
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of a heterogeneous Pd–Cu/zeolite Y Wacker catalyst for ethylene oxidation†
Jerick
Imbao
 ab,
Jeroen A.
van Bokhoven
*ab and
Maarten
Nachtegaal
*a
ab,
Jeroen A.
van Bokhoven
*ab and
Maarten
Nachtegaal
*a
aPaul Scherrer Institute, 5232 Villigen PSI, Switzerland. E-mail: maarten.nachtegaal@psi.ch
bInstitute for Chemistry and Bioengineering, ETH Zurich, Vladimir-Prelog-Weg 1, 8093 Zurich, Switzerland. E-mail: jeroen.vanbokhoven@chem.ethz.ch
First published on 7th January 2020
Abstract
Exchanging low amounts of palladium with excess copper in a heterogeneous zeolite Y catalyst leads to high Wacker activity and mitigates activity loss by reducing the extent of palladium sintering. Employing periodic regenerative treatments in oxygen to remove carbon deposits and reoxidize palladium results in the partial (but reversible) recovery of the high initial activity.
The homogeneous Wacker oxidation of olefins into synthetically versatile carbonyls is one of the most successful industrially applied processes after the Second World War.1,2 In particular, the palladium-catalyzed oxidation of ethylene remains industrially relevant for acetaldehyde production due to the continued growth in the demand for manufacturing its derivatives.3 However, the excessive use of hydrochloric acid leads to high corrosivity and formation of unwanted chlorinated side-products. Moreover, oxidizing longer-chained alkenes leads to products with boiling points above that of water, rendering their separation from the aqueous catalyst solution challenging.4 Consequently, there had been various attempts to heterogenize Wacker catalysts using chloride-free systems with varying co-catalysts. Stobbe-Kreemers et al.5 replaced the conventional copper co-catalyst with vanadia while Barthos et al.6 optimized this system with monolayer vanadia supported on silica and α-alumina. Nowinska et al.7 utilized the palladium salts of heteropolyacids supported on silica for the selective oxidation of ethylene to acetaldehyde. Espeel et al.8,9 and Arai et al.10 separately showed that palladium- and copper-exchanged zeolite Y (Pd–Cu/zeolite Y) is one of the most active heterogeneous Wacker catalysts. The activities of these heterogeneous catalysts (0.5–0.8 mmol acetaldehyde mmol Pd−1 min−1) are similar to those of the homogeneous systems (0.3–1.0 mmol acetaldehyde mmol Pd−1 min−1).11 The catalyst deactivates through the precipitation of copper oxalate in the homogeneous Wacker system1,4 and the irreversible formation of inactive zero-valent palladium in the heterogeneous Wacker system.4 Overall, the homogeneous system is still widely utilized commercially because the catalyst solution can be regenerated continuously and efficiently in high-purity oxygen (one-stage process developed by Farbwerke Hoechst) or air (two-stage process developed by Wacker-Chemie).2,4,12 Structural characterization of Pd–Cu/zeolite Y for the heterogeneous Wacker process is limited, preventing the rational design of better catalysts. Here, we show the influence of palladium and copper loading on the activity and stability of Pd–Cu/zeolite Y for Wacker oxidation of ethylene. We investigated the catalyst deactivation and reactivation mechanisms by determining the electronic and structural changes, deciphered by XAS and STEM, during the course of the reaction.
The catalysts were prepared through aqueous ion exchange of the sodium form of the zeolite Y (SiO2/Al2O3 = 5.1) denoted as Pd(a)Cu(b)–Y (a,b = wt%; Table S1, ESI†). The HAADF-STEM image and the corresponding elemental maps of Pd1Cu5–Y (Fig. S1, ESI†) illustrate that both palladium and copper are homogeneously distributed in the zeolite after ion exchange without any evident particle formation. The catalysts were pre-treated in oxygen at 378 K and their activity for Wacker oxidation was measured in a fixed-bed plug flow reactor at 378 K with a gas feed consisting of ethylene, water and oxygen (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:10 molar ratio) diluted in helium at constant flow (W/F0 = 0.86 kgcat s mol−1) to guarantee that ethylene conversion did not exceed 15%.
:7:10 molar ratio) diluted in helium at constant flow (W/F0 = 0.86 kgcat s mol−1) to guarantee that ethylene conversion did not exceed 15%.
Optimum Wacker activity and acetaldehyde selectivity were determined by testing catalysts containing varying amounts of copper and palladium (Fig. 1). Without palladium, no Wacker conversion was observed over Na–Y (not shown) and Cu7–Y (Fig. 1a). Without copper, low ethylene conversion was observed over Pd2–Y (Fig. 1d) with a selectivity of roughly 80% for acetaldehyde, which corresponds to an amount greater than the stoichiometric amount of palladium present. This is indicative of catalytic acetaldehyde formation in the absence of copper. The remaining ethylene was oxidized to carbon dioxide. Fig. 1a shows that the conversion of ethylene over Pd–Cu/zeolite Y with fixed copper loading (ca. 5 wt%) increased significantly from zero to ca. 9% in the presence of palladium (1 wt%, Cu/Pd = 6.7). Ethylene conversion continued to improve from 9% to 13.4% while the selectivity for acetaldehyde decreased slightly (from 91% to 85%, Fig. 1c) when the palladium loading was increased from 1 wt% (Cu/Pd = 6.7) to 4 wt% (Cu/Pd = 2.2). However, normalizing the activity to the amount of palladium (Fig. 1b) reveals that the rate of formation of acetaldehyde diminished as the palladium loading was increased from 1 wt% to 4 wt%. Moreover, only Pd1Cu5–Y reached steady-state activity after 4 hours while both Pd2Cu5–Y and Pd4Cu5–Y continued to deactivate thereafter.
 | ||
| Fig. 1 Effect of different metal loadings on ethylene oxidation. (a and d) Ethylene conversion, (b and e) rate of acetaldehyde formation and (c and f) product selectivities over Pd–Cu/Y with different Pd (a–c: fixed 5 wt% Cu) and Cu (d–f: fixed 1 wt% Pd) loadings. Hollow points in (a–c) represent Cu7-Y (without Pd) while those in (d–f) refer to Pd2–Y (without Cu). All values were determined after 4 hours of reaction (378 K, 0.86 kgcat s mol−1). Gas feed consists of C2H4:H2O:O2 at a 1:7:10 molar ratio. | ||
Next, we investigated the effect of varying the copper loading on the activity of Pd–Cu/Y with a fixed palladium loading of 1 wt% (Fig. 1d–f) since low palladium loading resulted in optimum Wacker activity. In comparison with Pd2–Y, a small improvement in conversion (from 1.6% to 2.2%) with a corresponding decrease in the selectivity for acetaldehyde (from 80% to 69%) was observed when the copper loading is 1 wt% (Cu/Pd = 1.7). When the copper loading was increased from 1 wt% to 5 wt% (Cu/Pd = 6.7), the ethylene conversion to acetaldehyde improved significantly, highlighting the co-catalytic activity of copper. However, a further increase in the copper loading from 5 wt% to 6 wt% (Cu/Pd = 9.4) did not lead to any significant variation in ethylene conversion but instead resulted in an increase in carbon dioxide selectivity at the expense of acetaldehyde selectivity. These results suggest that excess copper (Cu/Pd = 6.7) is needed for optimum Wacker activity as it ensures that there are enough copper ions in the proximity of each palladium ion. At higher copper loading (ca. 6 wt%), the additional copper ions participate in the total oxidation of ethylene.
Table S2 (ESI†) lists the properties of the catalysts having low (Pd1Cu5–Y) and high (Pd4Cu5–Y) palladium loading. Both catalysts exhibit lower surface area and total pore volume after 4 hours of Wacker oxidation relative to the as-prepared catalysts, which could be due to the accumulation of carbon deposits. The surface area and total pore volume of Pd4Cu5–Y are much lower than that of Pd1Cu5–Y after 4 hours under Wacker conditions, albeit having nearly similar amount of accumulated carbon deposits. In situ X-ray absorption spectroscopy (Methods, ESI†) was employed to monitor the changes in the electronic and local atomic structures of the catalysts under operating conditions. The XANES spectra of both pre-treated Pd1Cu5–Y and Pd4Cu5–Y (Fig. 2) are nearly similar suggesting that the difference in palladium loading does not lead to significant differences in the oxidation state and structure of palladium and copper. The Cu and Pd K-edge XANES spectra of both as-prepared Pd1Cu5–Y and Pd4Cu5–Y indicate that copper is present as hydrated Cu(II) (Fig. 2a: pre-edge feature at 8977.5 eV resulting from the 1s → 3d transition;13 continuum resonance peak at 8996 eV) and palladium as amminated Pd(II) (Fig. 2b: whiteline peak at 24372 eV, similar to the spectrum of the Pd(NH3)42+; Fig. S2, ESI†: weak IR band at 1310 cm−1 attributed to symmetric N–H deformation9). Pre-treatment in oxygen at 378 K resulted in the decrease of the whiteline intensity at 8996 eV (due to partial dehydration) in the Cu K-edge XANES spectra of Pd1Cu5–Y and Pd4Cu5–Y but there were no changes observed in the Pd K-edge XANES spectra. The Pd K-edge XANES of the Wacker-equilibrated Pd1Cu5–Y (Fig. 2b) exhibit a shift towards lower absorption edge energy at half-height relative to the pre-treated catalysts, which is indicative of the reduction of a fraction of Pd(II) to Pd(0). The Pd K-edge XANES spectrum of the Wacker-equilibrated Pd4Cu5–Y reveals a broader continuum resonance peak centered at 24372 eV and the appearance of a shoulder at ca. 24395 eV, suggesting the formation of more Pd(0) compared to what was observed for the Wacker-equilibrated Pd1Cu5–Y. The Cu K-edge XANES spectra of Pd4Cu5–Y after 4 hours under Wacker conditions (Fig. 2a) show the formation of a minor peak at 8983 eV, which has a higher intensity than that in the spectra of Wacker-equilibrated Pd1Cu5–Y. This indicates the formation of more Cu(I) species13 in the catalyst with higher palladium loading after 4 hours under Wacker conditions.
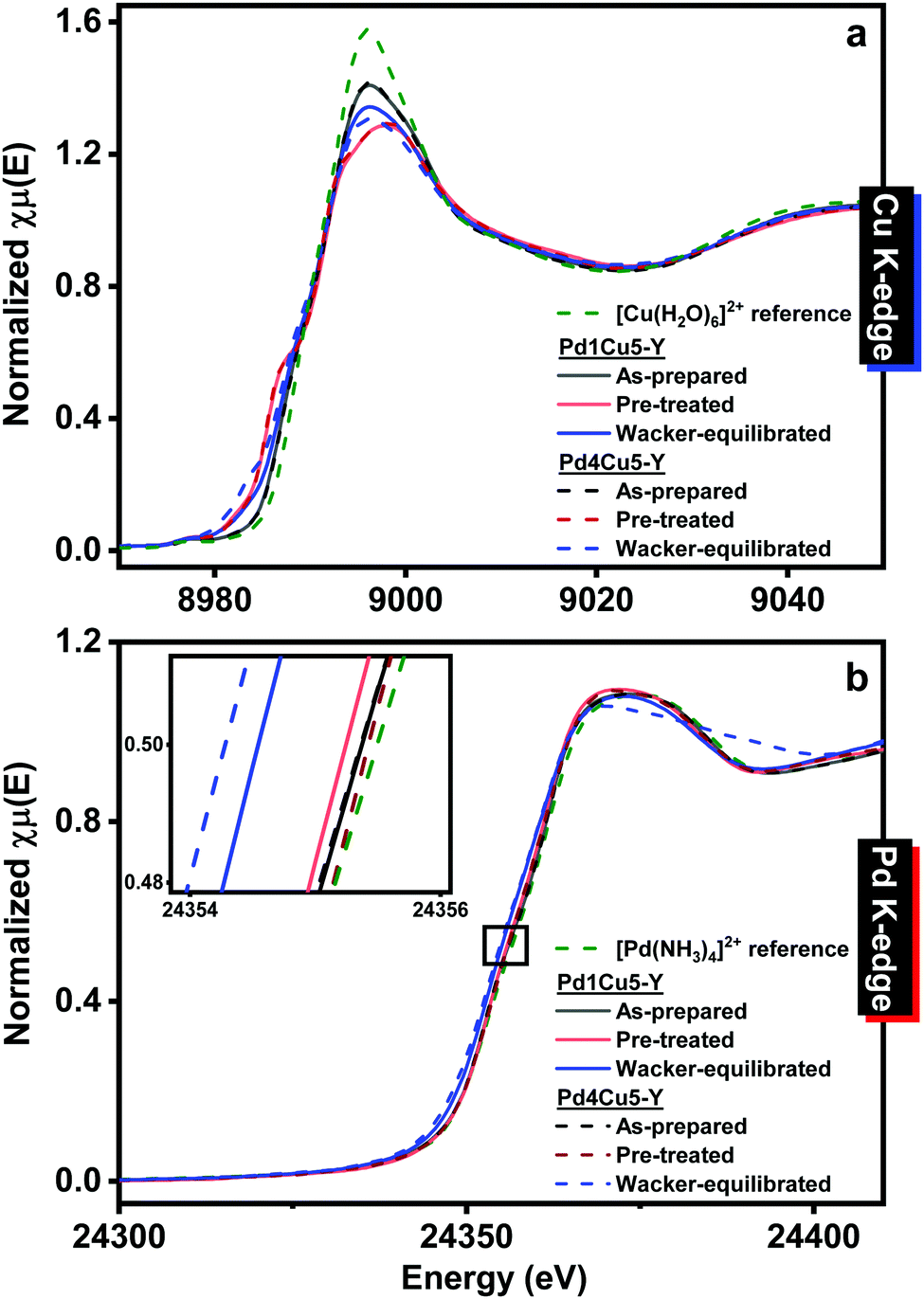 | ||
| Fig. 2 Normalized (a) Cu and (b) Pd K-edge XANES spectra of Pd–Cu/Y. Wacker conditions: 378 K, 0.86 kgcat s mol−1. Gas feed consists of C2H4:H2O:O2 at a 1:7:10 molar ratio. Inset: Magnification of (b) Pd K-edge XANES spectra at half-edge height. | ||
EXAFS analyses (Fig. 3, Fig. S3, S4, S8 and Table 1, Table S3, ESI†) reveal a decrease in the average Pd–O coordination number for Pd1Cu5–Y (from 3.5 to 3.1) and Pd4Cu5–Y (from 3.6 to 2.9) at a bond distance of 2.01 Å after 4 hours under Wacker conditions. Pd(II) → Pd(0) reduction is evident in the Wacker-equilibrated Pd4Cu5–Y due to the appearance of a Pd–Pd (metallic) coordination shell at 2.76 Å with a higher coordination number (2.6 Pd) than that of the Wacker-equilibrated Pd1Cu5–Y (1.5 Pd). This signifies that the extent of Pd(II) reduction in Pd4Cu5–Y is higher than that in Pd1Cu5–Y after 4 hours under Wacker conditions. The HAADF-STEM image and the corresponding EDX maps of spent Pd4Cu5–Y (Fig. S5, ESI†) illustrate the formation of big palladium particles (ca. 5–10 nm), which corroborate the Pd K-edge XANES and EXAFS spectra. Hence, high palladium loading leads to the formation of more Cu(I) species and inactive palladium metallic nanoparticles under Wacker conditions. This results in a lower Pd-normalized rate of acetaldehyde formation observed over Pd4Cu5–Y.
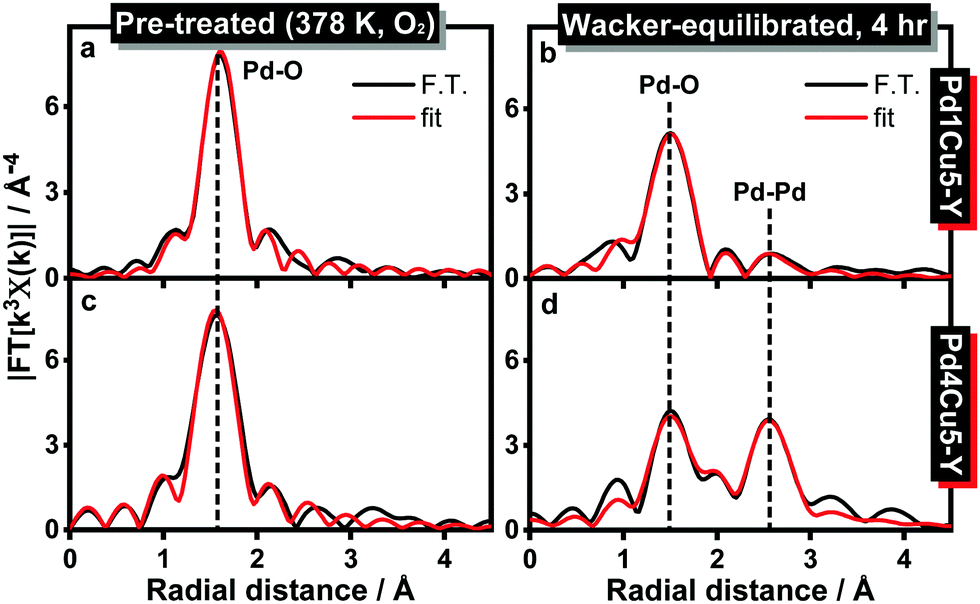 | ||
| Fig. 3 k 3-Weighted Fourier transform magnitude (non-phase shift corrected) of the Pd K-edge EXAFS spectra of (a and b) Pd1Cu5–Y and (c and d) Pd4Cu5–Y in different reaction conditions (FT performed in the 3–11 Å−1 range and fitted in R space in the 1–3 Å range; black: magnitude of FT, red: best fit). The corresponding k3-weighted Pd K-edge EXAFS spectra are shown in Fig. S8, ESI.† | ||
| Catalyst | Path | N | R (Å2) |
σ
2
(Å2) |
R fac |
|---|---|---|---|---|---|
a Number of nearest neighbors determined by fixing the amplitude reduction factor (S02) values obtained from the fits to Cu (0.82) and Pd (0.86) foil spectra.
b Interatomic distance.
c Pseudo Debye–Waller factor.
d
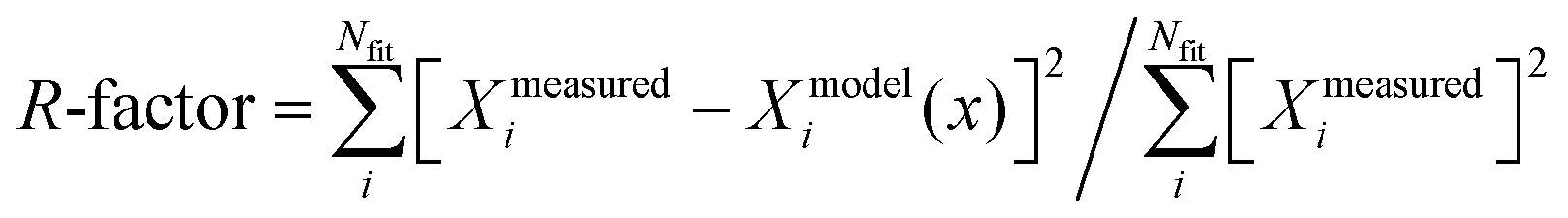 .
e Equilibrated in Wacker conditions (378 K, 0.86 kgcat s mol−1). .
e Equilibrated in Wacker conditions (378 K, 0.86 kgcat s mol−1).
|
|||||
| Pre-treated Pd1Cu5–Y | Pd–O/N | 3.5(0.2) | 2.01(0.01) | 0.004(0.001) | 0.010 |
| Wacker-eqe Pd1Cu5–Y | Pd–O/N | 3.1(0.3) | 2.01(0.01) | 0.004(0.001) | 0.009 |
| Pd–Pd | 1.5(0.9) | 2.76(0.05) | 0.018(0.003) | ||
| Pre-treated Pd4Cu5–Y | Pd–O/N | 3.6(0.2) | 2.01(0.01) | 0.004(0.001) | 0.011 |
| Wacker-eqe Pd4Cu5–Y | Pd–O/N | 2.9(0.5) | 2.04(0.01) | 0.006(0.001) | 0.017 |
| Pd–Pd | 2.6(0.7) | 2.75(0.01) | 0.006(0.001) | ||
The stability of the optimized catalyst, Pd1Cu5–Y, was then tested for Wacker oxidation under differential conditions. Fig. 4 depicts the first four hours of the reaction exhibiting two stages of activity loss or deactivation versus time-on-stream (TOS). Rapid deactivation was observed during the 1st hour of the reaction followed by a more gradual loss of activity before reaching steady-state conditions. After 4 hours under Wacker conditions, a partial loss of palladium–ammine coordination is evident from the decrease in the IR band intensity at 1310 cm−1 and the appearance of a band at 1450 cm−1 in the IR-ATR spectrum (Fig. S2, ESI†) of spent Pd1Cu5–Y.9 Fig. S6 (ESI†) shows that Pd1Cu5–Y is stable under Wacker conditions for up to 20 hours on stream with mild deactivation and an acetaldehyde selectivity of ca. 90%. If the catalyst deactivation is largely induced by the accumulation of carbon deposits and the reversible formation of Pd(0), the activity should be fully recovered after performing calcination to burn off all the carbon deposits and reoxidize Pd(0). The reactivation procedure involved heating at a higher temperature (623 K) than what was used in the standard activation and reaction conditions (378 K) because calcination of the spent catalyst at lower temperatures did not burn off all the carbon deposits. However, only 75% of the initial activity of Pd1Cu5–Y was regenerated by the reactivation procedure. Again, a fast and a slow deactivation of the catalyst were observed through time (Fig. 4, 1st cycle), albeit with a lower steady-state rate of acetaldehyde formation after 4 hours under Wacker conditions (Table 2). At temperatures higher than 573 K, the majority, if not all, of ammine ligands that were initially present are released from their coordination with Pd(II).14 Fig. S2 (ESI†) depicts the disappearance of the bands at 1310 and 1450 cm−1 in the IR-ATR spectrum of regenerated Pd1Cu5–Y, which is indicative of the loss of Pd–ammine coordination9 and the subsequent release and oxidation of ammonia at 623 K.14
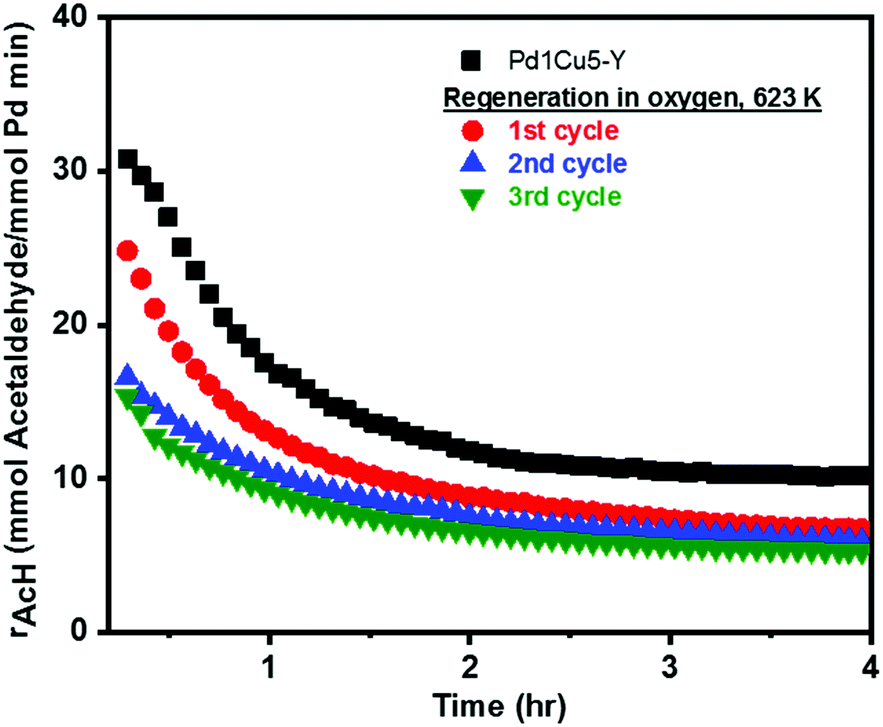 | ||
| Fig. 4 Ethylene oxidation over Pd1Cu5–Y and the regenerated catalyst (3 subsequent reactivation cycles) under standard Wacker conditions vs. time-on-stream (TOS). Wacker conditions: 378 K, 0.86 kgcat s mol−1. Gas feed consists of C2H4:H2O:O2 at a 1:7:10 molar ratio. | ||
| Catalyst | Initial reaction rate (mmol AcH mmol Pd−1 min−1) | Reaction rate after 4 h (mmol−1 AcH mmol Pd−1 min−1) |
|---|---|---|
| Pd1Cu5–Y | 30.8 | 10.2 |
| Regeneration in O2, 623 K | ||
| 1st cycle | 24.8 | 6.6 |
| 2nd cycle | 16.5 | 5.8 |
| 3rd cycle | 15.4 | 5.3 |
These lead to irreversible changes in the original catalyst as observed in the Pd and Cu K-edge EXAFS spectra of Pd1Cu5–Y after the 1st reactivation (Fig. S7, ESI†). EXAFS analyses (Table S3, ESI†) reveal the recovery of the initial intensity of Pd–O (2.02 Å) and Cu–O (1.95 Å) scattering peaks; the disappearance of metallic Pd–Pd (2.75 Å) scattering peak; and the emergence of Cu–(O)–Cu (2.91 Å) and two weak Pd–(O)–Pd coordination shells at 3.04 and 3.43 Å. These results signify that the reactivation procedure resulted in the reoxidation of Pd(0) and Cu(I) with the corresponding formation of palladium(II) oxide and copper(II) oxide particles. After 4 hours under Wacker conditions, the intensity of Pd–O and Cu–O scattering peaks decreased; the Pd–Pd (metallic) coordination shell reappeared at 2.75 Å; and the intensity of two Pd–(O)–Pd scattering peaks increased, which are indicative of the reduction of Pd(II) and Cu(II) and the formation of more palladium(II) oxide aggregates. Thus, we posit that the irrecoverable loss of initial activity in the 1st cycle of reactivation is associated largely with the release of ammine ligands from palladium coordination and the corresponding irreversible formation of inactive palladium and copper(II) oxide nanoparticles.
The reactivation procedure was done for a second time (Fig. 4, 2nd cycle) resulting in a nearly similar steady-state rate of acetaldehyde formation but only 67% of the initial activity was recovered relative to what was measured in the first cycle (Table 2). The third cycle of reactivation led to nearly full regeneration of Wacker activity in comparison with what was observed in the second cycle. In the 2nd and 3rd reactivation cycles, the loss of activity (ca. 10%) is reversible and can be mainly, if not totally, attributed to the formation of carbon deposits and zero-valent palladium. This regeneration procedure could possibly still be optimized further by employing periodic regeneration steps before the catalyst is maximally deactivated to take advantage of the high initial activity of the catalyst during the first 1 or 2 hours of the reaction before reaching steady-state conditions.
In summary, we show that optimum Wacker activity is achieved by exchanging low loadings of palladium with excess copper while high palladium loading was observed to induce more palladium sintering. In addition, accumulation of carbon deposits and formation of inactive Pd(0) were observed leading to the deactivation of the catalyst under steady-state Wacker conditions. The initial regeneration in oxygen at 623 K only resulted in a partial recovery of initial activity but became reversible in subsequent reactivation cycles. These results suggest that although deactivation of Pd–Cu/zeolite Y catalyst was not prevented, the heterogeneous process was optimized by diminishing the activity loss by utilizing less palladium and could be optimized further by including periodic regeneration steps, improving the stability.
We thank the Swiss National Science Foundation (159555) for the financial support and the SuperXAS beamline at the Swiss Light Source for the allocation of beamtime.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, R. Rüttinger and H. Kojer, Angew. Chem., 1959, 71, 176–182 CrossRef CAS.
- R. Jira, Angew. Chem., Int. Ed., 2009, 48, 9034–9037 CrossRef CAS PubMed.
- Acetaldehyde: A Global Strategic Business Report, 2016, Global Industry Analysts, Inc., San Jose, California.
- M. Eckert, G. Fleischmann, R. Jira, H. M. Bolt and K. Golka, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, Germany, 2000, 1, 191–207 Search PubMed.
- A. W. Stobbe-Kreemers, M. Makkee and J. J. F. Scholten, Appl. Catal., A, 1997, 156, 219–238 CrossRef CAS.
- R. Barthos, G. Novodárszki and J. Valyon, React. Kinet., Mech. Catal., 2016, 121, 17–29 CrossRef.
- K. Nowińska, D. Dudko and R. Golon, Chem. Commun., 1996, 277–279 RSC.
- P. H. Espeel, M. C. Tielen and P. Jacobs, Chem. Commun., 1991, 669–671 RSC.
- P. H. Espeel, G. De Peuter, M. C. Tielen and P. Jacobs, J. Phys. Chem., 1994, 98, 11588–11596 CrossRef CAS.
- H. Arai, T. Yamashiro, T. Kubo and H. Tominaga, J. Jpn. Pet. Inst., 1976, 18, 39–44 CAS.
- O. Bander et al. , US Pat., 3154586, 1964 Search PubMed; L. Hornig, et al., US Pat., 3149167, 1964 Search PubMed; W. Riemenschneider, et al., US Pat., 3301905, 1967 Search PubMed.
- H. J. Hagemeyer and U. b. Staff, Kirk Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2014, pp. 1–16 Search PubMed.
- L. S. Kau, E. I. Solomon and K. O. Hodgson, J. Phys. Colloques, 1986, 47, C8-289–C8-292 Search PubMed; L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- S. T. Homeyer and W. M. H. Sachtler, J. Catal., 1989, 117, 91–101 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc08835k |
| This journal is © The Royal Society of Chemistry 2020 |