents (Cu-salts in click-chemistry). Thus, fluorescent dye attachment was effected before the actual introduction of the amino acid.
Here, it was postulated that enzyme binding might be influenced by the fluorescent molecules, which tend to be quite bulky and often carry charges. To counter these effects, we attempted to increase the distance between dye and cofactor core. This distance can be acquired by adding aliphatic or oligo-ethylene glycol spacers. To evaluate the effect of the length and structure of the spacer, three different examples 3–5a were prepared: (1) no linker (2) an aliphatic extension of six carbons (3) extension with three ethylene glycol units, respectively. Chain extension proceeded smoothly when using N-Boc protected carboxylic acids and 1b. After removal of the protecting group, the free amine can be used in the next step without intermediate purification. Dye attachment was carried out through amide formation using similar reaction conditions as for the extension. In the presence of four equivalents of the β-lactone, the desired cofactors 3–5a (Fig. 2) were obtained in good conversions after reverse phase silica gel filtration.
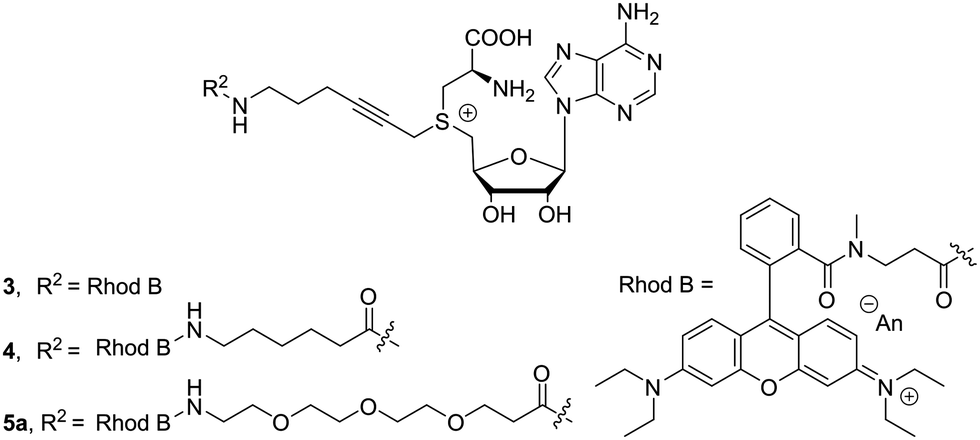 | ||
| Fig. 2 Fluorescent cysteine-based SAM analogues with various spacers. | ||
To further extend this product portfolio of chain extended direct cofactors, the dye repertoire was expanded and a small library, spanning the visible spectrum, was prepared (Fig. 3, Full structures in ESI†). To ensure solubility for cofactors conjugated to hydrophobic dyes, cofactor synthesis was started from the ethylene glycol extended molecule S2.
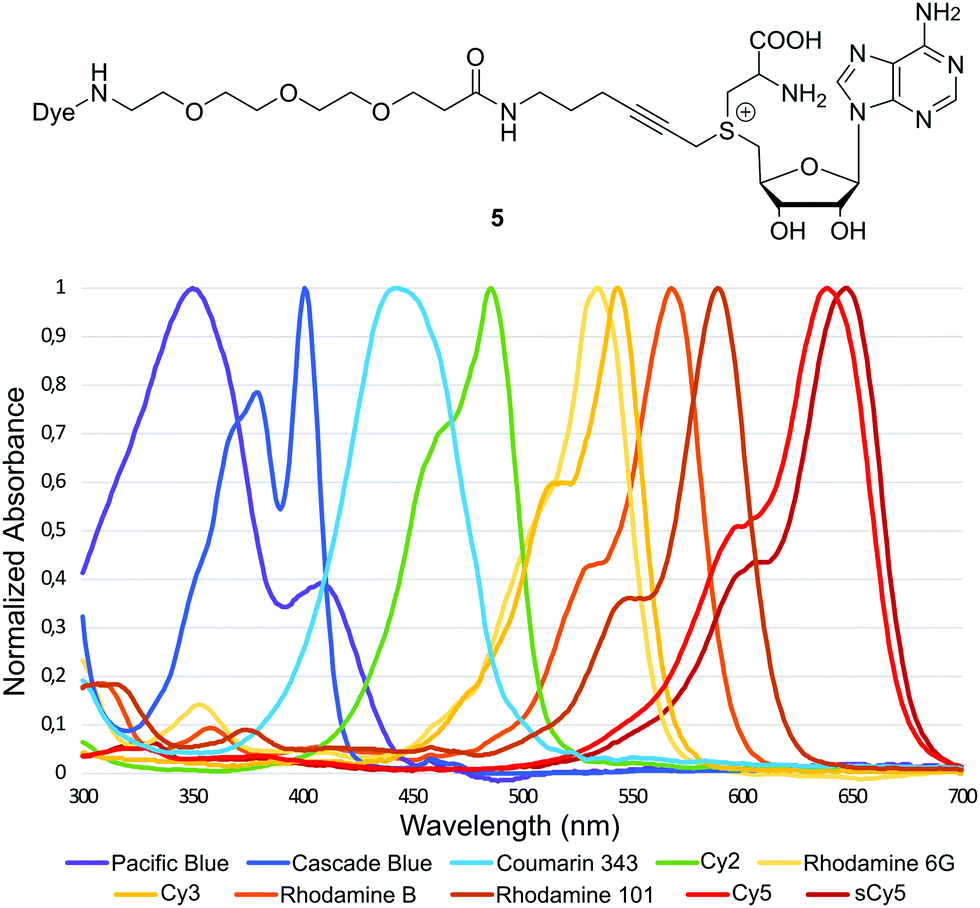 | ||
| Fig. 3 General structure and normalized absorbance spectra of the synthesized fluorescent cofactors 5a–5j. | ||
After successful synthesis of the fluorescent cofactors 3–5j, the compatibility in MTAse based labeling was validated through gel based DNA restriction analysis (Fig. 4 and ESI,† Fig. S4–S9). Interestingly, all extended cofactors 4–5 were accepted by the MTAse enzyme M.TaqI. In a next step, the labeling performance was assessed through a counting assay23 (ESI,† Fig. S10). Given their superior labeling performance, Rhodamine B containing cofactors 4 and 5a were selected to use in further optical mapping experiments.In short, DNA fragments of the bacteriophage lambda were labeled using the M.TaqI methyltransferase enzyme (recognition sequence 5′-TCGA-3′) and the fluorescent cofactor 4–5a. Labeled DNA is linearized on a zeonex coated coverslip using the “rolling droplet” technique37 and the fluorescence intensity trace (map) is extracted using Structured Illumination Microscopy (SIM). The obtained intensity profiles are cross-correlated to the theoretical profile and a matching score is assigned to each DNA strand (Fig. 5).38 DNA labeled by cofactors 4 and 5a shows excellent matching to the ground-truth species, indicating the applicability of these novel cofactor systems for optical mapping approaches.
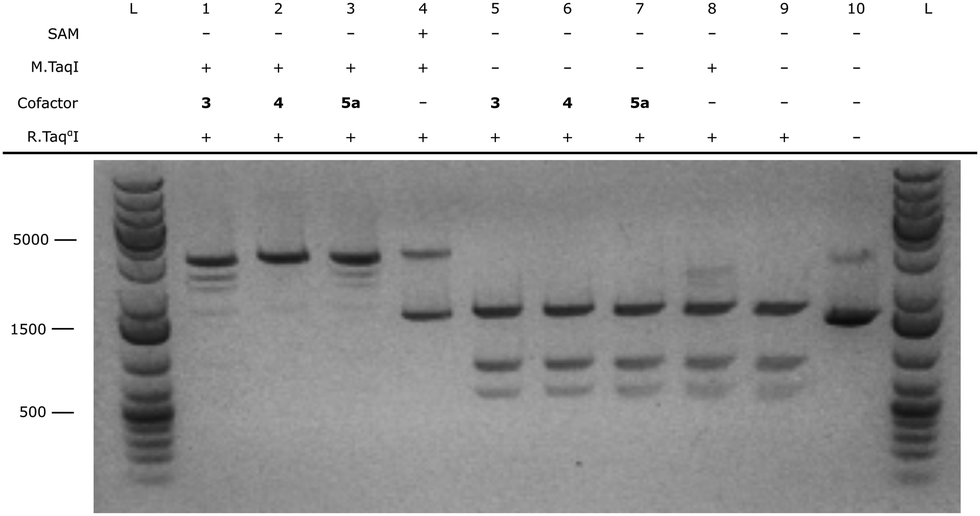 | ||
| Fig. 4 TaqI restriction assay on pUC19 DNA using oligonucleotide-treated M.TaqI (0.03 μg μl−1) with 3, 4 and 5a. All samples were reacted with TaqI restriction enzyme unless stated otherwise. From left to right: GeneRuler 1 kb plus (ladder); (1) M.TaqI with 50 μM 3, (2) M.TaqI with 50 μM 4, (3) M.TaqI with 50 μM 5a, (4) control sample with 50 μM natural SAM cofactor, (5) control sample without M.TaqI enzyme and with 50 μM 3, (6) control sample without M.TaqI enzyme and with 50 μM 4, (7) control sample without M.TaqI enzyme and with 50 μM 5a, (8) control sample without cofactor, (9) control sample without M.TaqI enzyme and without cofactor, (10) pUC19 DNA, GeneRuler 1 kb plus (ladder). | ||
 | ||
| Fig. 5 Matching results for 5a and 4. (A) Result obtained after matching 96 experimental maps (dashed line), obtained through 5a-mediated labeling of bacteriophage lambda. Out of 96 maps, 73 matched significantly (α = 0.05) to the ground truth species. (B) Field of view of 5a labeled bacteriophage lambda DNA obtained through SIM. (C) Cropped image from B as indicated by white dashed rectangle. (D) Result obtained after matching 47 experimental maps (dashed line), obtained through 4-mediated labeling of bacteriophage lambda. Out of 47 maps, 45 matched significantly (α = 0.05) to the ground truth species. (E) Field of view of 4 labeled bacteriophage lambda DNA obtained through SIM. (F) Cropped image from E as indicated by white dashed rectangle. 20 kilobasepairs was used as a size threshold, smaller molecules were not analyzed. | ||
In conclusion, cysteine based SAM analogues bring forward an easily accessible class of non-natural methyltransferase cofactors which are obtained through a high yielding pathway with limited byproduct formation. These compounds can be used to introduce various dyes onto DNA and provide excellent matching in optical mapping experiments. Despite a transfer efficiency that is somewhat reduced when compared to similar homocysteine cofactor analogues, their synthetic accessibility warrants widespread use.
This work was supported by the European Union's Horizon 2020 research and innovation program under grant agreement No. 686271, “AD-Gut” and European Union's Horizon 2020 research and innovation program under grant agreement No. 634890, “BeyondSeq”. J. H. gratefully acknowledges financial support of the Flemish government through long-term structural funding Methusalem (CASAS2, Meth/15/04). Mass spectrometry was made possible by the support of the Hercules Foundation of the Flemish Government (grant 20100225-7). L. D. gratefully acknowledges the Fonds voor Wetenschappelijk Onderzoek (FWO) Aspirant for a PhD fellowship (11D3718N). Open Access funding provided by the Max Planck Society.
Conflicts of interest
J. H. and V. L. are inventors on a patent application around the cofactor structures described in this manuscript.Notes and references
- A. Gottfried and E. Weinhold, Biochem. Soc. Trans., 2011, 39, 623–628 CrossRef CAS PubMed.
- R. K. Neely, P. Dedecker, J. Hotta, G. Urbanavičiūtė, S. Klimašauskas and J. Hofkens, Chem. Sci., 2010, 1, 453–460 RSC.
- R. K. Neely, J. Deen and J. Hofkens, Biopolymers, 2011, 95, 298–311 CrossRef CAS PubMed.
- M. Levy-Sakin and Y. Ebenstein, Curr. Opin. Biotechnol., 2013, 24, 690–698 CrossRef CAS PubMed.
- M. Mrksich, M. E. Parks and P. B. Dervan, J. Am. Chem. Soc., 1994, 116, 7983–7988 CrossRef CAS.
- N. T. Thuong and C. Hélène, Angew. Chem., Int. Ed. Engl., 1993, 32, 666–690 CrossRef.
- J. Deen, C. Vranken, V. Leen, R. K. Neely, K. P. F. Janssen and J. Hofkens, Angew. Chem., Int. Ed., 2017, 56, 5182–5200 CrossRef CAS PubMed.
- G. Pljevaljčić, F. Schmidt and E. Weinhold, ChemBioChem, 2004, 5, 265–269 CrossRef PubMed.
- C. Dalhoff, G. Lukinavičius, S. Klima
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) auskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed.
auskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed. - X. Cheng, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 293–318 CrossRef CAS PubMed.
- A. Grunwald, M. Dahan, A. Giesbertz, A. Nilsson, L. K. Nyberg, E. Weinhold, T. Ambjörnsson, F. Westerlund and Y. Ebenstein, Nucleic Acids Res., 2015, 43, e117 CrossRef.
- F. Kunkel, R. Lurz and E. Weinhold, Molecules, 2015, 20, 20805–20822 CrossRef CAS PubMed.
- L. Anhäuser, F. Muttach and A. Rentmeister, Chem. Commun., 2018, 54, 449–451 RSC.
- M. Heimes, L. Kolmar and C. Brieke, Chem. Commun., 2018, 54, 12718–12721 RSC.
- F. Muttach, F. Mäsing, A. Studer and A. Rentmeister, Chem. – Eur. J., 2017, 23, 5988–5993 CrossRef CAS PubMed.
- L. R. Comstock and S. R. Rajski, J. Am. Chem. Soc., 2005, 127, 14136–14137 CrossRef CAS PubMed.
- M. Pignot, C. Siethoff, M. Linscheid and E. Weinhold, Angew. Chem., Int. Ed., 1998, 37, 2888–2891 CrossRef CAS PubMed.
- R. L. Weller and S. R. Rajski, ChemBioChem, 2006, 7, 243–245 CrossRef CAS PubMed.
- G. Lukinavičius, V. Lapienė, Z. Staševskij, C. Dalhoff, E. Weinhold and S. Klimašauskas, J. Am. Chem. Soc., 2007, 129, 2758–2759 CrossRef PubMed.
- G. Lukinavičius, M. Tomkuvienė, V. Masevičius and S. Klimašauskas, ACS Chem. Biol., 2013, 8, 1134–1139 CrossRef PubMed.
- W. Peters, S. Willnow, M. Duisken, H. Kleine, T. Macherey, K. Duncan, D. Litchfield, B. Lüscher and E. Weinhold, Angew. Chem., Int. Ed., 2010, 49, 5170–5173 CrossRef CAS PubMed.
- C. Vranken, J. Deen, L. Dirix, T. Stakenborg, W. Dehaen, V. Leen, J. Hofkens and R. K. Neely, Nucleic Acids Res., 2014, 42, e50 CrossRef CAS PubMed.
- M. H. Lauer, C. Vranken, J. Deen, W. Frederickx, W. Vanderlinden, N. Wand, V. Leen, M. H. Gehlen, J. Hofkens and R. K. Neely, Chem. Sci., 2017, 8, 3804–3811 RSC.
- J. Jeffet, A. Kobo, T. Su, A. Grunwald, O. Green, A. N. Nilsson, E. Eisenberg, T. Ambjörnsson, F. Westerlund, E. Weinhold, D. Shabat, P. K. Purohit and Y. Ebenstein, ACS Nano, 2016, 10, 9823–9830 CrossRef CAS PubMed.
- J. Deen, S. Wang, S. Van Snick, V. Leen, K. Janssen, J. Hofkens and R. K. Neely, Nucleic Acids Res., 2018, 46, e64 CrossRef PubMed.
- N. O. Wand, D. A. Smith, A. A. Wilkinson, A. E. Rushton, S. J. W. Busby, I. B. Styles and R. K. Neely, Nucleic Acids Res., 2019, 47, e68 CrossRef CAS PubMed.
- I. R. Bothwell, K. Islam, Y. Chen, W. Zheng, G. Blum, H. Deng and M. Luo, J. Am. Chem. Soc., 2012, 134, 14905–14912 CrossRef CAS PubMed.
- I. R. Bothwell and M. Luo, Org. Lett., 2014, 16, 3056–3059 CrossRef CAS PubMed.
- S. Willnow, M. Martin, B. Lüscher and E. Weinhold, ChemBioChem, 2012, 13, 1167–1173 CrossRef CAS PubMed.
- T. D. Huber, F. Wang, S. Singh, B. R. Johnson, J. Zhang, M. Sunkara, S. G. V. Lanen, A. J. Morris, J. George, N. Phillips and J. S. Thorson, ACS Chem. Biol., 2016, 11, 2484–2491 CrossRef CAS PubMed.
- T. D. Huber, B. R. Johnson, J. Zhang and J. S. Thorson, Curr. Opin. Biotechnol., 2016, 42, 189–197 CrossRef CAS PubMed.
- B. W. Lee, H. G. Sun, T. Zang, B. J. Kim, J. F. Alfaro and Z. S. Zhou, J. Am. Chem. Soc., 2010, 132, 3642–3643 CrossRef CAS PubMed.
- S. Flade, J. Jasper, M. Gieß, M. Juhasz, A. Dankers, G. Kubik, O. Koch, E. Weinhold and D. Summerer, ACS Chem. Biol., 2017, 12, 1719–1725 CrossRef CAS PubMed.
- F. Wang, S. Singh, J. Zhang, T. D. Huber, K. E. Helmich, M. Sunkara, K. A. Hurley, R. D. Goff, C. A. Bingman, A. J. Morris, J. S. Thorson and G. N. P. Jr, FEBS J., 2014, 281, 4224–4239 CrossRef CAS PubMed.
- M. Pignot, G. Pljevaljcic and E. Weinhold, Eur. J. Org. Chem., 2000, 549–555 CrossRef CAS.
- L. D. Arnold, T. H. Kalantar and J. C. Vederas, J. Am. Chem. Soc., 1985, 107, 7105–7109 CrossRef CAS.
- J. Deen, W. Sempels, R. De Dier, J. Vermant, P. Dedecker, J. Hofkens and R. K. Neely, ACS Nano, 2015, 9, 809–816 CrossRef CAS PubMed.
- A. Bouwens, J. Deen, R. Vitale, L. D’Huys, V. Goyvaerts, A. Descloux, D. Borrenberghs, K. Grussmayer, T. Lukes, R. Camacho, J. Su, C. Ruckebusch, T. Lasser, D. V. De Ville, J. Hofkens, A. Radenovic and K. P. F. Janssen, NAR Genomics and Bioinformatics, 2020, 2, lqz007 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthetic protocols and characterization details. See DOI: 10.1039/c9cc08938a |
| This journal is © The Royal Society of Chemistry 2020 |