
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning coordination chemistry through the second sphere in designed metallocoiled coils†
Louise N.
Slope
a,
Michael G.
Hill
a,
Catherine F.
Smith
a,
Paul
Teare
a,
Felicity J.
de Cogan
b,
Melanie M.
Britton
a and
Anna F. A.
Peacock
 *a
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: a.f.a.peacock@bham.ac.uk
bInstitute of Microbiology and Infection, University of Birmingham, B15 2TT, UK
First published on 26th February 2020
Abstract
The metal hydration state within a designed coiled coil can be progressively tuned across the full integer range (3 → 0 aqua ligands), by careful choice of a second sphere terminal residue, including the lesser used Trp. Potential implications include a four-fold change in MRI relaxivity when applied to lanthanide coiled coils.
There is much interest in engineering metal sites within designed miniature protein scaffolds, as they allow us to mimic biologically relevant metal sites, recreate their function, and most excitingly, achieve new function. To fully realize this potential one needs to predictably program in any desired coordination features truly de novo. Nature achieves this with highly evolved sophisticated ligands, proteins, which exploit preorganization and sterics in both first and second coordination spheres,1 to realize this goal.
Promising designs often feature coiled coils, which offer the advantages of protein-like ligands but a simple robust scaffold.2,3 Second coordination sphere residues have been used to preorganize first coordination sphere ligands through favourable hydrogen bond formation.4 Of direct relevance to this work, the steric bulk of second coordination sphere residues has been used to modulate active site access and coordination chemistry,5 including hydration state.6–13 Furthermore, second coordination sphere effects have been found to be important in molecular inorganic chemistry, including, but not limited to, controlling catalysis,14,15 interactions with biological targets16 (which surprisingly receives little attention), and of relevance to the work presented herein, altering the stability,17 luminescent,18,19 or magnetic20 properties of lanthanide complexes. Despite the importance of second coordination sphere residues in molecular inorganic chemistry and biology, there has been no systematic study of second coordination sphere effects to control designed metal site hydration states beyond one water molecule within coiled coils. This is despite the control of hydration state being a key challenge in metalloprotein design, given how crucial it is for function (e.g. substrate binding, catalysis, MRI relaxivity). Herein we undertake the first such systematic study, and report for the first time that designed metal site hydration states (herein demonstrated for lanthanides) can be programmed to contain three, two, one or no bound water molecules, by selection of a terminal second coordination sphere residue.
We recently reported the first example of a gadolinium coiled coil and its promising MRI relaxivity.21 The hydration state of the bound lanthanide, which strongly correlates with MRI relaxivity, and as such represents a good model with which to investigate hydration state control, was sensitive to its linear location along the coiled coil. Terminal sites were highly hydrated, whereas sites buried centrally within the hydrophobic core were coordinatively saturated.22 This study starts with the highly hydrated Ln(OH2)3 site generated with MB1-1(2I), as a model with which we can explore hydration state tuning (as a general design principle), over a broad range. The MB1-1(2I) nomenclature refers to the MB1 binding site which features AsnAsp in adjacent layers, towards the N-terminus (1), with Ile (2I) in the non-coordinating second sphere terminal a layer above the binding site (position 2).‡ The terminal second sphere a residue identity was systematically altered (Table 1, Fig. 1 and Fig. S1–S5, ESI†), and its impact on metal hydration state assessed.§
| Relevant residues are bold and underlined. Sequences contain a single Trp/Tyr residue (see footnote ‡). |
|---|

|
 | ||
| Fig. 1 Pymol cartoon representations of the MB1-1(2X) peptides illustrating the terminal second coordination sphere a residue in position two through a space-filling representation. Backbone of single strands are represented as ribbons, key residues are shown in stick form (oxygen in red and nitrogen in blue) and Ln3+ as a sphere. Top-down view of proposed coordinatively saturated lanthanide binding site. Black dotted lines connect proposed bound atoms. | ||
It was important to establish whether altering the nature of this terminal second sphere a residue impacts its ability to bind the metal. Circular dichroism (CD) showed that the five apo MB1-1(2X) peptides were all well-folded, and that titration with Gd3+ led to a further signal enhancement at 222 nm, an indication of α-helicity, with binding curves in agreement with data previously reported for MB1-1(2I) (Fig. 2 and Fig. S7, ESI†). Furthermore, intact Tb(Pep)3 complexes were observed by mass spectrometry (Fig. S8, ESI†). Taken together these data are consistent with Ln3+ binding being retained on altering the second sphere layer.
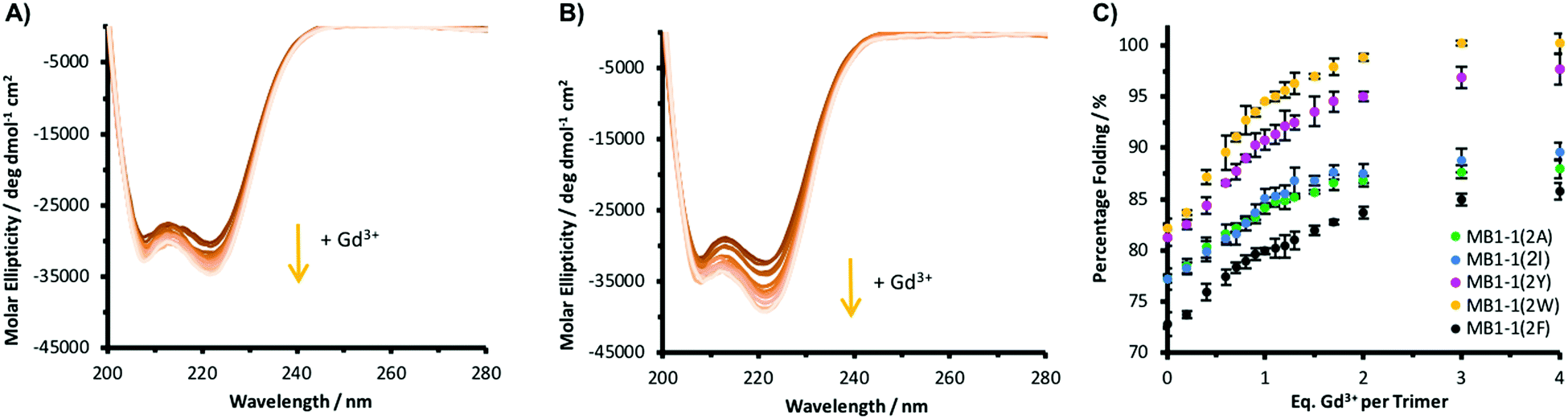 | ||
| Fig. 2 Representative Gd3+ binding titrations monitored by CD of 30 μM peptide monomer solutions of (A) MB1-1(2A) and (B) MB1-1(2W) in 10 mM HEPES buffer pH 7.0. (C) Plot of percentage folded, based on molar ellipticity at 222 nm as a function of Gd3+ equivalents per trimer, of 30 μM peptide monomer solutions of MB1-1(2A) (green), MB1-1(2I) (blue), MB1-1(2F) (black), MB1-1(2Y) (pink) and MB1-1(2W) (yellow). Data is a result of the average of three repeats where the error bars represent the standard deviation. | ||
Whether this terminal second sphere a residue can be used to tune the coordination chemistry of the bound Ln3+ was investigated. The hydration state of the Tb3+ complexes was determined by luminescence lifetime decay experiments in H2O/D2O (Fig. S9, ESI†). Our original design, which featured an Ile as the second sphere residue generates a highly hydrated site with three waters bound to the Tb3+.15 Replacing the relatively bulky Ile with the sterically undemanding Ala, did not increase the hydration state, as may have been expected.8,9 This suggests when the double AsnAsp binding site is presented, that the maximum Tb3+ hydration state is three. Though we previously found evidence of 3.6 waters bound when only the Asp layer is available.15
When the second sphere residue is Phe, the Tb3+ hydration state is reduced to two bound waters, which could be due to greater sterics of the benzyl side chain. On incorporation of Tyr, the data is consistent with only one bound water. However, care needs to be taken when interpreting this result, as Tyr sensitization induces a weaker Tb3+ emission signal and the Tyr side chain could: (1) be coordinating directly to the Tb3+; or (2) the OH group could be contributing to quenching of the Tb3+ emission. Finally, the introduction of Trp (its close proximity efficiently sensitizes Tb3+ emission), yields a coordinatively saturated Tb3+ with no evidence of bound exogenous water molecules (Table 2 and Table S1, ESI†). This would be consistent with Tb3+ bound through both the Asn and Asp side chains (see Fig. 1).
| Peptide | Percentage foldinga / % | Number aqua ligandsb | Relaxivityc | ||
|---|---|---|---|---|---|
| Apo | 1 eq. Gd3+ | r 1 / mM−1 s−1 | r 2 / mM−1 s−1 | ||
| a Percentage folding determined by CD spectroscopy. b Number of coordinated waters determined by luminescence emission of Tb3+ complex. c Relaxivity determined at 7 T for Gd3+ complex. d Data from ref. 15; nd – not determined. Errors are the standard deviation of three independent repeats. | |||||
| MB1-1(2A) | 77 ± 1 | 84 ± 1 | 3.0 ± 0.3 | nd | nd |
| MB1-1(2I) | 77 ± 1 | 85 ± 1 | 3.1 ± 0.2d | 10.0 ± 1.5d | 89.3 ± 16.8d |
| MB1-1(2F) | 73 ± 1 | 80 ± 1 | 2.1 ± 0.2 | nd | nd |
| MB1-1(2Y) | 81 ± 1 | 91 ± 1 | 1.3 ± 0.1 | nd | nd |
| MB1-1(2W) | 82 ± 1 | 95 ± 1 | 0.2 ± 0.1 | 3.9 ± 0.4 | 24.2 ± 2.4 |
| MB1-2d | 21 ± 3 | 62 ± 3 | 0.0 ± 0.1 | 4.2 ± 1.2 | 21.3 ± 2.6 |
| MB1-4d | 55 ± 6 | 70 ± 5 | 1.8 ± 0.4 | 7.5 ± 4.1 | 37.9 ± 4.0 |
| MB1-4(37W) | 67 ± 2 | 88 ± 2 | 0.1 ± 0.2 | nd | nd |
Intriguingly detection of the Tb(Pep)3 +6 ions in the mass spectra appear to be indirectly correlated with hydration state, with the most intense peak observed for Tb(MB1-1(2W))3, which could be indicative of the formation of a more stable site (Fig. S8, ESI†).
Replacing the terminal second sphere Ile with Trp converts a highly hydrated Ln(OH2)3 site into a coordinatively saturated site, which better resembles buried sites located centrally in the coiled coil (e.g. MB1-2).14 This profound change in coordination chemistry should be mirrored by the MRI properties of the Gd3+ complex.15 The longitudinal (r1) and transverse (r2) relaxivity of the Gd(MB1-1(2W))3 complex at 7 T (Fig, S10 (ESI†) and Table 2; r1 = 3.9 ± 0.4 mM−1 s−1 and r2 = 24.2 ± 2.4 mM−1 s−1) are the same within error to those reported previously for Gd(MB1-2)3 (Table 2),15 consistent with no bound water molecules (Table 2).15 Merely changing the non-coordinating second sphere residue directly above the Gd3+ binding site, Ile → Trp, leads to an almost four-fold reduction in transverse relaxivity.
One can account for this dramatic change in Ln3+ hydration state by considering the crystal structure of a comparable coiled coil with a similar Trp layer (i.e. the first hydrophobic position of the first heptad). The crystal structure shows that the Trp side-chain can be directed towards the center of the coiled coil, forming a well-packed layer,¶![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 thereby creating an effective barrier between a binding site and exogenous water. When mutated Trp → Ala, and to a lesser extent Trp → Ile, crude models yield a less efficiently packed and therefore more water penetrable layer (Fig. S11, ESI†).
23 thereby creating an effective barrier between a binding site and exogenous water. When mutated Trp → Ala, and to a lesser extent Trp → Ile, crude models yield a less efficiently packed and therefore more water penetrable layer (Fig. S11, ESI†).
To establish if this strategy could be applied more widely, we determined the impact of Trp at the C-terminus. The previously reported MB1-4 (Table 1) coordinates Tb(OH2)2 towards the C-terminus. The Ile directly below this site is more buried and in a d layer,|| so it was necessary to introduce an additional Trp a layer to yield MB1-4(37W) (Fig. S6, ESI,† and Table 1). CD titrations support Tb3+ binding (Fig. S12, ESI†), and though Trp is a third sphere residue, luminescence decay experiments are consistent with it yielding coordinatively saturated Tb3+ (Table 2 and Fig. S9, ESI†). These results suggest that a terminal a or d residue can be used to control water access at either termini. However, it is possible that introduction of the additional a layer could be responsible for preventing Tb3+ hydration.
Coiled coil designers generally avoid using Trp in the hydrophobic core,24 despite the importance of Trp as a second coordination sphere residue in biology25–27 and the report of a five stranded assembly with exclusively Trp in these positions.28 This likely stems from the antiparallel crystal structure of CoilSer, a three stranded coiled coil with Trp in the N-terminal hydrophobic a position.29 It was hypothesized that the steric bulk prevented three Trp packing in one layer in a parallel arrangement. However, despite various CoilSer derivatives subsequently crystalized as parallel assemblies,16,30–33 and reports of parallel designs with Trp/Tyr located in the same N-terminal a position,34,35 this misconception has continued to persist. In our case, it is inconceivable that the Tb(MB1-1(2W))3 complex features an antiparallel arrangement, as this could not provide an adequate Tb3+ ligand donor set, especially whilst preventing access of exogenous water. Hopefully, the work presented herein, together with the literature described, will lead to the greater exploitation of the Trp side chain sterics by the coiled–coil design community in terminal hydrophobic positions. Though a single Trp has been successfully introduced in the buried hydrophobic core of a parallel hetero three stranded coiled coil,36 it remains to be established whether multiple Trp can be tolerated in such sites.
This study has for the first time demonstrated that it is possible to truly control the hydration state of metal binding sites (from three, two, one and zero waters), by changing a terminal second sphere residue. With metal hydration often dictating the resulting chemistry, herein demonstrated with MRI, but also key for substrate access, “vacant” coordination sites, and catalysis more widely, this is a crucial goal that we have now realized. These findings will be of use to metallocoiled coil and protein designers more widely, for the truly de novo design of any desired metal site, the ultimate goal of metalloprotein design.
We thank the EPSRC (EP/L504907/1,EP/P511286/1), University of Birmingham and DSTL for support of this research.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Dudev, Y. L. Lin, M. Dudev and C. Lim, J. Am. Chem. Soc., 2003, 125, 3168 CrossRef CAS.
- F. Yu, V. Cangelosi, M. L. Zastrow, M. Tegoni, J. Plegaria, A. Tebo, C. Mocny, L. Ruckthorn, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495 CrossRef CAS PubMed.
- L. N. Slope and A. F. A. Peacock, Chem. – Asian J., 2016, 11, 660 CrossRef CAS PubMed.
- A. Lombardi, C. Summa, S. Geremia, L. Randaccio, V. Pavone and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6298 CrossRef CAS PubMed.
- K. J. Koebke, F. Yu, E. Salerno, C. van Stappen, A. G. Tebo, J. E. Penner-Hahn and V. L. Pecoraro, Angew. Chem., Int. Ed., 2018, 57, 3954 CrossRef CAS PubMed.
- S. Geremia, L. D. Costanzo, L. Randaccio, D. E. Engel, A. Lombardi, F. Nastri and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 17266 CrossRef CAS PubMed.
- L. Ruckthong, J. A. Stuckey and V. L. Pecoraro, Chem. – Eur. J., 2019, 25, 6773 CrossRef CAS PubMed.
- O. D. Monera, N. E. Zhou, P. Lavigne, C. M. Kay and R. S. Hodges, J. Biol. Chem., 1996, 271, 3995 CrossRef CAS PubMed.
- K.-H. Lee, M. Matzapetakis, S. Mitra, E. N. G. Marsh and V. L. Pecoraro, J. Am. Chem. Soc., 2004, 126, 9178 CrossRef CAS PubMed.
- L. Ruckthong, A. Deb, L. Hemmingsen, J. E. Penner-Hahn and V. L. Pecoraro, J. Biol. Inorg. Chem., 2018, 23, 123 CrossRef CAS PubMed.
- K.-H. Lee, C. Cabello, L. Hemmingsen, E. N. G. Marsh and V. L. Pecoraro, Angew. Chem., Int. Ed., 2006, 45, 2864 CrossRef CAS PubMed.
- A. F. A. Peacock, O. Iranzo and V. L. Pecoraro, Dalton Trans., 2009, 2271 RSC.
- A. F. A. Peacock, L. Hemmingsen and V. L. Pecoraro, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16566 CrossRef CAS PubMed.
- E. Haviv, D. Azaiza-Dabbah, R. Carmieli, L. Avram, J. M. L. Martin and R. Neumann, J. Am. Chem. Soc., 2018, 140, 12451 CrossRef CAS PubMed.
- P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2019, 58, 4504 CrossRef CAS PubMed.
- B. Therrien, Front. Chem., 2018, 6, 602 CrossRef CAS PubMed.
- N. E. Borisova, T. B. Sumyanova, A. V. Kharcheva, P. I. Matveev, A. V. Ivanov, E. A. Razumova and S. V. Patsaeva, Dalton Trans., 2018, 47, 16755 RSC.
- A. Pannwitz, S. Poirier, N. Bélanger-Desmarais, A. Prescimone, O. S. Wenger and C. Reber, Chem. – Eur. J., 2018, 24, 7830 CrossRef CAS PubMed.
- Y. Ning, Y.-W. Liu, Y.-S. Meng and J.-L. Zhang, Inorg. Chem., 2018, 57, 1332 CrossRef CAS PubMed.
- R. Herchel, P. Zoufalý and I. Nemec, RSC Adv., 2019, 9, 569 RSC.
- M. R. Berwick, D. J. Lewis, A. W. Jones, R. A. Parslow, T. R. Dafforn, H. J. Cooper, J. Wilkie, Z. Pikramenou, M. M. Britton and A. F. A. Peacock, J. Am. Chem. Soc., 2014, 136, 1166 CrossRef CAS PubMed.
- M. R. Berwick, L. N. Slope, C. F. Smith, S. M. King, S. L. Newton, R. B. Gillis, G. G. Adams, A. J. Rowe, S. E. Harding, M. M. Britton and A. F. A. Peacock, Chem. Sci., 2016, 7, 2207 RSC.
- D. S. Touw, C. E. Nordman, J. A. Stuckey and V. L. Pecoraro, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11969 CrossRef CAS PubMed.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79 CrossRef CAS PubMed.
- M. S. Rogers, E. M. Tyler, N. Akyumani, C. R. Kurtis, R. K. Spooner, S. E. Deacon, S. Tamber, S. J. Firbank, K. Mahmoud, P. F. Knowles, S. E. V. Phillips, M. J. McPherson and D. M. Dooley, Biochemistry, 2007, 46, 4606 CrossRef CAS PubMed.
- R. F. Abdelhamid, Y. Obara, Y. Uchida, T. Kohzuma, D. M. Dooley, D. E. Brown and H. Hori, J. Biol. Inorg. Chem., 2007, 12, 165 CrossRef CAS PubMed.
- A. K. Chaplin, C. Bernini, A. Sinicropi, R. Basosi, J. A. R. Worrall and D. A. Svistunenko, Angew. Chem., Int. Ed., 2017, 56, 6502 CrossRef CAS.
- J. Liu, W. Yong, Y. Deng, N. R. Kallenbach and M. Lu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16156 CrossRef CAS.
- B. Lovejoy, S. Choe, D. Cascio, D. K. McRorie, W. F. Degrado and D. Eisenberg, Science, 1993, 259, 1288 CrossRef CAS.
- S. Chakraborty, D. S. Touw, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, J. Am. Chem. Soc., 2010, 132, 13240 CrossRef CAS.
- L. Ruckthong, M. L. Zastrow, J. A. Stuckey and V. L. Pecoraro, J. Am. Chem. Soc., 2016, 138, 11979 CrossRef CAS.
- A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Angew. Chem., Int. Ed., 2009, 48, 7371 CrossRef CAS PubMed.
- M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Nat. Chem., 2012, 4, 118 CrossRef CAS.
- O. Iranzo, D. Ghosh and V. L. Pecoraro, Inorg. Chem., 2006, 45, 9959 CrossRef CAS PubMed.
- H. Wendt, C. Berger, A. Baici, R. M. Thomas and H. R. Bosshard, Biochemistry, 1995, 34, 4097 CrossRef CAS.
- A. Kashiwada, K. Ishida and K. Matsuda, Bull. Chem. Soc. Jpn., 2007, 80, 2203 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc08189e |
| ‡ MB1-1(2Y), MB1-1(2W) and MB1-4(37W) feature Gln in place of the external Trp residue (f position), so as to retain a single UV absorbing residue in the sequence. |
| § Tb3+ and Gd3+ are routinely used interchangeably given their almost identical coordination chemistry. Herein we take advantage of the visible emission afforded by Tb, but not Gd, to interrogate the binding site. |
| ¶ One Trp has been solved as being in two equal occupancy conformations, but only one conformation is shown here. |
| || Coiled coils are based on the heptad repeat HxxHxxx, where H corresponds to a hydrophobic amino acid, and the relative position within the heptad is referred to as abcdefg. Both a and d residues are located within the hydrophobic core of a three stranded coiled coil, but their side chains are directed differently relative to the helix backbone. |
| This journal is © The Royal Society of Chemistry 2020 |