
Metal organic frameworks as solid catalysts for liquid-phase continuous flow reactions
Amarajothi
Dhakshinamoorthy
 *a,
Sergio
Navalon
b,
Abdullah M.
Asiri
c and
Hermenegildo
Garcia
*cd
*a,
Sergio
Navalon
b,
Abdullah M.
Asiri
c and
Hermenegildo
Garcia
*cd
aSchool of Chemistry, Madurai Kamaraj University, Madurai-625 021, Tamil Nadu, India. E-mail: admguru@gmail.com
bDepartamento de Quimica, Universitat Politecnica de Valencia, Av. De los Naranjos s/n, 46022 Valencia, Spain
cCenter of Excellence for Advanced Materials Research, King Abdulaziz University, Jeddah, Saudi Arabia
dDepartamento de Quimica and Instituto Universitario de Tecnologia Quimica (CSIC-UPV), Universitat Politecnica de Valencia, Av. De los Naranjos s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es
First published on 18th November 2019
Abstract
Metal organic frameworks (MOFs) are widely used as solid catalysts in the liquid phase under batch mode conditions. Moving towards the development of industrial processes, data of the performance of MOFs under continuous flow operation would be desirable. This feature article describes the state of the art regarding the use of MOFs as catalysts of continuous flow processes, paying special attention to the issue of catalyst stability. The review is organized according to the type of bond that is formed in the reaction from C–C, to C–O to C–N bonds. Examples are presented of MOF catalysts that are stable under continuous flow operation, even for those structures that are not very stable such as Cu3(BTC)2. It can be anticipated that there will be a growth in the percentage of studies carried out under continuous flow with the final goal of implementing a commercial chemical process using MOFs as a catalyst.
 Amarajothi Dhakshinamoorthy | Amarajothi Dhakshinamoorthy obtained his PhD, degree in 2009 from Madurai Kamaraj University, Tamil Nadu, India. Later, he worked as a postdoctoral researcher with Prof. Hermenegildo Garcia at the Technical University of Valencia for four years. Then, he returned to India and joined as a UGC-Assistant Professor in June 2013 at School of Chemistry, Madurai Kamaraj University. His research interests include catalysis by metal–organic frameworks and graphene-related materials. He is the recipient of the Young Scientist Award 2014 for Chemical Sciences by The Academy of Sciences, India. He was also awarded an INSA-DFG international bilateral exchange award to visit Germany in 2015. |
 Sergio Navalon | Sergio Navalon was born in Valencia (Spain) in 1979. He graduated in Chemical Engineering in 2003 and obtained his PhD in 2010 at the Technical University of Valencia (UPV). Currently, he is working as an Associate Professor at the Chemistry Department of UPV. His research area includes the development of heterogeneous (photo)catalysts based on graphene, porous materials and nanoparticles, as well as green chemistry processes. He has co-authored around 60 publications. |
 Abdullah M. Asiri | Abdullah M. Asiri received a PhD from University of Wales, College of Cardiff, UK in 1995. He has been the Head of the Chemistry Department at King Abdulaziz University since October 2009 and he is the founder and the Director of the Center of Excellence for Advanced Materials Research. He is a Professor of Organic Photochemistry. He is the Editor-in-Chief of King Abdulaziz University Journal of Science. He holds 10 granted USA patents, more than 1242 Publications in international Journals (WoS), ten book Chapters, and 28 Books. He won the best researcher award in KAU for many successive years. |
 Hermenegildo Garcia | Hermenegildo Garcia is a full Professor at the Instituto de Tecnologıa Quimica of the Technical University of Valencia and Honorary Adjunct Professor at the Center of Excellence in Advanced Materials Research of King Abdulaziz University. Prof. Garcia has been active in the field of heterogeneous catalysis working with porous catalysts and nanoparticles, has published over 750 papers and has filed over 25 patents. Prof. Garcia is Doctor Honoris Causa from the University of Bucharest and the recipient of the 2011 Janssen-Cilag award given by the Spanish Royal Society of Chemistry and the 2008 Alpha Gold of the Spanish Society of Glass and Ceramics. |
1. Introduction
Metal organic frameworks (MOFs), in which metallic nodes of one or a few metal cations are coordinated to rigid organic linkers, are becoming increasingly used in heterogeneous catalysis for liquid phase reactions under moderate conditions.1–5 The main reasons to explain why these porous crystalline materials have gained such large importance as solid catalysts include their large surface area and porosity,6,7 easy and reliable preparation and synthesis by design,8,9 and the large proportion of transition metals in their composition. There are many examples of MOFs in which the metal nodes contain some coordination positions not compromised with the construction of the lattice. These coordination positions are typically occupied by solvent molecules or other exchangeable ligands that can be easily removed, upon activation, generally by thermal treatment under vacuum, resulting in coordinatively unsaturated positions10,11 around the metal clusters that can act as Lewis sites.12 In addition to exchangeable ligands, MOFs always contain a certain density of defects in which the metal ions are not well coordinated and these defects can behave also as catalytic sites.13–16Compared to other porous solids used in heterogeneous catalysis,17–20 and zeolites21 in particular, MOFs offer considerable flexibility in design and the possibility to select structures with large pore dimensions in the range of mesopores and low framework density.22,23 MOFs are considered as the materials with the highest open porosity and the record of free empty space in the structure often is over 50% volume.24
Another especially important feature of MOFs is the large variety of transition metals and ligands that can be employed in their preparation.25–27 Moreover, by knowing the directionality of the coordination bonds around the metal clusters and the geometry and dimensions of the organic linkers, the topology of the voids in MOFs can be predicted.28,29 This possibility to anticipate pore size and pore geometry in MOFs has led to the concept of synthesis by design meaning that there is a considerable predictive understanding of the porosity in MOFs.30–32
For gas phase reactions, particularly in the refining and petrochemical industry, zeolites and porous aluminosilicates are the preferable solid catalysts.33–35 However, due to the limited pore size of conventional zeolites, the activity of these crystalline aluminosilicates in a liquid phase reaction is considerably much lower19 and there was an interest in the development of other classes of porous solid catalysts for this type of reaction, particularly for the production of fine chemicals36,37 where the size of the molecules is larger than normal alkanes found in refining and petrochemistry. In this context, since the first reports of MOF synthesis, it was clear that these materials should be very promising as solid catalysts, complementing zeolites, particularly in the liquid phase.38
However, in spite of the previously commented positive structural features exhibited by MOFs, one of the limiting factors that has hampered applications of MOFs at industrial scale has been the poor structural stability of many MOFs.5,39,40 In contrast to zeolites that can be reactivated by combustion of the organic matter at temperatures above 500 °C under air,41,42 MOFs cannot stand thermal treatments above 400 °C and in many cases, even at much lower temperature. Besides thermal stability, MOFs can also be unstable in the presence of certain acidic/basic conditions, aggressive reagents or even solvents.43 For instance, MOFs can stand aqueous solutions in a certain range of pH value and amines and even carboxylic acids can deteriorate the structure of MOFs.
However, although the lack of stability is well known in the area, there are certain structures that are remarkably robust and stable both upon heating and in the presence of chemical reagents. Examples of these stable MOFs include certain materials of the MIL series44,45 (MIL: Materials Institute of Lavoisier), such as MIL-100(Cr) and MIL-101(Cr), and different MOFs of Zr-based nodes, particularly UiO-669,46,47 (UiO: University of Oslo). In the last case, it is possible to heat at 400 °C causing a reversible dehydration of the Zr6O4(OH)4 nodes, but without causing the collapse of the crystal structure. In the case of MIL-101, it is possible to perform harsh reactions at the organic linker such as sulfonation with highly corrosive chlorosulfonic acid without altering the crystallinity of the sample. The crystal structure of UiO-66 consists of a cubic framework of cationic Zr6O4(OH)4 nodes and 1,4-benzenedicarboxylate (BDC) as linkers.48 UiO-66 is one of the most robust MOFs. UiO-66 is often synthesised in the presence of modulators to introduce structural defects or increase crystallinity.49 Besides, it can also be subjected to post-synthetic modifications50 to tune further the active sites. The combination of these interesting properties together with high surface area and large pore size makes UiO-66 an attractive material for catalytic applications.47,51 In many other cases, experimental evidence has shown that the crystal structure can stand the reaction conditions and they are acting as catalytically stable materials.52Table 1 summarizes the most commonly used MOFs in catalysis indicating their metal ions/node, linkers, typical BET surface area and pore volume.
| Catalyst | Metal ion/node | Linker | BET surface areaa (m2 g−1) | Pore volumea (cm3 g−1) |
|---|---|---|---|---|
| a These values are typical data reported in the literature and they may vary depending on the measurements. | ||||
| MIL-101(Cr) | Cr3+ | BDC | 2750 | 2.2 |
| MIL-100(Fe) | Fe3+ | 1,3,5-Benzenetricarboxylic acid (BTC) | 2000 | 1.2 |
| Cu3(BTC)2 | Cu2+ | BTC | 1200 | 0.7 |
| UiO-66 | Zr6O4(OH)4 | BDC | 1250 | 0.5 |
However most of the data on catalyst stability reported so far are based on batch conditions, evaluating the performance of the material in a series of consecutive reuses of the same sample.53 Reusability tests typically consider the initial reaction rates, conversions and selectivity values of the same sample submitted to consecutive runs. Catalytic tests are combined with complimentary analytical and structural characterization of the used solid showing that the crystallinity is preserved upon successive reuses and that the metal content of the materials has remained unchanged.
In spite of the considerable information gathered for the activity of MOFs as solid catalysts in liquid phase reactions, application of these materials for large scale reactions in industry still remains elusive. Implementation of commercial processes using MOFs as catalysts for the production of fine chemicals in the liquid phase is a long sought goal pursued in this field that will serve to demonstrate the superiority of MOF solid catalysts in liquid phase reactions over other alternative solid catalysts.54,55
Towards this final goal, one necessary step is to determine the catalytic behaviour of MOFs under continuous flow operation.56,57 Industrial processes typically prefer continuous flow operation versus batch reactions, since the former represents an intensive process, resulting in higher productivity.58–60 Compared to batch conditions, operation under continuous flow provides unique information about catalyst stability and the main deactivation pathways.61 In this context, it is remarkable that there is a paucity of information about the behaviour of MOFs as catalysts under continuous flow conditions in the liquid phase. Studies of the catalytic behaviour of MOFs under continuous flow can surely serve to demonstrate their stability under reaction conditions.
Compared to batch reactions, continuous flow presents significant advantages, including a high ratio of surface-to-volume, enhanced heat transfer and precise temperature control, higher reaction rates, operation under extreme conditions, safer handling of highly exothermic, explosive, or toxic reagents, good control of the residence time, easy automation and scale-up and cost effectiveness.
Considering these beneficial advantages of continuous flow reactions compared to batch processes, many industrial processes, including the manufacture of pharmaceutical products, are being performed under continuous flow mainly due to the easy recovery of the products from the catalysts and the remarkable process intensification in terms of production per unit of time. On the other hand, efficient separation of the reaction mixture from the solid catalyst presents the two-fold advantages of isolating the final product in high purity and minimizing catalyst deactivation by poisoning. Furthermore, selectivity can be higher under continuous flow conditions by arresting consecutive reactions that could occur in the primary products.
Solid catalyst stability in batch reactions is commonly ascertained by recovering the solid after the reaction and reusing it in a subsequent run, measuring initial reaction rates and final conversions at a certain time. Although it is considered as the most convenient way to study catalyst stability in a batch reaction, this method suffers from several disadvantages including unavoidable catalyst loss and tedious and time consuming workup. On the other hand, the stability of a solid catalyst can easily be monitored under continuous flow operation by measuring the decay in conversion over the time on stream. Chart 1 shows examples of continuous flow reactors namely, capillary, packed-bed, honeycomb and monolithic reactors. These reactors are classified based on the dimensions of the inner diameter varying between micro (10–500 μm) or mesofluidic (500 μm up to few mm) reactors and on the arrangement of the reactor channels.62,63 It should be noted at this point that the previous classification has been often employed for a continuous flow process with solid catalysts and that the number of reports using MOFs as solid catalysts for liquid-phase continuous flow is still very limited, particularly compared to the use of MOFs under batch conditions.
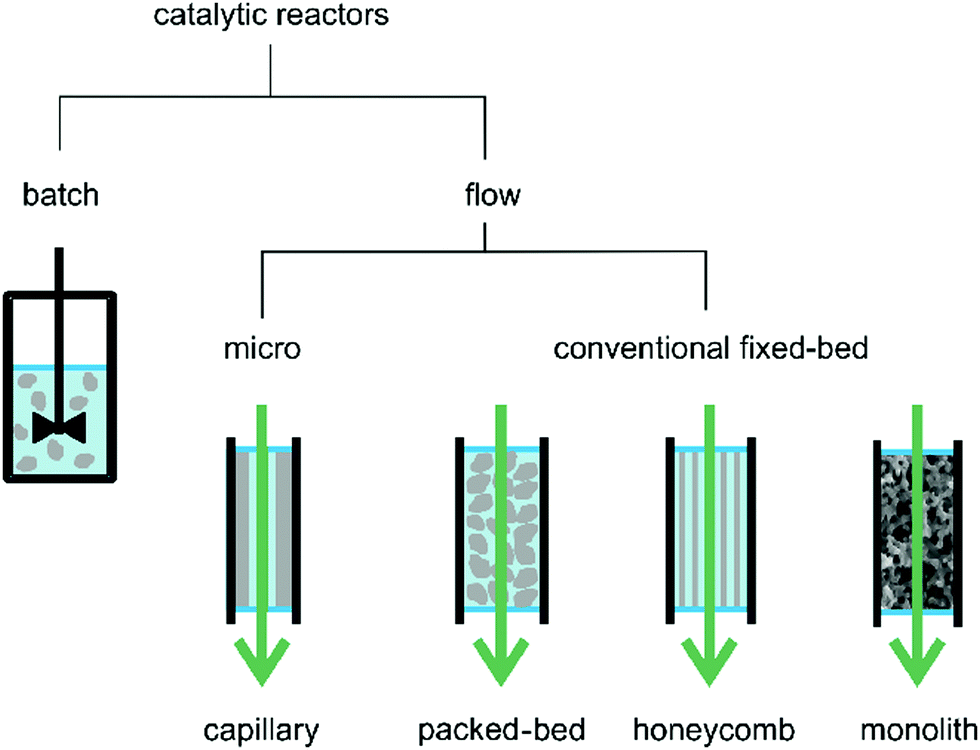 | ||
| Chart 1 A representation of most common reactor types for batch and continuous-flow reactions. Particles, layer or body of the catalyst are shown in grey. Reproduced with permission from ref. 64 Copyright Bentham 2017. | ||
Development of continuous flow reactors is an important technological aspect for both synthetic chemists and process engineering.62,65–73 Interestingly, various continuous flow reactors and processes have been reported for the synthesis of drug molecules through cross coupling reactions, including Suzuki–Miyaura, Sonogashira and Heck,74,75 as well as for hydrogenation of alkynes64 and aerobic oxidations.76 Recently, Pd@MIL-101-NH2 was packed in a micro-flow reactor and employed as a catalyst for the preparation of a series of biaryls of commercial importance under continuous flow, exhibiting a remarkable stability up to 54 h.77
Very recently, the main achievements, challenges and limitations regarding the synthesis of fine chemicals, pharmaceuticals and bulk chemicals under continuous flow has been reviewed and readers are encouraged to refer to this article for a detailed overview.78 In another review article, the use of MOFs and covalent organic frameworks packed in columns as a stationary phase in chromatographic separation as well as continuous flow synthesis was discussed.79 In this context, the main objective of the present article is to show the relevance and opportunity for developing liquid-phase continuous flow reactions using MOFs as catalysts to achieve the final goal of implementing commercial synthetic processes based on these porous materials.
Due to the significant advances in the field of micro- and mesoreactors, several examples of the synthesis of drugs and fine chemicals using continuous flow processes have recently been described. Some of the main reasons that make continuous flow preferable to batch processes include higher productivity for a given period of time, but also better yields or selectivity and safer process together with less environmental impact. Although batch reactions are more versatile and flexible being adequate for the first stages of catalytic studies at a laboratory scale, they are less adequate than continuous flow processes for scaling up and present practical complications to implement adequate heat and mass transfer, which may heavily hinder productivity. Continuous flow processes are also better suited for automation and require less manpower. Thus, continuous flow systems are characterised by reduced costs and maximize the economic viability of new drugs and products.80–83 For these reasons, studies under continuous flow conditions for liquid phase reaction generally correspond to reactions of potential industrial interest and are based on stable catalysts.
The purpose of the present review is to cover the existing literature on the use of MOFs as catalysts under continuous flow operation in liquid phase reactions. The existing literature has been reviewed placing particular emphasis on the most recent achievements, especially in the use of MOFs in enantioselective continuous flow processes.84 It will be shown that in several cases, remarkably stable MOFs catalysts for certain reactions under continuous flow such as acetalization, cyclocondensations, CO2 insertion and alcohol oxidation have been reported, illustrating the possibility to go one step further towards the implementation of MOFs as catalysts for large scale processes. Recently, Lin and co-workers have published a perspective on the various strategies employed in gas-phase reactions using MOFs as solid catalysts, discussing the various limiting factors in this type of process.85 Hence, the present review is exclusively focussed on the use of MOFs as catalysts in continuous flow reactions in the liquid phase, focussing mainly on activity and stability. Emphasis will be made in comparing the performance of MOFs as catalysts with other solid catalysts. Unfortunately, given the incipient development level of MOFs in continuous flow, the current lack of information makes it not possible to provide a fair comparison for every reaction.
The following sections are organized based on the type of bond formed in the final product. Emphases are given to the structural aspects of the MOF catalysts employed for the continuous flow processes, reactant conversion level, product selectivities, productivity data and catalyst stability based on catalytic data and on characterization of the catalyst after extensive use by analytical or microscopic techniques. Table 2 provides an overview of reactions that have been screened up to now under continuous flow in the liquid phase using MOFs as solid catalysts, the reactor type employed and the activity and stability reached.
| Catalyst | Reactor type | Reaction | Activity | Stability | Ref. |
|---|---|---|---|---|---|
| a L1 structure in Scheme 1; DMA: N,N-dimethylacetamide. b BIT: Beijing Institute of Technology. c BTC: 1,3,5-benzenetricarboxylate. d Silica monolith. e OPH: organophosphorous hydrolase. | |||||
| [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMAa | Stainless steel column filled with finely grounded solid | Friedel–Crafts reaction | 89–92% yield with 91–94% ee after 12 h time on stream (TOS) | Reused seven times | 86 |
| Nano-BIT-58 filmb | 70 wt% nano BIT-58 in polyacrylonitrile fabricated by electrospinning | Knoevenagel condensation | 100% conversion | Reused three times | 87 |
| ZrOTf-BTC | Stainless steel column packed with ZrOTf-BTC and SiO2 | Diels–Alder, ring opening of epoxide and Friedel–Crafts reactions | TON > 2700 | Thirty cycles | 88 |
| Cu3(BTC)2c | Mixed matrix membranes of Cu3(BTC)2 and cellulose acetate | Acetalization | 0.59 mmol min−1 gCu3(BTC)2−1 | TOS of 24 h | 89 |
| Cu3(BTC)2 | Packed-bed | Acetalization | 3.12 h−1 | Reuse, XRD, BET | 90 |
| Copper hydroxysulfate@Cu3(BTC)2 | Flow | Acetalization | 87% yield and 99% selectivity | — | 91 |
| MIL-101(Sc) | Packed-bed | CO2 cycloaddition to propylene oxide | Turnover frequency (TOF) 87 h−1 | Stable up to 5 h, XRD | 92 |
| Co(CO)4⊂MIL-101(Cr) | Packed-bed | Carbonylation of β-lactones | 1300 molAnhydride molCo−1 after 6 h | — | 93 |
| UiO-66(Zr) | Packed-bed | Methyl levulinate to γ-valerolactone | 92.3 mmolGVL g−1 h−1 | TOS = 1 h BET, TEM | 94 |
| Cu3(BTC)2-MonoSild | Packed-bed (2 cm length × 6 cm diameter) | Friedlander reaction | 826 g of the desired product per g of Cu3(BTC)2 per day | — | 95 |
| Pd@MIL-88B-NH2@nano-SiO2 | Packed-bed | Alcohol oxidation | Conversion at around 80% | Conversion almost stable for 7 days | 96 |
| UiO-66(Zr,Ti) | Tubular glass column | Oxidative desulfurization of thioanisole | <1 ppm of sulphur | — | 97 |
| OPH@MIL-100(Fe)e | Plug-flow | Cascade degradation of organophosphate nerve agents to 4-aminophenol | Retained more than 50% of the initial activity after nine cycles | Retained more than 50% of the initial activity after nine cycles | 98 |
2. C–C bond formation
One of the fundamental challenges in heterogeneous catalysis is the appropriate design of an efficient catalyst featuring high activity and recyclability without deactivation exhibiting an exquisite selectivity. In this regard, stereoselective and enantioselective reactions correspond to the most challenging case, due to the structural similarity of the products. In a study of catalytic asymmetric induction, Cui and co-workers have recently developed high-performance heterogeneous asymmetric catalysts using a ligand design strategy. Three porous chiral MOFs with the general formula [Me2NH2][Mn2(Li)(H2O)2]·2H2O·nDMA (for Li see the structure in Fig. 1, n = 2 or 3) were prepared from enantiopure phosphono-carboxylate ligands of 1,1′-biphenol that are functionalized with 3,5-bis(trifluoromethyl)-, bismethyl-, and bisperfluorophenyl substituents at the 3,3′-position.86 The carboxylate and phosphonate groups were coordinated to metal ions to form crystalline porous networks in which the metal nodes act as Lewis acids, while the 3,3′-substituents generate a chiral environment that dictates the stereochemical and electronic control over the organic reactions. TGA indicated that these solids are stable up to 380 °C. Chemical stability experiments with acids and bases revealed that the stability increases in the order of [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA > [Me2NH2][Mn2(L2)(H2O)2]·2H2O·3DMA > [Me2NH2][Mn2(L3)(H2O)2]·2H2O·3DMA which follows the same order as the increase in the size of the bulky phenyl substituent at the 3,3′-position of the 1,1′-biphenyl ligands. | ||
| Fig. 1 (a) Structure of the organic ligands H5L1–H5L3 employed in the synthesis of chiral MOFs; (b) construction of enantioselectively pure MOFs from dimeric [Mn2(CO2)4(PO4)(H2O)2] linked by H5L to give 3D networks with fns topology; (c) framework structures of the resulting chiral Mn MOFs with channel diameters of ∼2.5 × 2.5 nm2 and ∼0.68 × 0.68 nm2 along the c-axis (Mn, green polyhedra; O, red; P, yellow; C, black; F, light blue. H atoms are omitted for clarity). Reproduced with permission from ref. 86 Copyright American Chemical Society 2017. | ||
The activity of these chiral solid catalysts was studied in the Friedel–Crafts reaction of N-methylindole with methyl(E)-2-oxo-4-phenylbut-3-enoate (Scheme 1). The experimental results have shown that [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA exhibits much higher activity and stereoselectivity induction ability than the other two solids. [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA gave in chloroform at 30 °C the 1,4-addition product in 91% yield with 96% ee. Analogously, a wide range of β,γ-unsaturated α-keto esters with various electronic properties and different ring sizes were reacted with N-methyl or N-benzylindole, achieving the expected products in 84–95% yields with 91–99.9% ee. The 1,4-addition product was the only product observed in all substrates studied, the 1,2-addition isomer not being formed. In comparison, the addition of N-methylindole to α-keto esters in the presence of [Me2NH2][Mn2(L2)(H2O)2]·2H2O·3DMA and [Me2NH2][Mn2(L3)(H2O)2]·2H2O·3DMA resulted in 73–82% and 79–84% yields and 55–74% and 71–85% ee of the products, respectively, which are lower values than with [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA under identical conditions. The enhanced activity of [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA is proposed to arise from the o-disubstitution of the aromatic ring with strong electron withdrawing –CF3 groups which should favour the increase of both the steric encumbrance around the sites and the Lewis acidity of metal ions, thereby resulting in a notable enhancement of the reaction rate and the ee values. In contrast, the activity of the ligand as well as the metal nodes in solution was lower in terms of yield and ee values under identical conditions. In addition, [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA was reused for ten cycles without any decrease in its activity, regioselectivity and enantioselectivity. The powder XRD pattern and BET surface area of the ten-times reused solid remained unchanged compared to the fresh solid. Hot filtration experiments showed also the absence of leaching of Mn. All these characterization data indicate the stability of the catalyst under reaction conditions. Furthermore, the Friedel–Crafts reaction of trans-β-nitrostyrene with pyrrole (Scheme 1) was also catalyzed by [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA solid affording 87% yield with 97% ee in chloroform at 0 °C. The substrate scope of this solid catalyst was further exploited with other aryl-substituted nitroalkenes possessing electron-donating and electron-withdrawing groups on the aromatic rings, achieving excellent to high yields (79–93%) and high ee values (62–97%). [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA MOF promoted pyrrole alkylation with stereoselectivities close to the values reached with other homogeneous and heterogeneous catalysts based on binaphthyl derived phosphoric acids (Table 3).
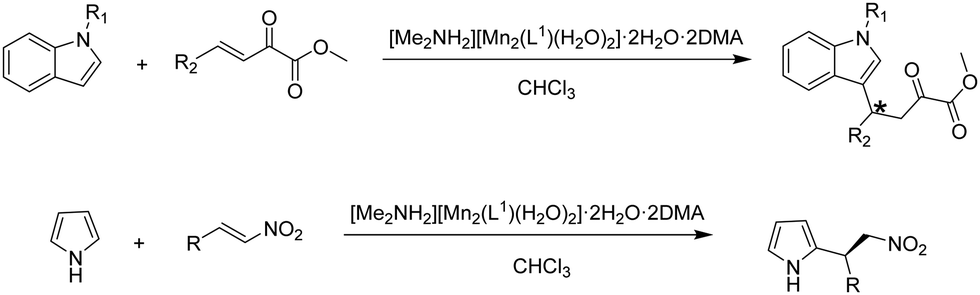 | ||
| Scheme 1 Enantioselective Friedel–Crafts alkylation reactions catalyzed by chiral [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA. | ||
The performance of [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA was also evaluated under continuous flow set up as shown in Fig. 2. The continuous flow reaction of N-methylindole with α-keto esters was performed in a stainless steel column filled with finely ground [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA (0.4–5 μm) mixed with quartz sand. Under optimized flow conditions, a complete conversion of α-keto esters towards the formation of the 1,4-addition product in 89–92% yield with 91–94% ee after a residence time of 12 h was achieved. The comparison of the batch and flow processes shows that the flow process afforded a slight increase in the yield, but a slight decrease in enantioselectivity. This lower ee value for the continuous flow process was ascribed to the negative effect of the silica needed for packing the catalyst bed in the reactor that introduces adventitious sites deteriorating the enantioselectivity.
 | ||
| Fig. 2 Experimental set up for the fixed bed reactor used for the enantioselective Friedel–Crafts reactions between N-methylindole and β,γ-unsaturated α-ketoesters under continuous flow conditions using [Me2NH2][Mn2(L1)(H2O)2]·2H2O·2DMA as a catalyst. Yields of 89, 91, and 92% with 94, 91, and 94% ee were obtained for R2 with 4-MePh, 4-BrPh and 4-thienyl, respectively. Reproduced with permission from ref. 86 Copyright American Chemical Society 2017. | ||
One of the key findings in this study is the proposal that the availability of a large pore size in an MOF catalyst assists the distribution of the heat generated during the reaction, avoiding coke formation generated by local over heating as observed with zeolites.103 This is one of the beneficial advantages of using MOFs as heterogeneous catalysts compared to other porous solids with smaller pore size and lower thermal conductivity like zeolites.
The catalyst of the flow system was reused for seven cycles (each a 24 h cycle) without any decrease in activity and selectivity. This is one of the first reports of MOF-based heterogeneous asymmetric catalysts for continuous-flow reactions and illustrates the vast potential that MOFs offer in this area.
Knoevenagel condensation between aldehyde and an active methylene compound is a very versatile reaction widely used in organic synthesis. In this context, a coordination modulation method was developed for the synthesis of nano-sized Ce-MOF through defect engineering and an electrospinning process (Fig. 3). Nano-sized Ce-MOF was used as a catalyst in Knoevenagel condensation.87 The Ce-BTB MOF (BIT-58; BIT: Beijing Institute of Technology) was obtained by coordination between Ce3+ as the metal center and 1,3,5-tris(4-carboxyphenyl)benzene (BTB) as a ligand. The crystal size and defects of BIT-58 were controlled by the presence of various nitrogen heterocycles such as 1-methylimidazole acting as a modulator during the synthesis of the material.104 BIT-58 possesses a one-dimensional channel system with pore diameters of about 1 nm. Ce exists in a BIT-58 structure as a trivalent ion as revealed by XPS and coordinates to nine oxygen atoms as a CeO9 cluster. BIT-58 showed very high chemical and thermal stability. BIT-58 was stable at temperatures as high as 490 °C under N2 and 400 °C under air. The crystallite size of BIT-58 powders was adjusted by treating with 1-methylimidazole as a modulator as well as a monodendate ligand that competes with the coordination sites and controls the growth of MOF crystals.105 The average particle size of BIT-58 was greatly reduced from 25 μm to 30 nm, while preserving the crystallinity, rendering nano-BIT-58 samples.
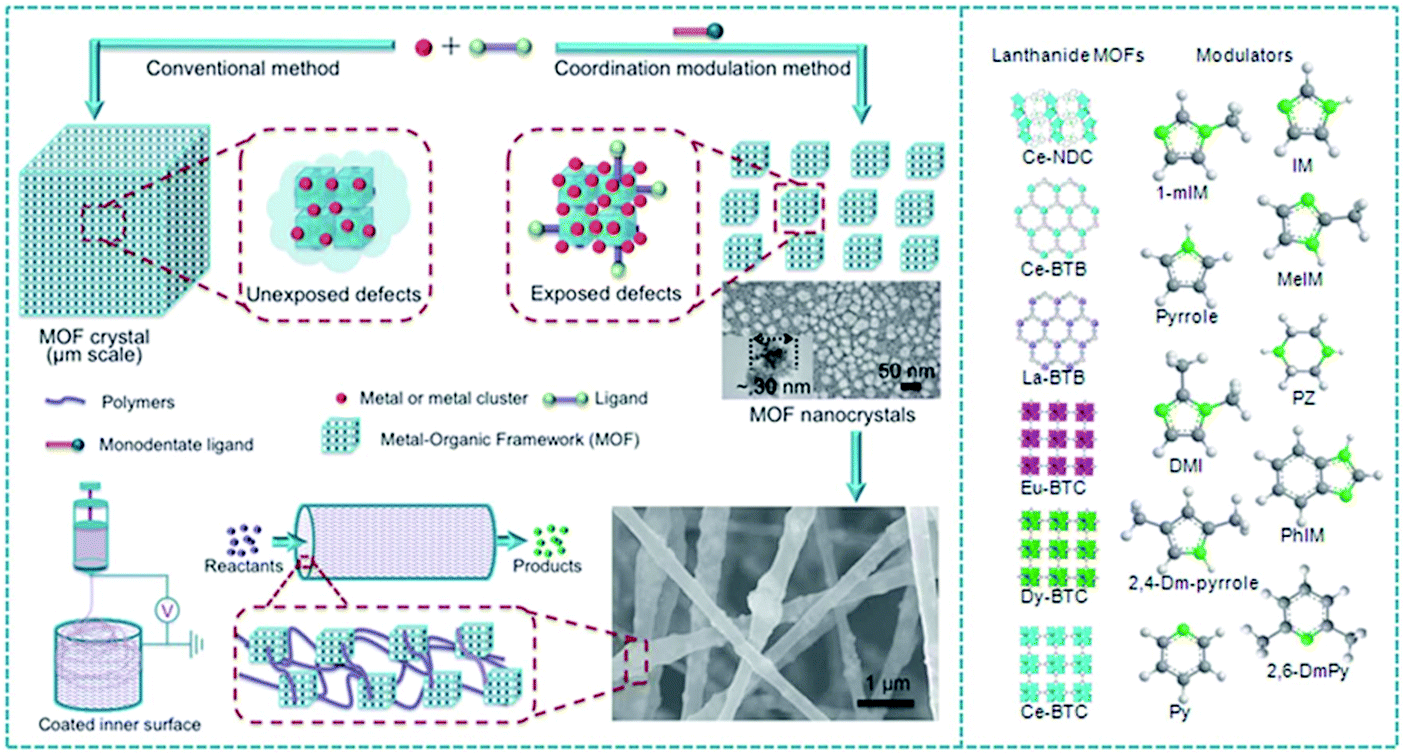 | ||
| Fig. 3 Representation of the coordination modulation concept for lanthanide MOF synthesis of NPs and fabrication of a catalyst film by electrospinning. Reproduced with permission from ref. 87 Copyright Royal Society of Chemistry 2018. | ||
Gas adsorption measurements indicated that nano-BIT-58 showed a slightly higher BET surface area of 1169 m2 g−1 than BIT-58 (1075 m2 g−1). Although, the micropore volume of nano-BIT-58 (0.39 cm3 g−1, <2 nm) is similar to that of BIT-58 (0.40 cm3 g−1, <2 nm), the mesopore volume of nano-BIT-58 (0.35 cm3 g−1, 2–40 nm) was around seven times bigger than that of BIT-58 (0.05 cm3 g−1, 2–40 nm). This higher mesopore volume derives from the assembly of the nanoparticles (NPs) and is highly beneficial for avoiding mass transport limitation of substrates to the active sites. Furthermore, NH3-TPD titration curves revealed that nano-BIT-58 contains a total acid site amount of 7.06 sites per gram which is 10.5 times higher than the micrometric BIT-58 sample (0.67 sites per gram). This enhancement is due to the higher density of Lewis acid sites arising from defects (e.g., unsaturated metal sites) and higher population of Brønsted acid sites (e.g. uncoordinated carboxy groups) present in the crystal structure upon treatment with 1-methylimidazole. It is unclear, however, what exactly the number of 7.06 sites per gram corresponds to in terms of mmols of acid sites per gram.
The activity of BIT-58 was tested in the Knoevenagel condensation reaction under continuous flow conditions. The conversion efficiency for the reaction between benzaldehyde and malononitrile with nano-BIT-58 was 100%, while the activity of BIT-58 was only 78% which clearly indicates the superior activity of the former catalyst due to its higher acidity derived from defects. A similar reactivity trend was observed for the other substrates tested. Particularly, the conversion of 1-naphthaldehyde was under similar reaction conditions 31 and 90% with BIT-58 and nano-BIT-58, whereas the conversion of 9-anthraldehyde was 3 and 42% for BIT-58 and nano-BIT-58, respectively. These experimental results nicely agree with lesser diffusion limitations for nano-BIT-58 compared to BIT-58, resulting in a higher activity of the former.
A nano-BIT-58 film (70 wt% nanoBIT-58 in polyacrylonitrile (PAN)) was fabricated by electrospinning with intact internal topology of the nano MOF (Fig. 4). This film displayed a nanofiber-interweaved network, where nano-BIT-58 particles are uniformly dispersed. The porosity of the nano-BIT-58 film was 409 m2 g−1 as evidenced by BET analysis. The catalytic efficiency of this film was studied under continuous flow, observing 100% conversion for the Knoevenagel condensation of benzaldehyde and malononitrile after 6 h. The catalytic film was recycled at least three times without any decay in activity.
 | ||
| Fig. 4 Experimental set up and photographs of a beaker before and after the coating with the nano-BIT-58 film used for Knoevenagel condensation between benzaldehyde and malononitrile at 60 °C for a residence time of 6 h. Reproduced with permission from ref. 87 Copyright Royal Society of Chemistry 2018. | ||
In this context, it is worth commenting that besides the importance in organic synthesis to form C–C bonds, the Knoevenagel condensation is one of the favourite probe reactions that has also been carried out with many other solid catalysts under continuous flow reactions, such as amino groups immobilized on mesoporous silicas,106 zeolites107 and organic polymers,108 among others,109 and the present data do not allow a clear picture of the possible advantages of the use of MOFs as solid catalysts for this process.
Recently, Zr-BTC (MOF-808) was post-synthetically modified to obtain ZrOTf-BTC MOF with enhanced Lewis acidity compared to MOF-808 by attaching triflate in the metal node (Scheme 2a).88 The catalytic activity of ZrOTf-BTC in a series of reactions including Diels–Alder reaction, epoxide ring-opening reaction and Friedel–Crafts acylation was significantly higher than that of Sc(OTf)3. Furthermore, ZrOTf-BTC was supported on SiO2 and this composite was packed in a stainless steel column and employed as a solid Lewis acid catalyst under continuous flow conditions. A mixture consisting of benzoquinone in dichloromethane and cyclohexa-1,3-diene (0.5 M) with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6 molar ratio was purged at a flow rate of 10 mL h−1 to achieve quantitative conversion of benzoquinone (Scheme 2b) and its experimental set up is shown in Scheme 2c. The TOF and turnover number (TON) values were 100 h−1 and 1700 after 17 h, respectively. The catalyst retained its activity for 17 cycles without any decay in the yield in the flow process. The epoxide ring-opening reaction between styrene oxide and aniline was also performed under continuous flow processes (Scheme 2d). Styrene oxide was kept as the limiting reagent and aniline in dichloromethane (0.5 M) was flowed through the column at a flow rate of 60 mL h−1 to obtain the desired product in 93% yield. The product yields were obtained consistently (83–93%) for 30 cycles and the total TON was >2700 in 30 h. Furthermore, a solution of 2-methoxynaphthalene (0.05 M) in Ac2O/CH2Cl2 (1:4, v/v) was passed through with a flow rate of 30 mL h−1 to produce 1-acetyl-2-methoxynaphthalene in high yields (85–99%) for the first five runs (Scheme 2e). However, the product yield was dropped to 65% in the sixth, but the activity of the catalyst was regained by flushing the column with CH3CN/CH2Cl2 (1:9, v/v). A total TON of 326 was achieved after fifteen consecutive runs and the catalyst performance was significantly higher under flow mode than a batch process.
:1.6 molar ratio was purged at a flow rate of 10 mL h−1 to achieve quantitative conversion of benzoquinone (Scheme 2b) and its experimental set up is shown in Scheme 2c. The TOF and turnover number (TON) values were 100 h−1 and 1700 after 17 h, respectively. The catalyst retained its activity for 17 cycles without any decay in the yield in the flow process. The epoxide ring-opening reaction between styrene oxide and aniline was also performed under continuous flow processes (Scheme 2d). Styrene oxide was kept as the limiting reagent and aniline in dichloromethane (0.5 M) was flowed through the column at a flow rate of 60 mL h−1 to obtain the desired product in 93% yield. The product yields were obtained consistently (83–93%) for 30 cycles and the total TON was >2700 in 30 h. Furthermore, a solution of 2-methoxynaphthalene (0.05 M) in Ac2O/CH2Cl2 (1:4, v/v) was passed through with a flow rate of 30 mL h−1 to produce 1-acetyl-2-methoxynaphthalene in high yields (85–99%) for the first five runs (Scheme 2e). However, the product yield was dropped to 65% in the sixth, but the activity of the catalyst was regained by flushing the column with CH3CN/CH2Cl2 (1:9, v/v). A total TON of 326 was achieved after fifteen consecutive runs and the catalyst performance was significantly higher under flow mode than a batch process.
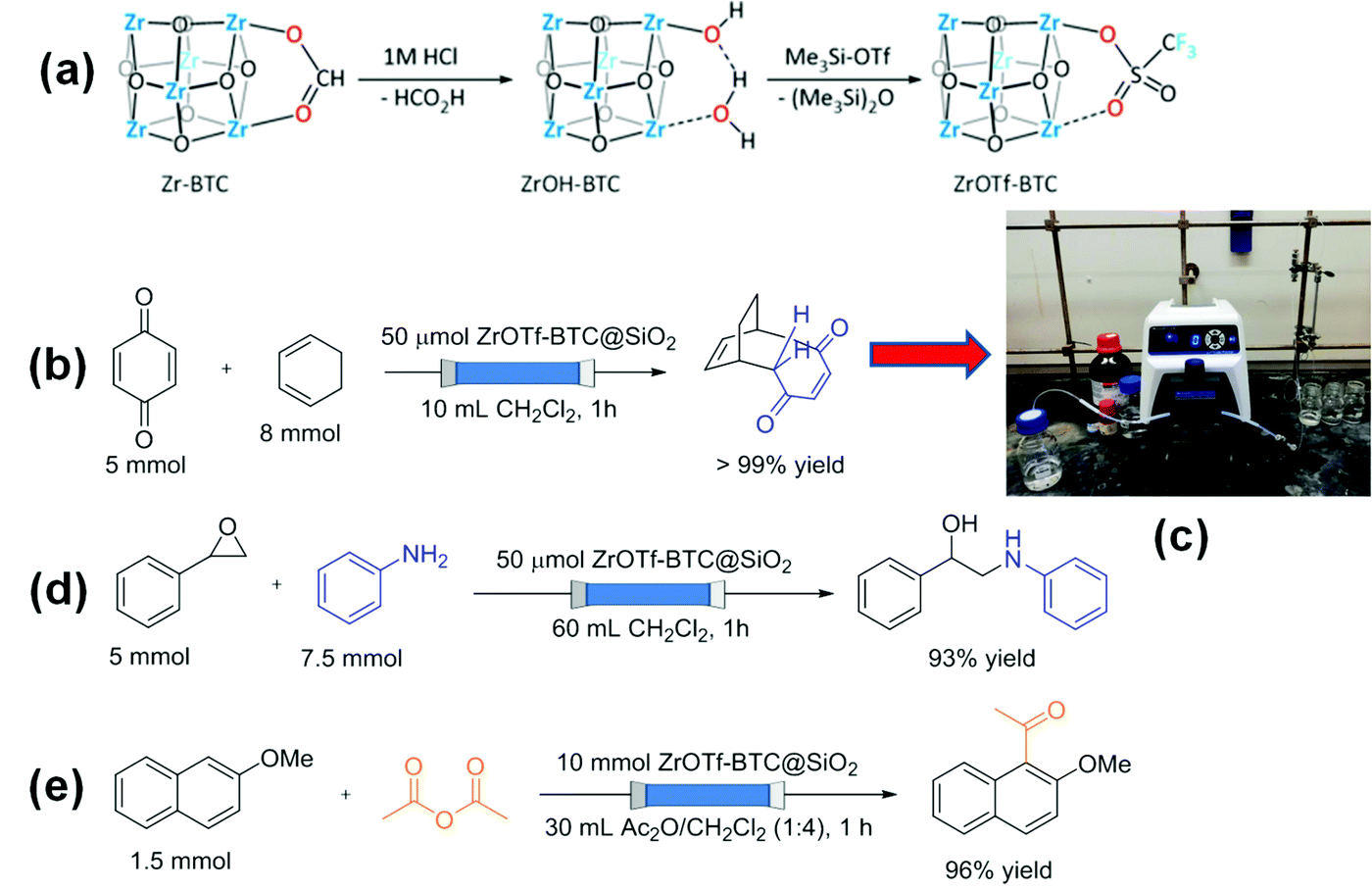 | ||
| Scheme 2 (a) Synthesis of ZrOTf-BTC through stepwise activation of Zr-BTC with 1 M HCl and treatment with Me3SiOTf; (b) Diels–Alder reaction under flow mode; (c) experimental setup for the Diels–Alder reaction under a continuous flow process; (d) ring opening reaction and (e) Friedel–Crafts alkylation under flow mode. Reproduced with permission from ref. 88 Copyright American Chemical Society 2019. | ||
3. C–O bond formation
Polymer membranes have been extensively employed in continuous flow catalytic processes.110 Although these organic membranes have high fluxes, they generally suffer from short lifetime, low stability and insufficient selectivity.111 On the other hand, mixed matrix membranes (MMMs) have been reported to overcome some of the limitations of polymeric membranes by introducing inorganic or organic fillers in the polymer matrix.112 MOFs have been identified as one of the possible fillers for polymeric membranes due to their large surface area, high porosity and tunable structures.113 One example of these MMMs containing MOFs is the combination of Cu3(BTC)2 and cellulose acetate (CA) that exhibits catalytic activity for the continuous production of acetals through acetalization of benzaldehyde.89 CA serves as the matrix and Cu3(BTC)2 as the filler with catalytic activity. In this study, a series of catalytic membranes with different Cu3(BTC)2 loading were prepared. SEM images revealed that CA fibres are twisted in the membrane and that the size of the Cu3(BTC)2 particles is around 15–20 μm. It was proved that the MOF component was uniformly distributed on the CA membrane. Furthermore, Cu3(BTC)2-based MMMs exhibit the characteristic XRD peaks corresponding to CA and to Cu3(BTC)2 MOF.Acetalization of benzaldehyde was performed under continuous flow for 2 h as shown in Fig. 5, reaching a benzaldehyde conversion of 94% and an acetal selectivity of 99%, with a productivity value of 0.59 mmol min−1 gCu3(BTC)2−1 in the steady state regime. These values were maintained for 24 h, indicating the long-term stability of the catalytic membrane (Fig. 6). Furthermore, upon increasing the benzaldehyde flow rate, the yields of benzaldehyde diethyl acetal were also increased. For example, the conversion of benzaldehyde was found to be 79, 87 and 94% for flow rates of 0.8, 1.6 and 3.2 μL min−1, respectively. No conversion of benzaldehyde was observed with CA. Under the same conditions, using 3.2 μL min−1 of benzaldehyde, the conversion of benzaldehyde was 78% with 99% selectivity and 99% conversion with 82% selectivity using methanol and ethylene glycol, respectively. Under identical conditions, a Cu3(BTC)2 catalyst showed 16% conversion with 99% selectivity towards diethyl acetal, revealing the advantages of the MMM. This protocol was also extended to the synthesis of a series of acetals, reaching 68 to 94% conversion values at a selectivity of 99%.
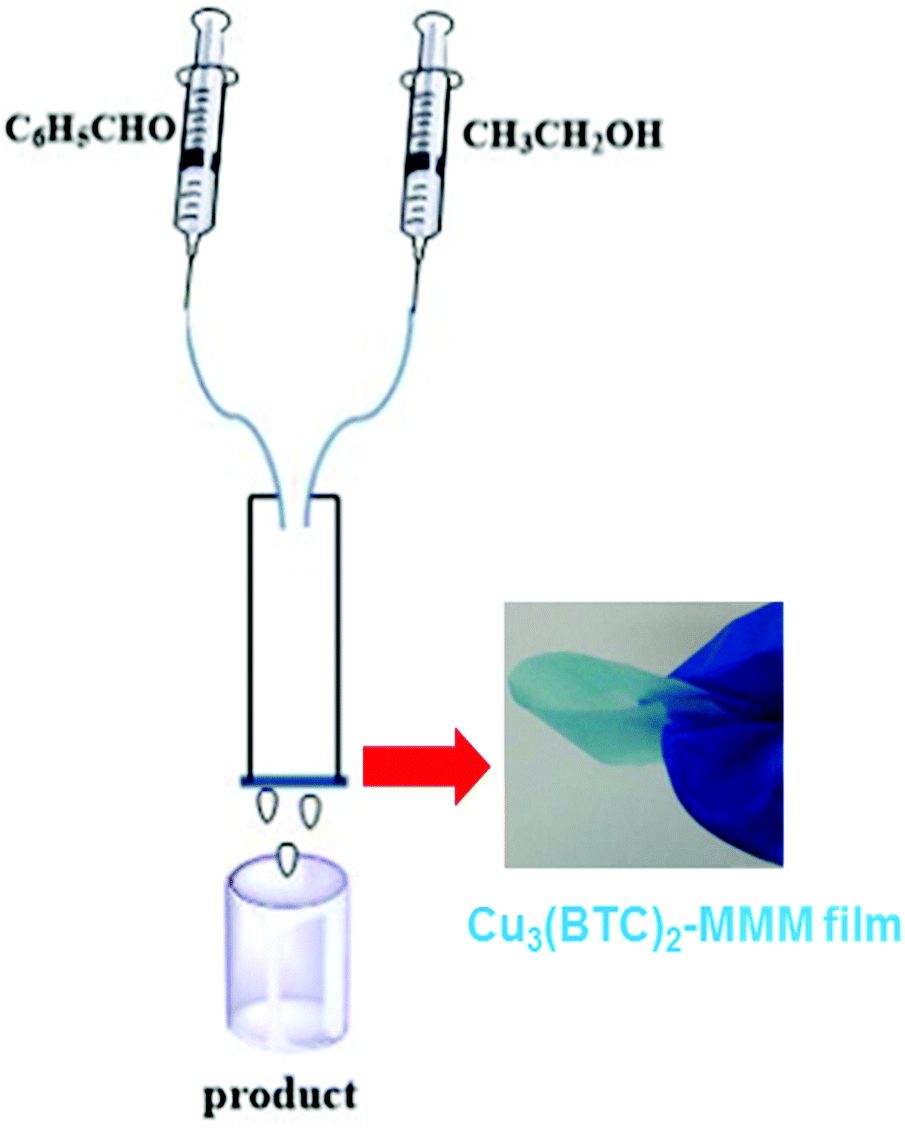 | ||
| Fig. 5 Experimental set-up used for continuous flow acetalization of benzaldehyde. Reproduced with permission from ref. 89 Copyright Royal Society of Chemistry 2017. | ||
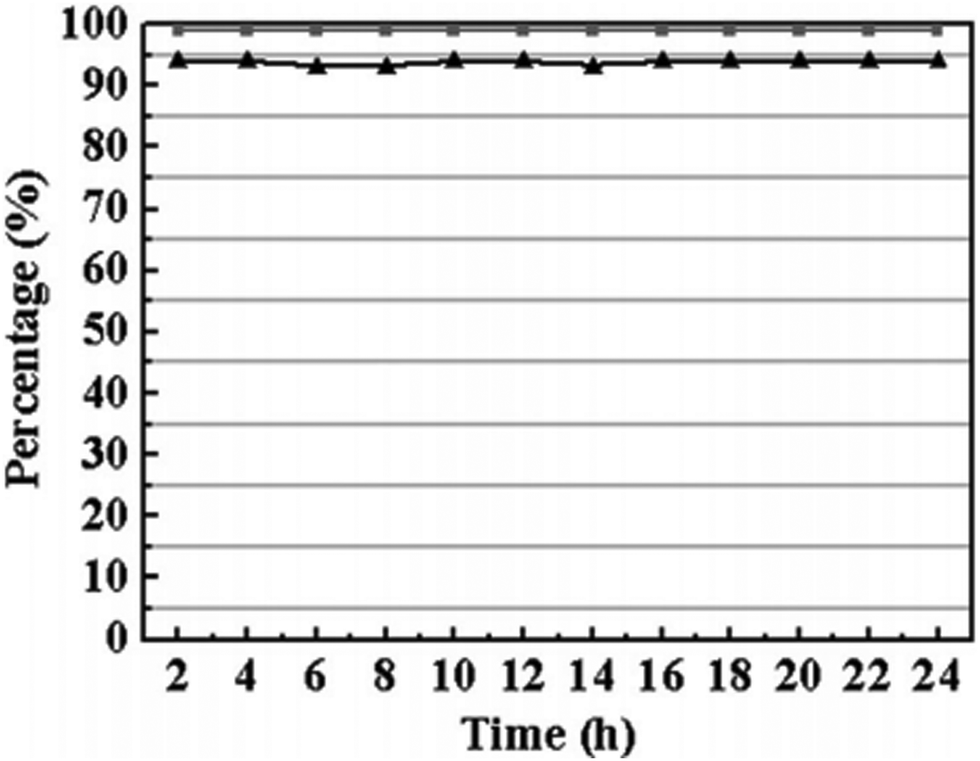 | ||
| Fig. 6 TOS conversion (triangle) and selectivity (square) for acetalization of benzaldehyde with ethanol on the Cu3(BTC)2. Reproduced with permission from ref. 89 Copyright Royal Society of Chemistry 2017. | ||
Shape-selective acetalization could also be performed with Cu3(BTC)2 MMM, providing indirect evidence that the reaction takes place inside the pores of the Cu MOF. Thus, acetalization of benzaldehyde (0.53 × 0.66 nm2) was 94% since its dimensions were smaller than the pore size of Cu3(BTC)2. In contrast, acetalization of 2-naphthaldehyde (0.77 × 0.66 nm2) gave 56% yield, while the yield of 9-anthraldehyde (1.03 × 0.66 nm2) was only 18% under identical conditions.
In another precedent, acetalization of benzaldehyde with methanol was reported using Cu3(BTC)2 as a heterogeneous catalyst, both under batch as well as continuous flow conditions (Fig. 7).90 The focus of this study was to investigate catalyst stability during batch and continuous flow conditions. The productivity rate for benzaldehyde dimethyl acetal using Cu3(BTC)2 was 5.85 × 10−2 h−1 at room temperature after 24 h, while the rate was 6.83 × 10−1 h−1 after 10 h at 70 °C. Besides temperature, the productivity rate could also be enhanced to 3.12 h−1 at 70 °C after 10 h by using ground fine particles of Cu3(BTC)2. A significant decay in the catalytic activity was observed after recycling the solid catalyst in repeated batch reactions. Under continuous flow tests a sudden deactivation occurs at initial minutes of TOS, but after this period the catalytic activity grows moderately. Powder XRD and BET surface area analysis revealed the occurrence of structural as well as morphological changes, decreasing the intensities of the peaks and diminishing the surface area and pore volume, respectively. Interestingly, fine particles of Cu3(BTC)2 exhibit the highest continuous flow activity and also present an increase in the catalytic activity over the TOS after the initial deactivation (Fig. 8).
 | ||
| Fig. 7 Acetalization of benzaldehyde with methanol using Cu3(BTC)2 as a solid catalyst under continuous flow conditions. | ||
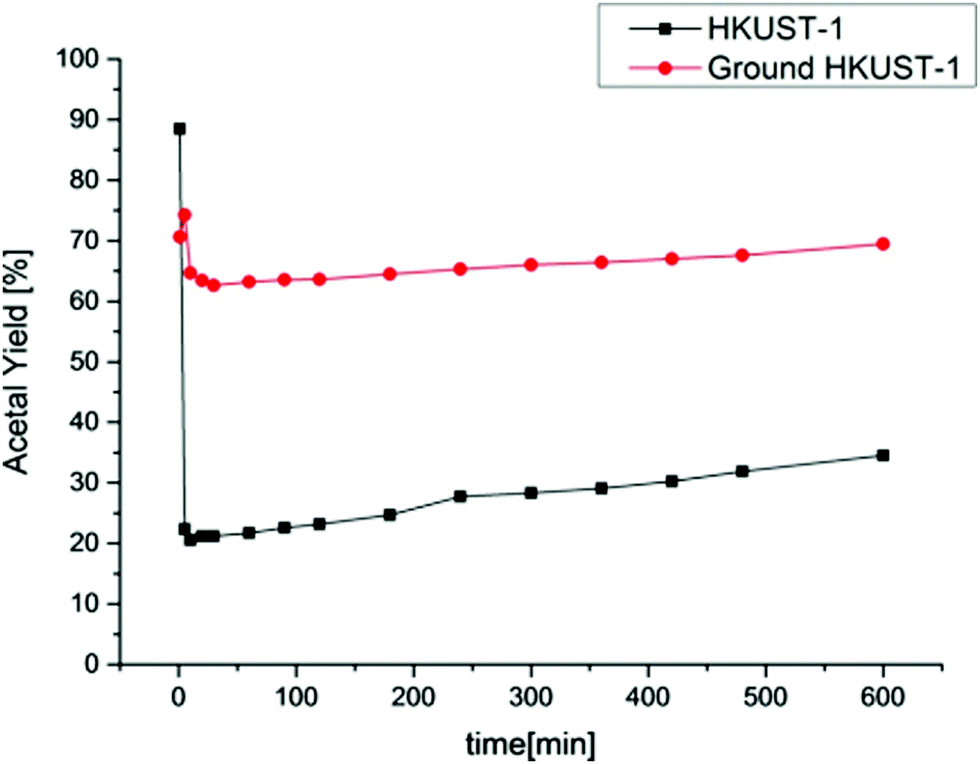 | ||
| Fig. 8 Yield of benzaldehyde dimethyl acetal versus time for the continuous flow tests. Reproduced with permission from ref. 90 Copyright Wiley 2016. | ||
A hierarchical yolk/shell copper hydroxysulfate@Cu3(BTC)2 (CHS@Cu3(BTC)2) was obtained at room temperature starting from a homogeneous yolk/shell copper hydroxysulfate (CHS) template composed of a reactive shell and a stabilized core.91 The active shell contributed as the source of Cu2+ ions that were in situ transformed to a well-defined MOF crystal shell upon reacting with BTC present in the solution. As a result, the stabilized core retained during the formation of the MOF shell its own CHS nature. The process is illustrated in Fig. 9. In the resulting CHS@Cu3(BTC)2, the core was constructed of tiny nanoplatelets with a thickness of ∼10 nm, and the outer shell is assembled of closely packed nanosheets, each with a thickness of ∼100 nm. The activity of CHS@Cu3(BTC)2 was tested in the room temperature acetalization of benzaldehyde with ethanol. Benzaldehyde was converted to benzaldehyde diethyl acetal in 88% yield with 99% selectivity. A blank run in the absence of catalyst showed under identical conditions no benzaldehyde conversion, while the use of either pure CHS template or pure Cu3(BTC)2 as catalyst reaches a benzaldehyde conversion of 80 and 17%, respectively. The superior activity of yolk/shell CHS@Cu3(BTC)2 has been attributed to the CHS core that primarily promotes benzaldehyde acetalization, while the Cu3(BTC)2 shell with large surface area behaves as a reactor impeding further agglomeration of the CHS core and provides multiple access channels for the reactants to reach the active CHS surface. The CHS@Cu3(BTC)2 core–shell provides a close intimacy of reactants that should accelerate the reaction rate. CHS@Cu3(BTC)2 was reused five times without observing any notable change in its efficiency and selectivity. In contrast, the activity of the CHS catalyst drastically decreased to 28%. Since the morphology and crystal structure of the used CHS and CHS@Cu3(BTC)2 remained unchanged, the significant deactivation of CHS was attributed to benzaldehyde adsorption on its surface. This proposal was supported by GC-MS analysis. Powder XRD indicated that the phase of the three-months stored CHS was transformed from Cu2.5SO4(OH)3·2H2O to Cu4SO4(OH)6 and the morphology is transformed from yolk/shell microspheres to stacked microsheets. On the other hand, the phase and morphology of three months stored CHS@Cu3(BTC)2 were retained without any change, thus indicating its stability.
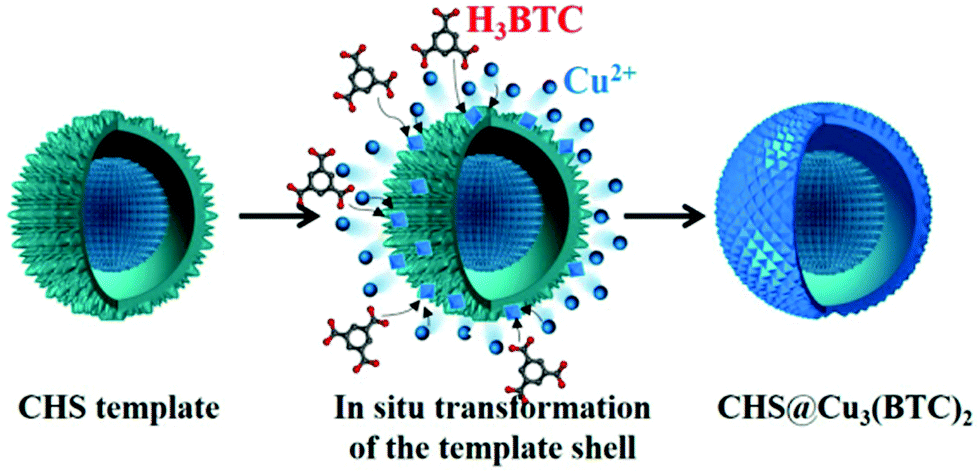 | ||
| Fig. 9 Preparation of a yolk/shell CHS@Cu3(BTC)2 catalyst. Reproduced with permission from ref. 91 Copyright American Chemical Society 2015. | ||
A flow reactor was developed using CHS@Cu3(BTC)2 as an immobilized stationary-phase catalyst (Fig. 10). The reactant solution was injected into the reactor and the product was recovered at the exit. Under continuous flow conditions, benzaldehyde was effectively converted to benzaldehyde diethyl acetal in 87% yield and 99% selectivity in a single pass, however, no details on residence time were provided. In addition, the catalyst stability under continuous flow conditions (TOS) and its activity in terms of TOF remain to be determined.
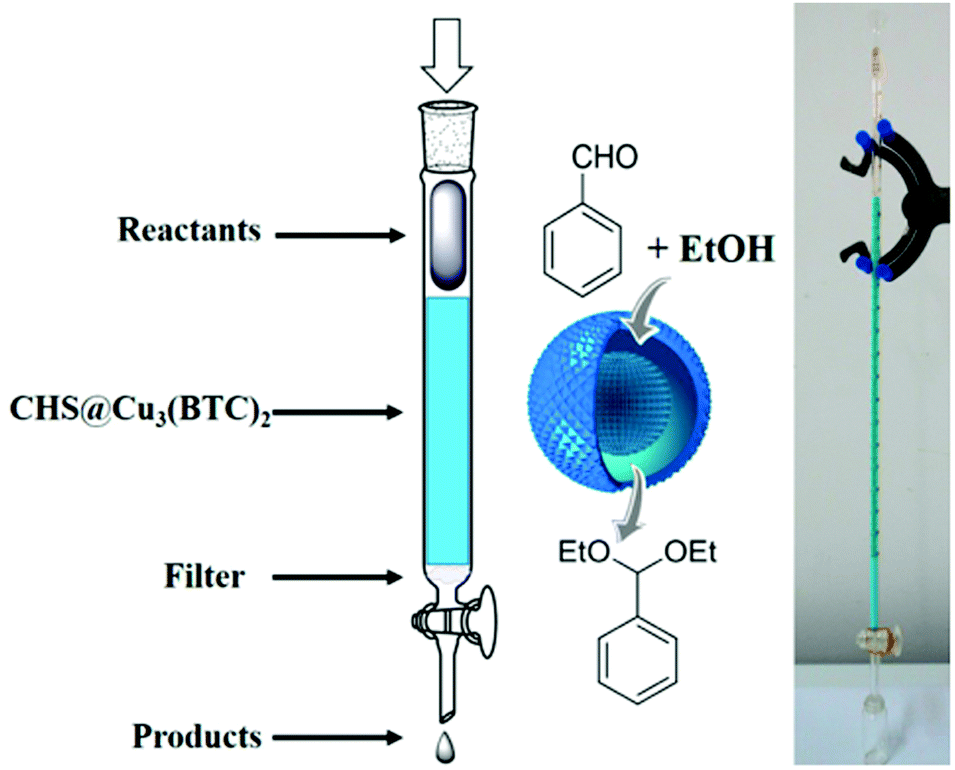 | ||
| Fig. 10 Schematic representation and photograph of the flow reactor using a self-supported CHS@Cu3(BTC)2 catalyst with an aldehyde and ethanol molar ratio of 1:80 at a flow rate of 0.05 mL min−1 at 25 °C. Reproduced with permission from ref. 91 Copyright American Chemical Society 2015. | ||
As in the case of the Knoevenagel reaction, there is a large variety of solid acids that have been reported as solid catalysts for the continuous flow acetalization of aldehydes in the liquid phase, particularly for their application as biofuels including sulfonic acid resins,114,115 zeolites116 and mesoporous silicas having grafted sulfonic acid groups.117 Some of the reported materials do not undergo deactivation upon TOS and, therefore, there is still a need to find stable MOFs that do not undergo the initial strong decline in their catalytic activity.
Cyclic carbonates are valuable commodity chemicals that find application as solvents for Li-ion batteries (propylene carbonate), as well as monomers for polycarbonate synthesis.118 One of the conventional syntheses of cyclic carbonates is the reaction of phosgene with the corresponding diol (Scheme 3).119 An attractive alternative strategy for the synthesis of these compounds is the cycloaddition of CO2 to epoxides to yield the corresponding cyclic carbonate (Scheme 3),120 a reaction with complete atom economy, and the use of inexpensive and convenient substrates, avoiding phosgene as a reagent.121 Recently, MIL-101(Cr) has been reported as an effective catalyst for the CO2 cycloaddition to propylene oxide to give propylene carbonate under flow conditions without the need of a halide co-catalyst (Fig. 11).92 Under steady-state operation after 1 h, the propylene carbonate was obtained with a TOF of 20 h−1 using TBAB as a co-catalyst. This TOF value is equivalent to 0.033 mmol min−1 of propylene carbonate in a single pass, corresponding to 80% yield (Fig. 12). Later, the reaction was performed without TBAB under identical flow conditions, reaching under these conditions 54% yield in a single pass, which is a remarkable activity in the absence of a bromide co-catalyst. Although these results are promising, the residence time is not provided which is very crucial to determine the activity of a solid under flow conditions. The effect of synthesis conditions on the catalytic performance of the resulting MIL-101(Cr) was studied by preparing MIL-101(Cr) using a series of acids, including hydrochloric acid, hydrofluoric acid, acetic acid and also under acid-free conditions. These four MIL-101(Cr) samples showed similar catalytic activity with single pass yield ranging between 33 and 41%. In addition to MIL-101(Cr), analogous isostructural Fe- and Sc-based MOFs were also prepared and their activity under identical flow conditions tested. MIL-101(Cr) MOF prepared under acid free conditions afforded 41% yield at steady state operation with a TOF of 21 h−1. In comparison, MIL-101(Fe) showed a very low activity of only 1–2% yield under analogous conditions. In contrast, the more oxophilic Sc-based catalyst, MIL-101(Sc) exhibited higher initial activity than MIL-101(Cr), with 53% yield of propylene carbonate and a TOF value of 87 h−1 for MIL-101(Sc) after 1 h. Unfortunately, the activity of MIL-101(Sc) decreased rapidly to <10% in 3 h. This result indicates catalyst deactivation under flow conditions. This was confirmed by recording the powder XRD pattern of the spent MIL-101(Sc) catalyst that showed a notable crystallinity loss after 3 h under the reaction conditions. In contrast, a minimal loss of crystallinity was seen for MIL-101(Cr) under analogous conditions, thus showing superior stability of MIL-101(Cr) with TOS. Interestingly, in contrast to the performance of MIL-101(Sc), the activity of MIL-100(Sc) was found to be superior than MIL-101(Cr) by affording a product yield of 57% and a TOF of 28 h−1 at steady state operation under the standard conditions. Furthermore, the activity of MIL-100(Sc) was maintained up to 5 h without any decay in its activity.
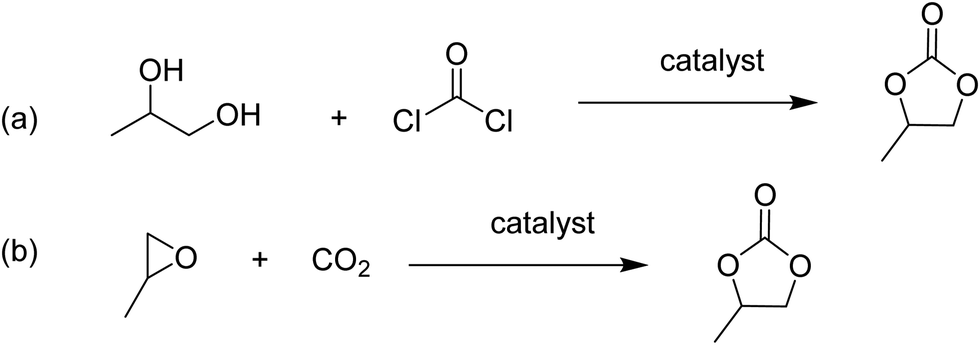 | ||
| Scheme 3 Phosgene/diol (a) and CO2/epoxide (b) routes to propylene carbonate. | ||
 | ||
| Fig. 11 Schematic illustration of the pressurized packed-bed flow reactor used for the cycloaddition of CO2 to epoxides. The flow reaction was performed at 100 °C, and 5 bar of CO2 with a flow rate of 4 sccm min−1. A stock solution of 165 mM propylene oxide and 8.4 mM TBAB in chlorobenzene was passed at a rate of 0.25 mL min−1. Reproduced with permission from ref. 92 Copyright Royal Society of Chemistry 2018. | ||
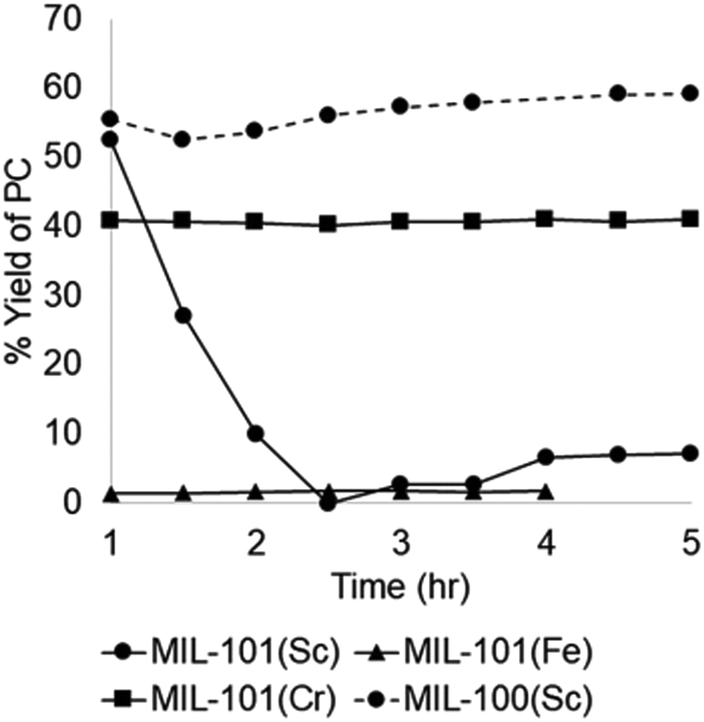 | ||
| Fig. 12 Contrasting catalytic activity of different MOFs for the conversion of propylene oxide to propylene carbonate. Reproduced with permission from ref. 92 Copyright Royal Society of Chemistry 2018. | ||
Fig. 11 shows the experimental set-up for the packed bed flow reactor. A HPLC pump and a high-pressure mass flow controller were used to supply epoxide stock solution of CO2 gas, at the required pressure. A four-way dynamic mixer was used to ensure the homogeneous mixing of reactants. The oven was constructed from a copper pipe covered in a silicone-wrapped heating tape connected to a temperature controller. The catalyst bed was made up of a glass tube fitted with Vespel/graphite ferrules and packed with the MOF catalyst. A thermocouple was connected to the outside surface of the bed for regulating the temperature. An ice bath was attached to control the evaporation of solvent.
The structure activity relationship in Sc-based MOFs was studied by preparing several analogues with different crystal structures and node geometries which include MIL-88D(Sc) and MIL-68(Sc). MIL-88D(Sc) exhibits a yield of 11% and a TOF value of 12 h−1. Furthermore, the activity of MIL-68(Sc) was even lower, affording <1% yield of propylene carbonate under similar conditions. These results indicate that the presence of accessible Lewis acidic sites on the metal nodes in these MOF-based catalysts is a key requirement for exhibiting activity.
Recently, NaCo(CO)4-incorporated on MIL-101(Cr) [Co(CO)4⊂MIL-101(Cr)] was reported as the first heterogeneous catalyst for the selective ring-expansion carbonylation of β-lactones to succinic anhydrides.93 The main reasons to select MIL-101(Cr) as a solid support for the deposition of Co(CO)4− were its high surface area, large pore openings (12 and 16 Å) and pore diameters (29 and 34 Å) that should allow diffusion of reagents to the active sites. Besides porosity, high chemical and thermal stability were other factors for the selection of this material.122 Initially, the catalytic activity of Co(CO)4⊂MIL-101(Cr) was tested in the carbonylation of β-butyrolactone at 80 °C in toluene with 15 bar of CO for 24 h, reaching full conversion. A similar reactivity was also observed for a homogeneous catalyst [(salph)Al(THF)2][Co(CO)4] (salph: N,N′-o-phenylenebis(3,5-di-tert-butylsalicylideneimine); THF: tetrahydrofuran).123 Unfortunately, product analysis indicated the formation of the desired product methylsuccinic anhydride together with unwanted poly(3-hydroxybutyrate) as a major side product.
Later, the carbonylation activity of Co(CO)4⊂MIL-101(Cr) was studied for β-propiolactone (Scheme 4), targeting the synthesis of the commercially desirable unsubstituted succinic anhydride. Gratifyingly, Co(CO)4⊂MIL-101(Cr) efficiently promoted the carbonylation of β-propiolactone at room temperature in toluene with 15 bar of CO to give exclusively succinic anhydride in 92% yield without observing the formation of the poly(3-hydroxypropionate) byproduct. This activity corresponds to an overall site time yield (the number of molecules of a specified product made per catalytic site and per unit time) of 16 h−1 which is higher than the value provided by the homogeneous [(salph)Al(THF)2][Co(CO)4] catalyst (12 h−1) under analogous conditions. MIL-101(Cr) and Na[Co(CO)4] exhibited 0 and 12% yields, respectively. This poorer catalytic performance of the individual components was attributed to the lack of Co(CO)4− active species required for CO insertion in one case and to the absence of strong Lewis acids to activate the lactone in the other one (Fig. 13). Furthermore, an equimolar mixture of MIL-101(Cr) and Na[Co(CO)4] as catalysts gave 43% yield, which is, also, higher compared to the use of either precursor individually. In any case, the activity of the physical mixture was much inferior to that of Co(CO)4⊂MIL-101(Cr) with well-defined Cr/Co sites which can efficiently activate both the substrate and CO that become inserted into the lactone. A hot filtration experiment verified the heterogeneity of the process. Powder XRD of the spent Co(CO)4⊂MIL-101(Cr) showed the identical crystalline structure to that of the fresh solid, thus confirming the stability under the reaction conditions.
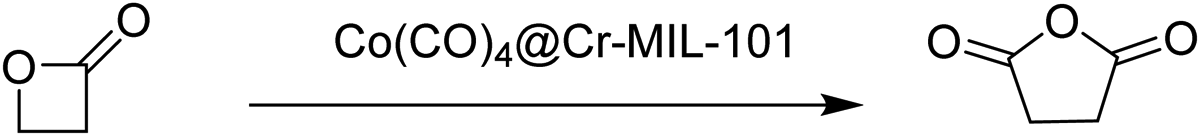 | ||
| Scheme 4 Carbonylation of β-propiolactone. | ||
 | ||
| Fig. 13 (A) Proposed mechanism for the ring-expansion carbonylation of β-lactones requiring the combination of [Lewis acid] and [Co(CO)4]−; (B) proposed active sites in Co(CO)4⊂MIL-101(Cr) with coordinated tetrahydrofuran molecules. Reproduced with permission from ref. 93 Copyright American Chemical Society 2018. | ||
A laboratory-scale packed-bed reactor was designed to examine the activity of Co(CO)4⊂MIL-101(Cr) under continuous flow conditions (Fig. 14). Using Co(CO)4⊂MIL-101(Cr) as a solid catalyst operating at 0.1 mL min−1 flow of 0.1 M β-propiolactone in CH2Cl2, equivalent to a weight hourly space velocity of 1200 h−1, with an excess CO flow of 30 mL min−1 at 45 bar renders succinic anhydride as the sole product reaching at room temperature a productivity of 1300 molAnhydride molCo−1 after 6 h on stream (Fig. 15). Among all catalysts reported, the activity reached by Co(CO)4⊂MIL-101(Cr) is one of the best values achieved for β-carbonylation. Similarly, Co(CO)4⊂MIL-101(Cr) was also efficient to carbonylate β-butyrolactone to the desired methylsuccinic anhydride as the only product with an activity at 40 °C of 360 molAnhydride molCo−1 after 60 h on stream (Fig. 15). One of the main advantages of the packed-bed reactor is that solvent evaporation affords succinic anhydride crystals of very high purity. This process based on Co(CO)4⊂MIL-101(Cr) greatly contrasts with alternative procedures consisting on poorly-selective hydrocarbon oxidation or microbial fermentation processes that require NH3/H2SO4-assisted succinate precipitation or NaOH-assisted electrodialysis or amine-assisted reactive extraction.124
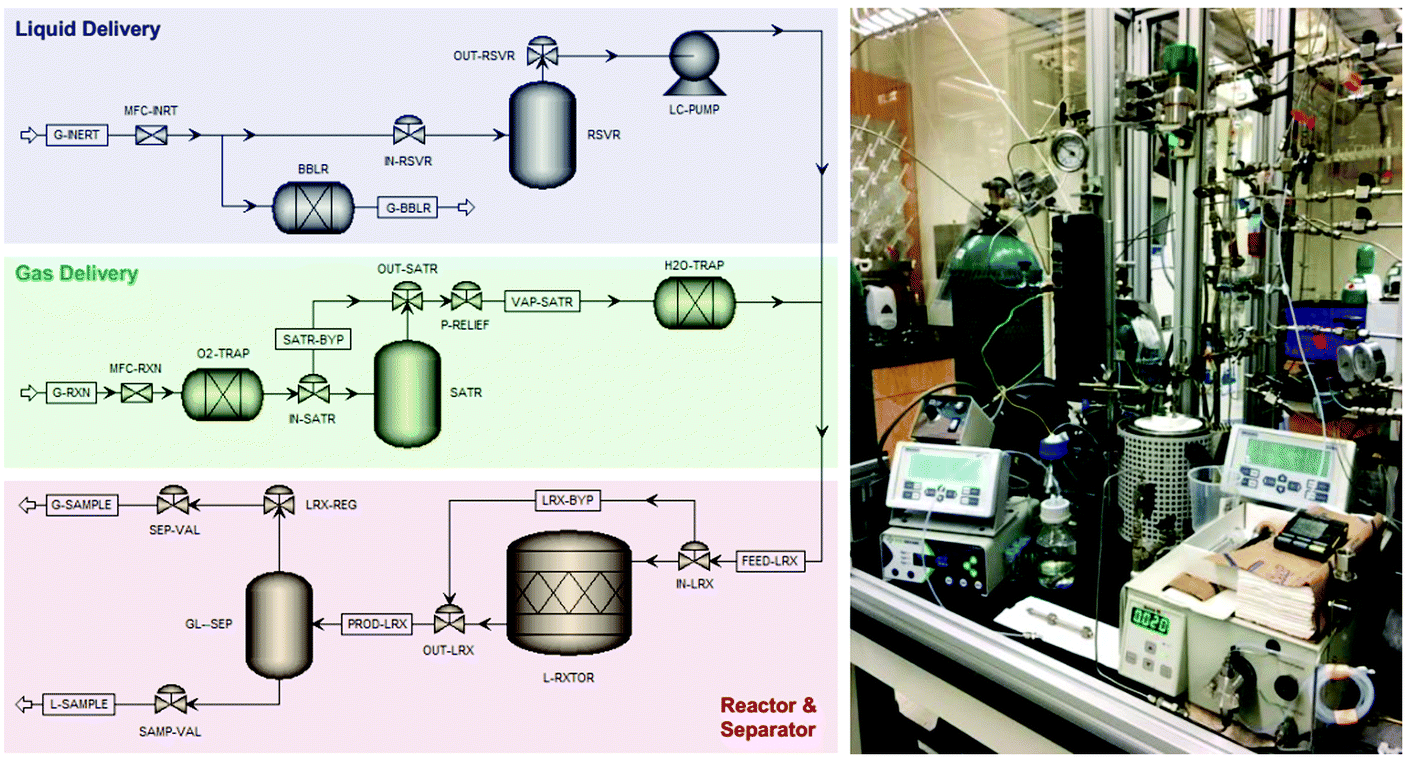 | ||
| Fig. 14 Process flow diagram and photograph of the experimental setup of the packed bed reactor employed for the continuous-flow β-lactone carbonylation. Reproduced with permission from ref. 93 Copyright American Chemical Society 2018. | ||
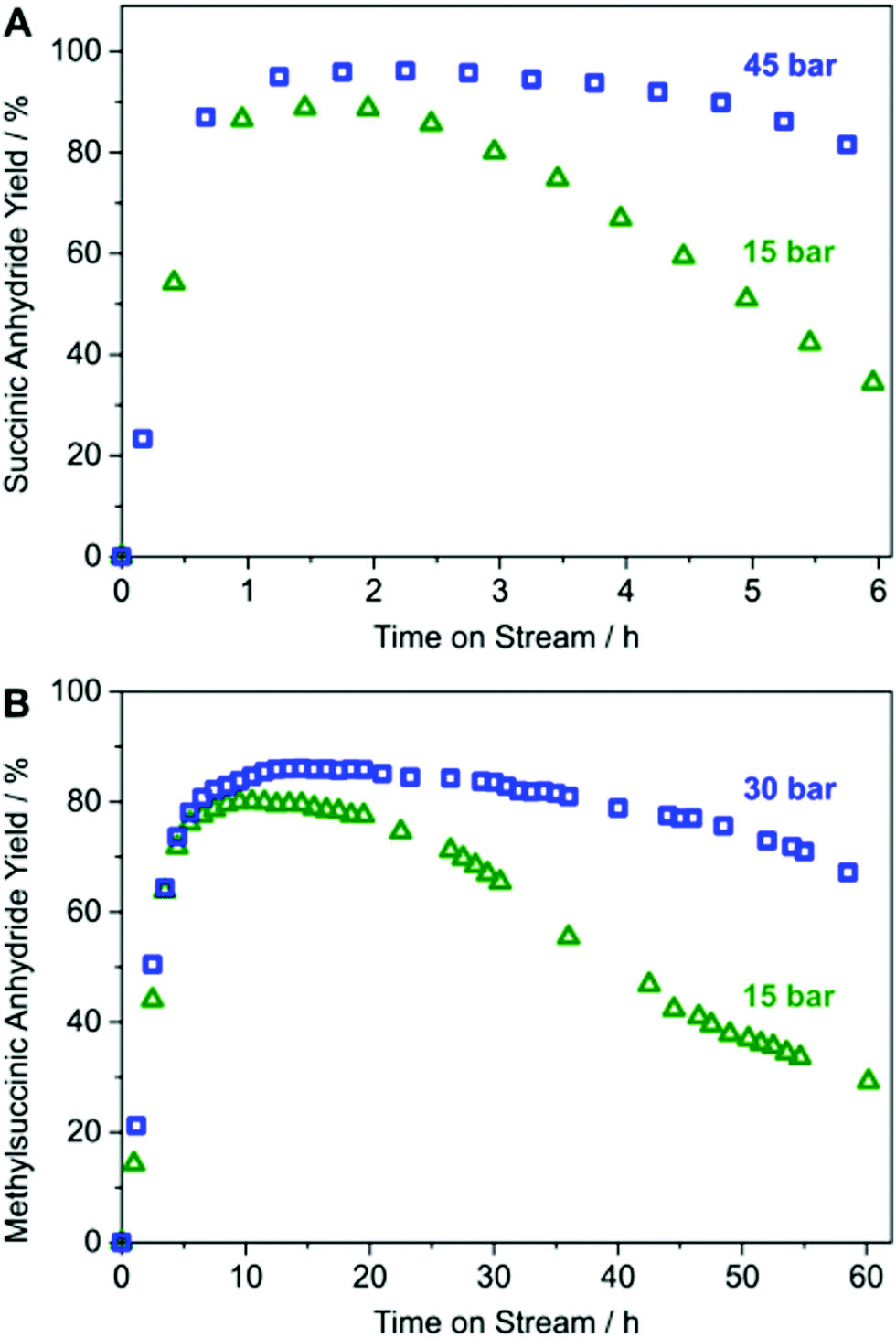 | ||
| Fig. 15 (A) Yield-TOS plot for continuous-flow carbonylation of β-propiolactone by Co(CO)4⊂MIL-101(Cr) and (B) continuous flow carbonylation of β-butyrolactone by Co(CO)4⊂MIL-101(Cr). Reproduced with permission from ref. 93 Copyright American Chemical Society 2018. | ||
A UiO-66(Zr) sample with 1147 m2 g−1 and a total pore volume of 0.58 cm3 g−1 was tested as a heterogeneous catalyst for the conversion of methyl levulinate (ML) to γ-valerolactone (GVL) through catalytic transfer hydrogenation under continuous flow conditions.94 The conversion of ML to GVL under the batch reaction was optimized employing 0.23 g of UiO-66(Zr) as a catalyst and 2.4 M of ML in isopropanol at 240 °C under 35 bar. This study was also extended by performing a continuous flow reaction as shown in Fig. 16. The results obtained are given in Fig. 17. The catalytic activity of UiO-66(Zr) was considerably stable up to 9 h on stream, observing a slight decrease of ML conversion at longer times. ML conversion reached to 56% after 30 h TOS with GVL productivity of 58.94 mmolGVL g−1 h−1 and very high selectivity to GVL. The initial GVL productivity using UiO-66(Zr) was 92.3 mmolGVL g−1 h−1 (TOS = 1 h), while it decreased to 58.9 mmolGVL g−1 h−1 after 30 h on stream. These values are much superior than with UiO-66(Zr) as a catalyst under batch conditions (1 mmolGVL g−1 h−1) as well as with other earlier mixed oxide Zr-FeO (1:1)-300 catalysts (39.2 mmolGVL g−1 h−1) (Table 4).125 Furthermore, the catalytic performance of UiO-66(Zr) for the production of GVL under continuous flow was also compared with a series of heterogeneous solid catalysts under batch conditions and these catalytic data clearly indicate that UiO-66(Zr) exhibits better performance under continuous flow than under batch conditions. These results clearly reveal the efficiency of UiO-66(Zr) as a catalyst under continuous flow providing very high productivity of GVL. The decrease in the conversion of ML was due to catalyst decomposition, a proposal that was based on the detection of diisopropyl terephthalate by GC-MS analysis. Furthermore, the BET surface area of the catalyst recovered after continuous flow reaction decreased considerably to 109 m2 g−1 compared to the value of the fresh sample. This low surface area value suggests the collapse of the crystalline framework, this being compatible with the detection of the BDC ester derivative. TEM images of fresh and used UiO-66(Zr) indicated that the surface of fresh UiO-66(Zr) becomes transformed from smooth to rough after continuous flow operation which can be due to the loss of organic linkers and possibly formation of an amorphous ZrO2 phase. These results indicate that further modification of UiO-66(Zr) is still necessary, together with milder reaction conditions to develop a continuous flow process in which UiO-66(Zr) can enjoy stability.
 | ||
| Fig. 16 Experimental set up for the conversion of ML to GVL using UiO-66 as a catalyst under continuous flow conditions. | ||
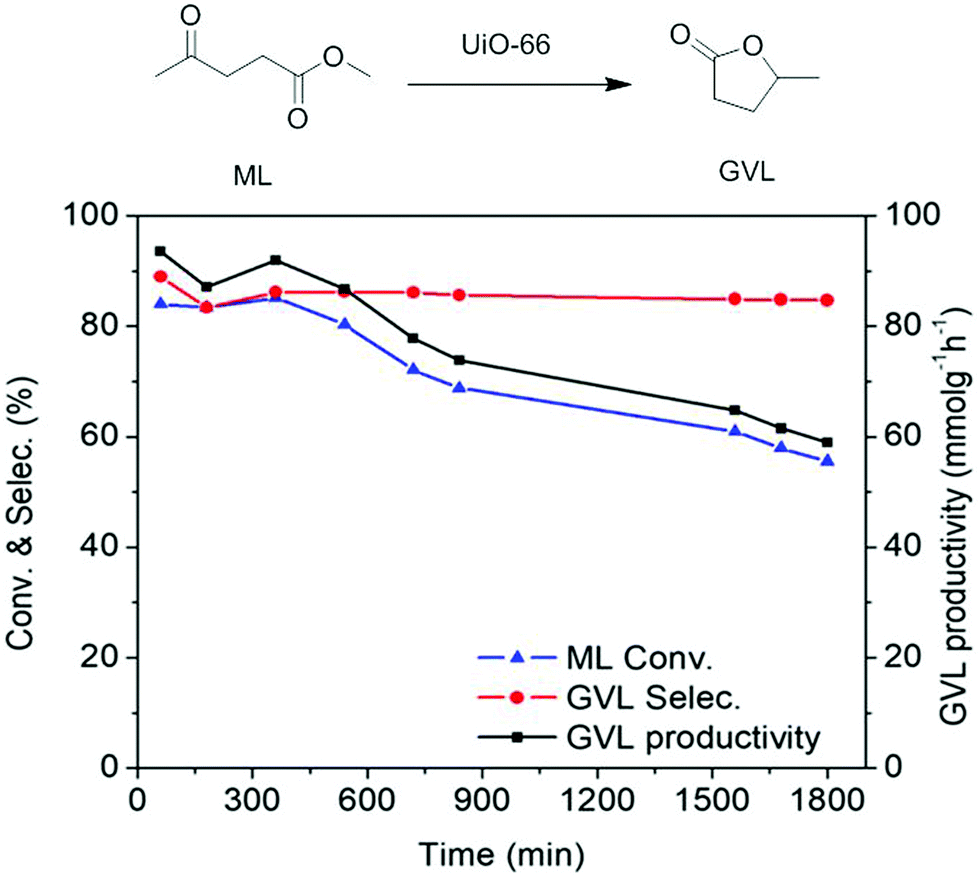 | ||
| Fig. 17 Continuous flow cyclization of ML to GVL catalyzed by UiO-66(Zr). Reproduced with permission from ref. 94 Copyright American Chemical Society 2018. | ||
| Catalyst | Conditions | Conversion (%) | Selectivity (%) | GVL productivity (mmolg−1 h−1) | Ref. |
|---|---|---|---|---|---|
|
a S60 represents 60 mol% sulfonated ligand.
b Molar ratio Al/Zr is 7:3 and the sample was calcined at 300 °C.
c Molar ratio Zr/Fe is 1:1 and the sample was calcined at 300 °C.
d EE: ethyl levulinate.
|
|||||
| UiO-66(Zr) | 0.23 g of catalysts, 2.4 M ML in isopropanol, flow rate = 0.2 mL min−1, 240 °C, 35 bar, TOS = 1 h | 83 | 89 | 92.3 | 94 |
| UiO-66(Zr) | 0.22 g of catalysts, 4 mmol of EEd in 400 mmol of isopropanol, 130 °C, t = 3 h (batch reaction) | 43.3 | 18.5 | 0.534 | 126 |
| MOF-808 | 0.22 g of catalysts, 4 mmol of EEd in 400 mmol of isopropanol, 130 °C, t = 3 h (batch reaction) | 100 | 85 | 5.66 | 126 |
| UiO-66(Zr) | 0.1 g of catalysts, 1 mmol of ML in 5 mL of 2-butanol, 140 °C, Ar, 0.5 MPa, t = 9 h (batch reaction) | 70 | 51 | 0.4 | 127 |
| UiO-66-S60a | 0.1 g of catalysts, 1 mmol of ML in 5 mL of 2-butanol, 140 °C, Ar, 0.5 MPa, t = 9 h (batch reaction) | 98 | 82 | 0.889 | 127 |
| Al7Zr3-300b | 0.072 g of catalysts, 1 mmol of EEd in 5 mL of isopropanol, 220 °C, t = 4 h (batch reaction) | 95.5 | 87.1 | 2.89 | 128 |
| Zr(OH)4 | 1 g of catalysts, 2 g of EEd in 38 g ethanol, 240 °C, t = 1 h (batch reaction) | 89.1 | 84.5 | 10.7 | 129 |
| ZrFeO(1:1)-300c |
0.2 g of catalysts, 0.65 g of EEd in 11.8 g of isopropanol, 230 °C, t = 0.5 h (batch reaction) | 94.2 | 92 | 39.2 | 125 |
4. C–N bond formation
Powder Cu3(BTC)2 of large sized grains around 20 μm has been reported as a heterogeneous catalyst for a wide range of organic reactions under batch conditions. A continuous flow process in a fixed-bed reactor packed with large MOF grains has drawback of the high pressure drop throughout the reactor due to the very high resistance against flow. Deposition or in situ growth of MOFs forming a 10–30 μm thick layer on supports such as conventional millimetric pore monoliths or non-porous alumina beads have been reported under batch conditions.130–132 Non-conventional silica monoliths offer many advantages like a hierarchical macro- and mesoporosity which is an important prerequisite for catalysis in flow to avoid mass transfer limitations between the liquid and the solid phases. Furthermore, these non-conventional monoliths are highly promising due to their highly interconnected and isotropic network of macropores (5 μm) with small internal diffusion paths (3 μm skeleton thickness), and high surface area (605 m2 g−1). Hence, Cu3(BTC)2 was synthesized in situ as NPs within the mesopores of these silica monoliths to be used for continuous flow in the liquid phase Friedlander condensation.95 The in situ synthesis of Cu3(BTC)2 NPs was evidenced by powder XRD and TEM images. Also, a control Cu3(BTC)2 catalyst was prepared with 1 μm size crystals. Gas adsorption isotherm measurements of bulk Cu3(BTC)2 showed a BET surface area of 1812 m2 g−1 and a micropore volume of 0.71 mL g−1. In addition, the in situ synthesis of Cu3(BTC)2 on the silica monolith (Cu3(BTC)2-MonoSil) was supported by EDX analysis. SEM images did not show any growth of Cu3(BTC)2 crystals on the monolith macropores and TEM images showed the formation of 7–12 nm nanocrystals located inside the monolith structure and few 50 nm NPs at the rims of the monolith. In addition, gas sorption measurements provide convincing evidence in support of the existence of Cu3(BTC)2 NPs inside the mesopores of the silica monolith by decreasing the mesopore volume from 1.24 to 0.77 mL g−1 and concomitantly showing an increase in the microporosity from 0.04 to 0.23 mL g−1 as well as showing higher BET surface area from 605 to 971 m2 g−1 compared to the parent silica monolith. The catalytic performance of Cu3(BTC)2-MonoSil was tested in the Friedlander reaction as shown in Fig. 18 under continuous flow conditions as shown in Fig. 19. The Cu3(BTC)2-MonoSil was covered with a heat shrinkable clad and placed in a chamber heated at 80 °C. The solution of reactants, 2-aminobenzophenone and acetylacetone, was passed through the monolith at a flow rate of 0.5 mL min−1. The experimental results showed that the initial activity reaches 90% conversion and a steady-state value of 85% was attained after 4 h on flow and maintained for 24 h. The productivity at the steady-state was 0.5 mmol min−1 gMonolith−1 or 2.2 mmol min−1 gCu3(BTC)2−1. This value is about 2.5 times superior to the activity reported for commercial Cu3(BTC)2 powder (10–20 μm) in a batch reactor that reaches a conversion rate of 0.86 mmol min−1 gCuBTC−1.133 In another report, the Friedlander reaction between 2-aminobenzophenone and acetylacetone condensation was almost quantitative after 1.5 h with Cu3(BTC)2, while 75 and 52% yields134 were obtained with H-BEA and Cu-BEA catalysts, respectively. The enhanced activity of Cu3(BTC)2 was attributed to its low energy barrier for the annulation step and the favourable geometry of reaction precursors adsorbed on two adjacent Cu2+ sites in Cu3(BTC)2. On the other hand, Cu3(BTC)2 possessed high population of active sites together with the concerted effect of adjacent active sites. These two factors are much less important in the case of H-BEA or Cu-BEA catalysts.134 A continuous flow process operated successfully with Cu3(BTC)2-MonoSil as a catalyst to produce 826 g of the desired product per g of Cu3(BTC)2 per day. These results clearly establish the superior activity of Cu3(BTC)2 under continuous flow operation provided that the active MOF is suitably deposited on the reactor.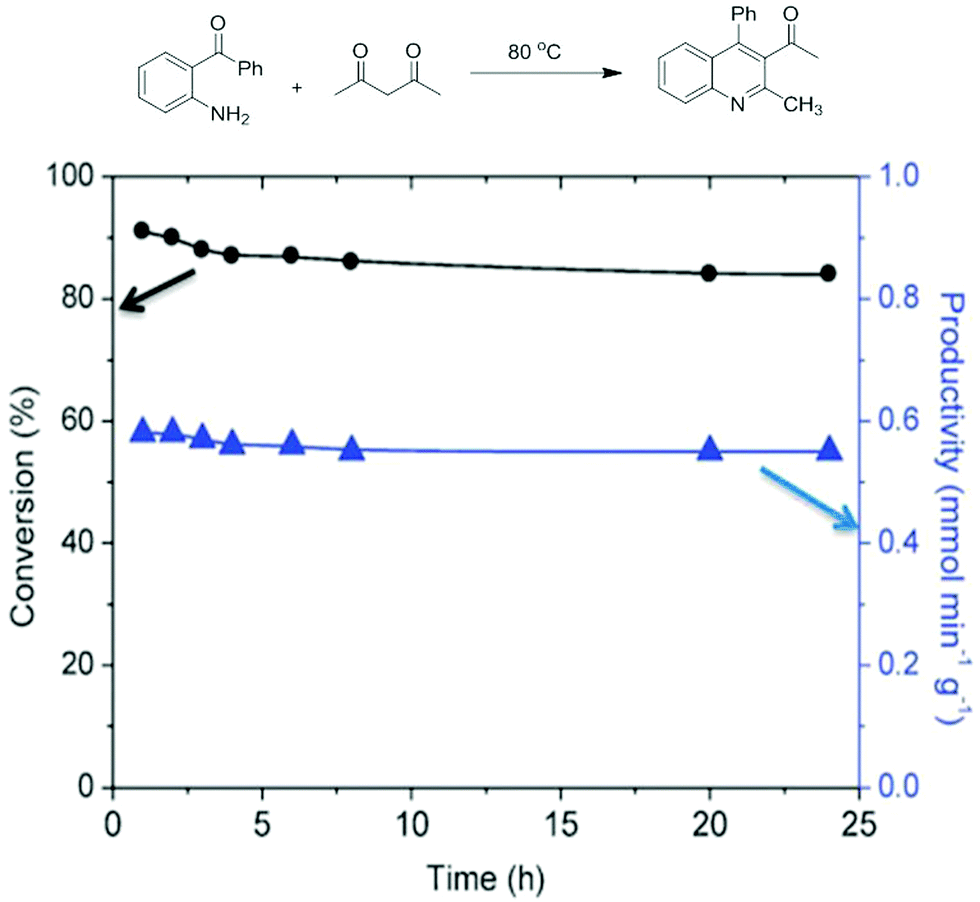 | ||
| Fig. 18 Conversion and productivity for the Friedlander reaction between 2-aminobenzophenone and acetylacetone as a function of time on Cu3(BTC)2-MonoSil under continuous flow conditions. Reproduced with permission from ref. 95 Copyright Royal Society of Chemistry 2012. | ||
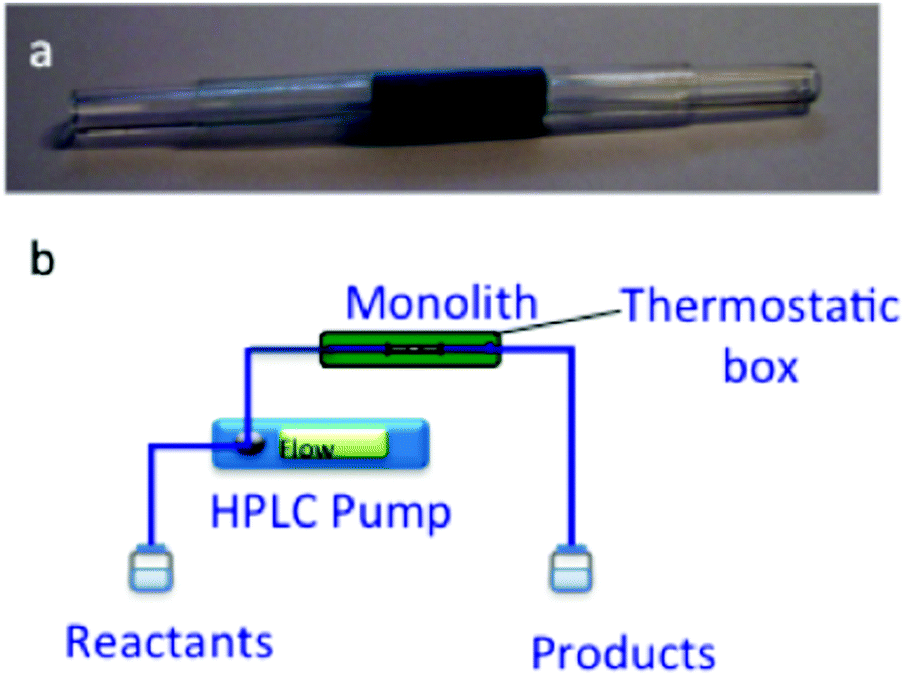 | ||
| Fig. 19 Experimental set up for the Friedlander reaction with cladded Cu3(BTC)2-MonoSil of 2 cm length and 6 mm diameter (a) and flow reactor design for in-flow catalysis (b). Reproduced with permission from ref. 95 Copyright Royal Society of Chemistry 2012. | ||
5. Oxidation reactions
Aerobic oxidations using molecular oxygen at quasi ambient conditions as the terminal oxidant are gaining considerable importance due to the greenness of the process, but require highly selective catalysts. Noble metals such as Pd are typically active centres for this type of oxidation.In this context, Pd NPs were encapsulated within the pores of MIL-88B-NH2 to achieve Pd@MIL-88B-NH2 (8 wt% Pd). This was later protected by SiO2 NPs to obtain Pd@MIL-88B-NH2@nano-SiO2 (Scheme 5)96 with the purpose to increase catalyst stability. The activity of this solid was tested in the oxidation of alcohols using air as the oxidant at ambient pressure under batch as well as continuous flow conditions. Powder XRD ascertained no changes in the structural integrity of MIL-88B-NH2 during Pd NP encapsulation. The average particle size of Pd NPs was 2–3 nm and they were uniformly distributed in the MOF matrix as shown by TEM images. Gas adsorption measurements indicated that the BET surface area and pore volume for Pd@MIL-88B-NH2@nano-SiO2 are 603 m2 g−1 and 1.82 cm3 g−1, respectively. The activity of Pd@MIL-88B-NH2@nano-SiO2 for the oxidation of 1-phenylethanol using air as an oxidant in p-xylene was tested at 150 °C, reaching 98% yield. Interestingly, a wide range of secondary alcohols could also be oxidized to their respective ketones in high yields (68–97%) under identical conditions. The superior stability of Pd@MIL-88B-NH2@nano-SiO2 was established by performing comparative reusability experiments with Pd@MIL-88B-NH2, without a silica protecting shell. Due to the small particle size and the tedious recovery through centrifugation, a tea-bag system was developed by taking 200 mg of 0.51 Pd wt% Pd@MIL-88B-NH2@nano-SiO2 (1 mol% respect to the substrate) tied with a Teflon wire and dipped into 1-phenylethanol in p-xylene solution. The catalyst introduced inside the tea-bag was pulled out and subsequently immersed in a fresh reaction batch. This batch process was repeated for four cycles observing a significant catalyst deactivation from 85 to 38%. One of the main causes for catalyst deactivation was Pd leaching. 1-Phenylethanol was selected as a substrate to measure the oxidation activity in a continuous flow process using synthetic air (20.9 ± 1% pure O2) as an oxidizing agent. The packed-bed reactor was assembled in a fritted-glass chromatography column loaded with 0.51 wt% Pd@MIL-88B-NH2@nano-SiO2 (800 mg, 0.0385 mmol of Pd, ca. 40 mm bed height) thermostated at 110 °C (Fig. 20) with the residence time of around 10 min. After 6 h, the conversion was almost stable at around 80% until the experiment was stopped after 7 days (168 h) (Fig. 21). Selectivity was also constant during the run forming acetophenone and ethylbenzene in 75 and 5% yields, respectively. These results are, certainly, remarkable and show the possibility to develop industrial processes using MOFs as catalysts. Although metal NPs have been frequently used as catalysts for continuous flow benzylic alcohol oxidation,135 including metal NPs supported on various MOFs,136 the activity of Pd@MIL-88B-NH2@nano-SiO2 is an exclusive example of efficient aerobic oxidation of 1-phenylethanol under continuous flow conditions using Pd NPs. On the other hand, Au/MIL-101(Cr) (29300 h−1)137 and Au/MIL-101(Cr)-NH2 (103 h−1)138 have also shown to be effective solid catalysts in promoting alcohol oxidation under batch conditions. Among the probable reasons for the enhanced activity of Pd@MIL-88B-NH2@nano-SiO2 under continuous flow conditions those that have been proposed include Pd NP stabilization by amino groups and the silica coating protecting effect minimizing Pd leaching/deactivation.
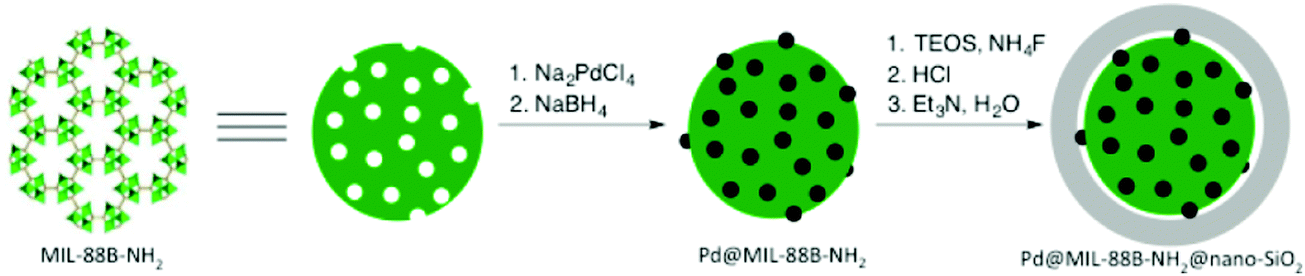 | ||
| Scheme 5 Synthetic procedure for Pd@MIL-88B-NH2@nano-SiO2. Reproduced with permission from ref. 96 Copyright American Chemical Society 2015. | ||
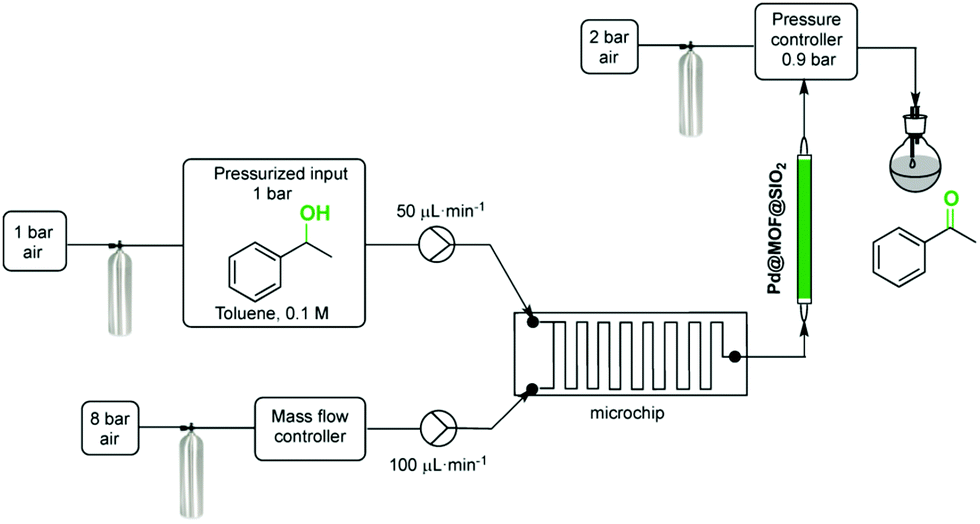 | ||
| Fig. 20 Process for the aerobic oxidation of 1-phenylethanol under continuous flow. Reproduced with permission from ref. 96 Copyright American Chemical Society 2015. | ||
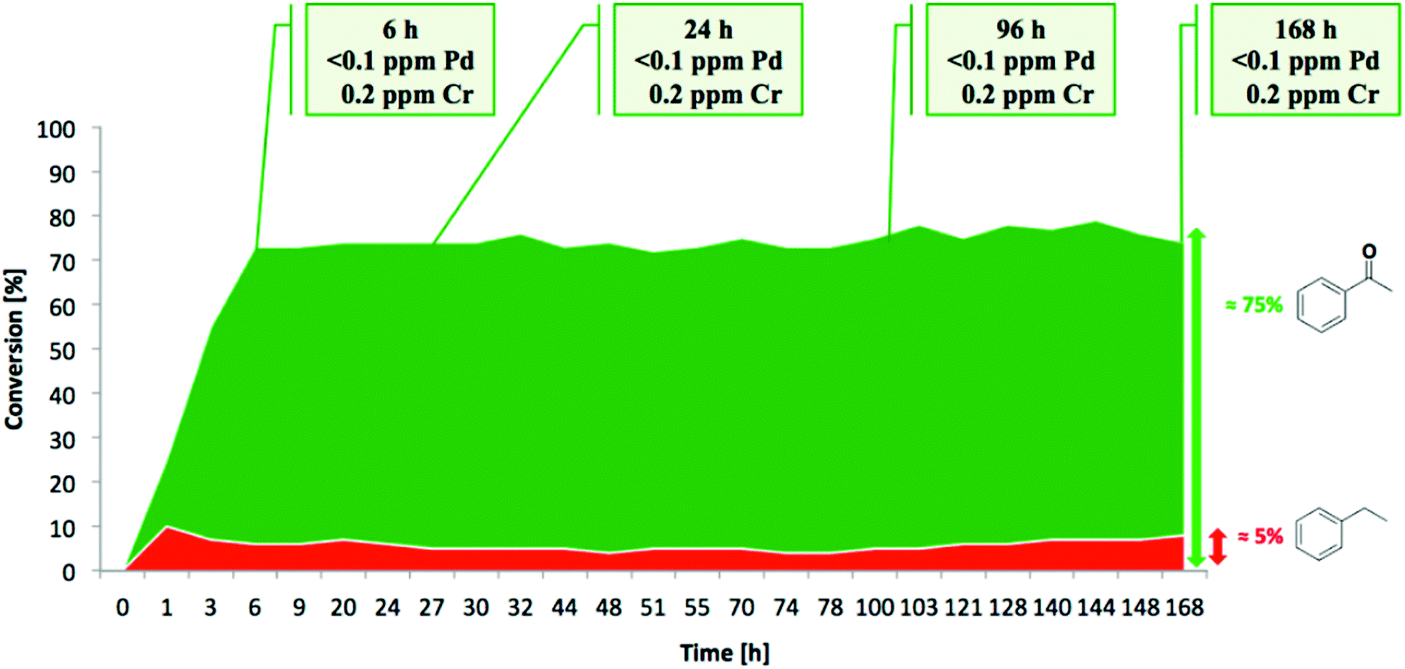 | ||
| Fig. 21 Conversion and metal leaching in the aerobic oxidation of 1-phenylethanol catalyzed by Pd@MIL-88B-NH2@SiO2 for 7 days. Reproduced with permission from ref. 96 Copyright American Chemical Society 2015. | ||
Very recently, a crystalline UiO-66 solid was functionalized with Ti to obtain UiO-66(Zr, Ti) that was tested in the oxidative desulfurization (ODS) of diesel.97 The parent UiO-66 was prepared using HCl as a crystallization agent without any modulator. TiIV was anchored to the μ3 hydroxyl groups located on the faces of the octahedral UiO-66 nodes to provide UiO-66(Zr, Ti) (Fig. 22) without altering the structural integrity of UiO-66 as evidenced by powder XRD. The introduction of Ti at the nodes of UiO-66 slightly reduced the BET surface area of the parent UiO-66(Zr) from 1328 to 1229 m2 g−1. The activity of UiO-66(Zr) and UiO-66(Zr, Ti) was established in the ODS of a solution of dibenzothiophene (Scheme 6) in dodecane (1 mL; 1000 ppm of sulphur) with 1 mL of 9:1 mixture of acetonitrile/30% aqueous H2O2 at 60 °C (Fig. 23). Analysis of the mixture showed that the model fuel still contained after the treatment 82 ppm of residual sulphur with UiO-66(Zr), while the sulphur content was 75 ppm with UiO-66(Zr, Ti). Under continuous flow conditions, the suphur content in the ODS process of dibenzothiophene using UiO-66(Zr) and UiO-66(Zr, Ti) was 535 and 276 ppm, respectively. These results imply that there is not much difference in the sulphur removal efficiency between batch versus continuous flow processes. One of the probable reasons for these results would be the limited diffusion of dibenzothiophene to the active sites. In order to confirm this hypothesis, a smaller size sulphur molecule like thioanisole (Scheme 6) was selected as a sulphide source. UiO-66(Zr) as a catalyst gave 21 ppm of residual sulphur, a value close to the specification for ULSD (<10 ppm). Interestingly, UiO-66(Zr, Ti) as a catalyst showed complete desulfurization with <1 ppm of sulphur in the model fuel under identical continuous flow conditions. These results clearly indicate the remarkable beneficial impact of the Ti atoms on the catalytic process under continuous flow conditions, in agreement with the well-known activity of Ti as a Lewis acid to activate H2O2.139,140
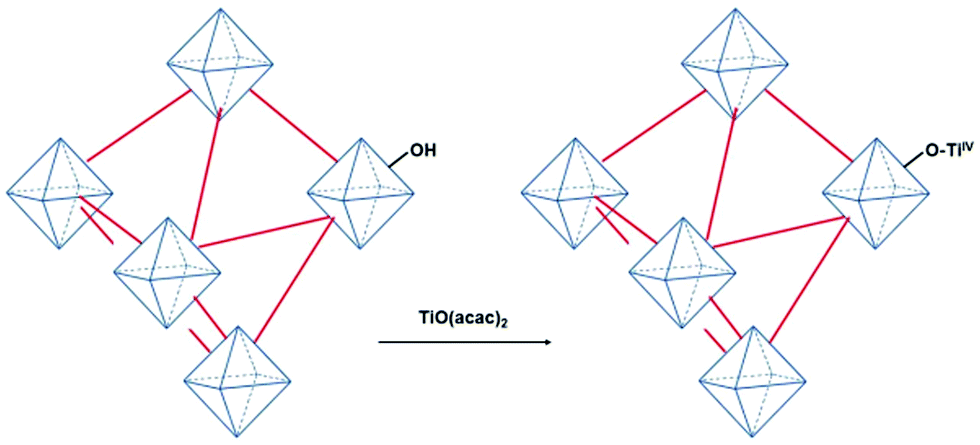 | ||
| Fig. 22 Synthesis of UiO-66(Zr, Ti) from UiO-66(Zr) using TiO(acac)2 as a Ti precursor. Reproduced with permission from ref. 97 Copyright Wiley 2019. | ||
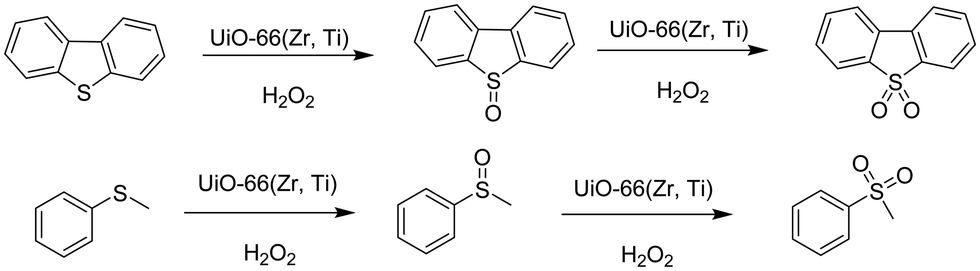 | ||
| Scheme 6 ODS of dibenzothiophene and thioanisole catalyzed by UiO-66(Zr, Ti). | ||
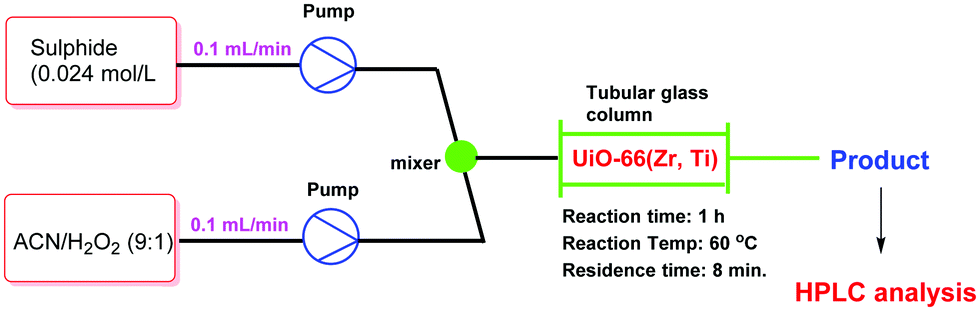 | ||
| Fig. 23 Continuous flow set up for the ODS reaction using UiO-66(Zr,Ti) as a catalyst. | ||
6. Reduction reactions
Recently, organophosphorous hydrolase (OPH) was encapsulated within the framework of MIL-100(Fe) to obtain an integrated nanocatalyst denoted as OPH@MIL-100(Fe) and its activity was tested in the cascade degradation of organophosphate nerve agents to 4-aminophenol.98 OPH is responsible for the hydrolysis of organophosphate nerve agents into 4-nitrophenol, while MIL-100(Fe) provides active sites for the reduction of 4-nitrophenol to 4-aminophenol (Scheme 7). Fluorescence experiments and acid treatment clearly confirmed that OPH is covalently attached to MIL-100(Fe) rather than adsorbed as a physical mixture. The optimal OPH loading was found to be 53 mg gMIL-100(Fe)−1. The activity of OPH@MIL-100(Fe) was studied by constructing a simple plug-flow reactor by dispersing 20 mg of OPH@MIL-100(Fe) in Tris–HCl solution and loaded onto a commercial polymer membrane by filtration as shown in Fig. 24. The solid catalyst OPH@MIL-100(Fe) retained more than 50% of the initial degradation activity after nine cycles for four organophosphate nerve agents (Fig. 25). Furthermore, the crystalline structure of OPH@MIL-100(Fe) remained unchanged after nine cycles compared to the fresh solid. A total leaching of 0.55 wt% of the initial Fe content was detected. Furthermore, OPH maintained approximately 96 and 95% of its original activity after being incubated, respectively, in a solution of 4-nitrophenol and 4-aminophenol for 90 min. Fig. 26 clearly indicates that the hydrolytic activity of OPH and the hydrogenation activity of MIL-100(Fe) were reduced after nine catalytic cycles to 53% and 95% of the original activity, respectively. These results suggest that the decrease in the degradation activity of this integrated catalyst was mainly due to the effect of NaBH4 on the hydrolytic activity of OPH. On the other hand, the degradation performance of OPH@MIL-100(Fe) was improved by reducing the contact time between the solid and NaBH4. Hence, NaBH4 was added to the reaction after the hydrolysis step. A complete degradation of methyl parathion to 4-aminophenol was observed in 45 min, which was higher activity than 85% degradation at 90 min, when NaBH4 was added at the beginning of the cascade reaction. Similarly, three other tested parathions were completely degraded by this procedure.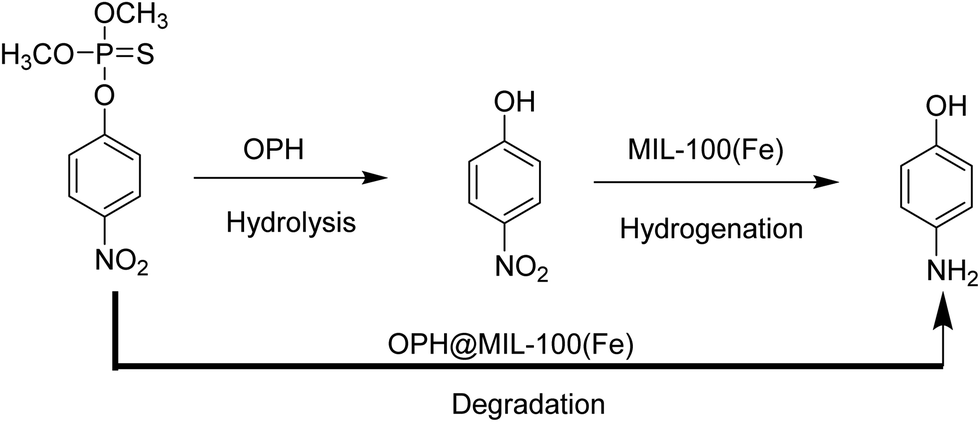 | ||
| Scheme 7 The schematic illustration of a cascade methyl parathion degradation using OPH@MIL-100(Fe) as a catalyst. | ||
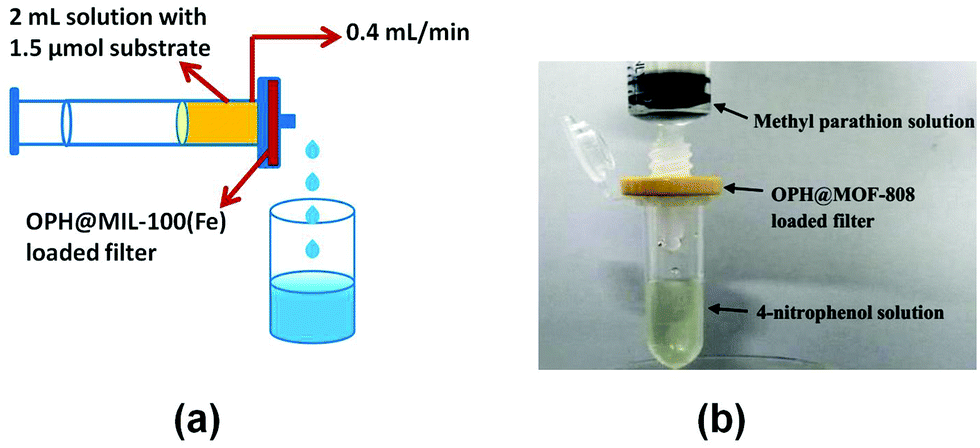 | ||
| Fig. 24 (a) Schematic illustration of the OPH@MIL-100(Fe) plug-flow reactor and (b) photograph of the OPH@MOF-808-based plug-flow reactor for hydrolysis of methyl parathion into 4-nitrophenol. Reproduced with permission from ref. 98 Copyright Royal Society of Chemistry 2018. | ||
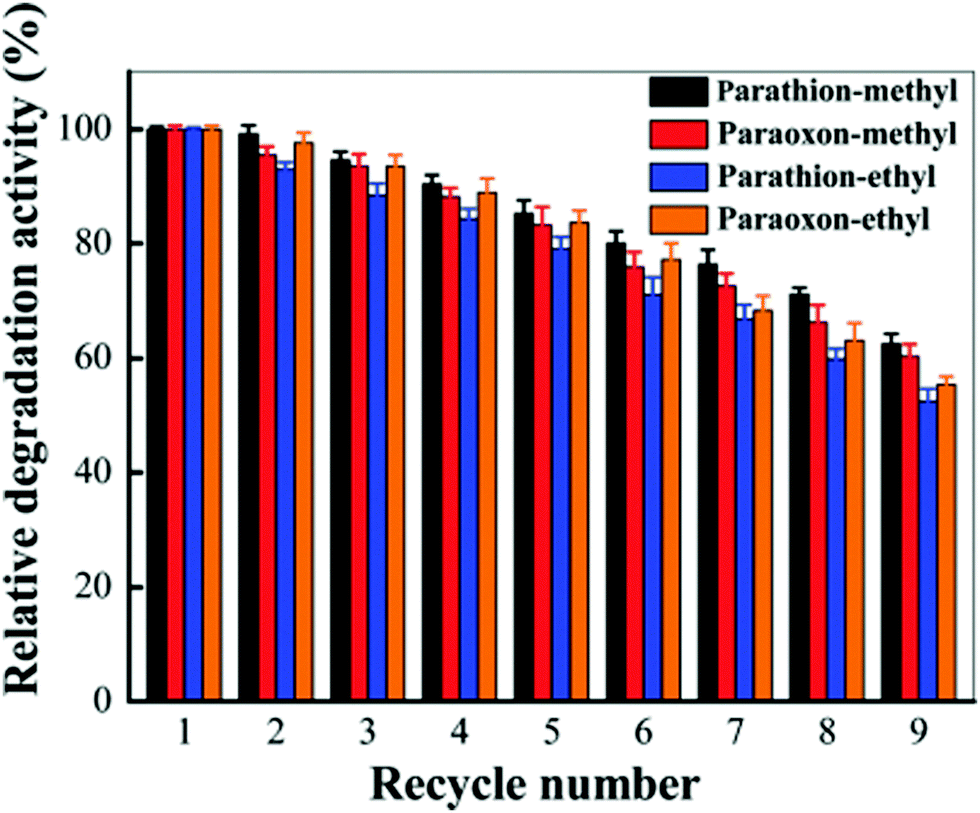 | ||
| Fig. 25 Relative degradation activity of OPH@MIL-100(Fe) with four substrates after multiple degradation cycles through a plug-flow reactor. Reproduced with permission from ref. 98 Copyright Royal Society of Chemistry 2018. | ||
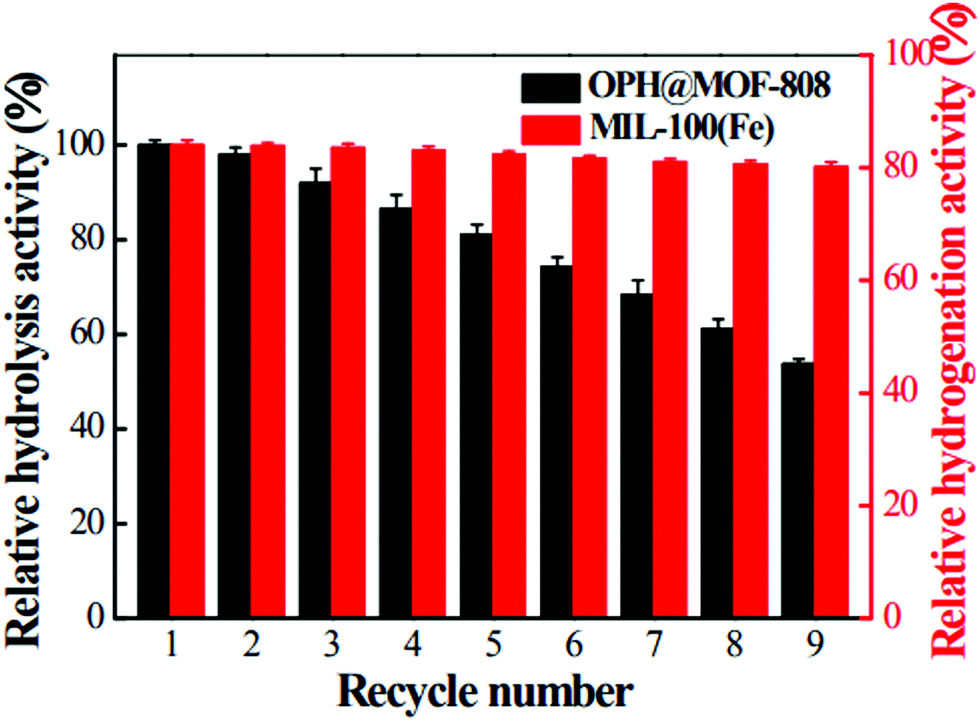 | ||
| Fig. 26 Relative hydrolysis activity of OPH and relative hydrogenation activity of MIL-100(Fe) after multiple degradation cycles. Reproduced with permission from ref. 98 Copyright Royal Society of Chemistry 2018. | ||
7. Summary and outlook
As commented in the introduction, MOFs are intensively studied as solid catalysts for liquid phase reactions, but scarce attention has been paid to their use under continuous flow reactions. Continuous flow operation requires stable catalysts that do not undergo deactivation under reaction conditions. Since this operation mode represents a high level of process intensification compared to batch-like processes, they are widely used in industrial processes. The purpose of the present review was to spur the interest of researchers in this area in performing continuous flow reactions using MOFs as catalysts, by showing that there are certain reaction types in which the stability has allowed the use of MOFs as catalysts. The reactions include acetalizations, CO2 insertion to epoxides, carbonylations, aerobic oxidations, reductions and even enantioselective Michael additions. It is remarkable that several of the examples reported use Cu3(BTC)2 as a catalyst. This Cu MOF exhibits certainly a wide range of catalytic activities as Lewis acid and oxidation catalysts, but it is not the most stable material. On one hand, the reports on the use of Cu3(BTC)2 under continuous flow illustrate that even those MOFs that are less robust can enjoy stability in a window of experimental conditions that could permit continuous flow operation. On the other hand, the current prevalence of studies using Cu3(BTC)2 as a catalyst is, in our opinion, a reflection of the lack of maturity of this field and it can be predicted that the area will be finally dominated by structurally robust MOFs.The long term goal is the implementation of MOFs as catalysts for industrial processes, probably replacing metal salts. Compared to metal salts, MOFs offer much higher activity of the fresh material due to the high surface area and porosity that makes the metals accessible to substrates and reagents. Towards this objective of commercial preparation of fine chemicals in the liquid phase using MOFs as catalysts, a possible way to proceed is the scaling up of processes carried out at smaller scale under continuous flow. Thus, it can be predicted that considering the proven activity under batch conditions, there will be in the near future an increasing interest in developing continuous flow reactions using MOFs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. D. thanks the University Grants Commission, New Delhi, for the award of an Assistant Professorship under its Faculty Recharge Programme. A. D. also thanks the Department of Science and Technology, India, for the financial support through Extramural Research Funding (EMR/2016/006500). Financial support by the Spanish Ministry of Economy and Competitiveness (Severo Ochoa and RTI2018-890237-CO2-R1) and Generalitat Valenciana (Prometeo 2017-083) is gratefully acknowledged.References
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC.
- L. Zhu, X.-Q. Liu, H.-L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS.
- P. Silva, S. M. F. Vilela, J. P. C. Tomé and F. A. Almeida Paz, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. O. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS.
- R. L. Martin and M. Haranczyk, Chem. Sci., 2013, 4, 1781–1785 RSC.
- Y. He, B. Li, M. O'Keeffe and B. Chen, Chem. Soc. Rev., 2014, 43, 5618–5656 RSC.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef.
- J. Park, H. Kim, S. S. Han and Y. Jung, J. Phys. Chem. Lett., 2012, 37, 826–829 CrossRef.
- J. G. Vitillo, L. Regli, S. Chavan, G. Ricchiardi, G. Spoto, P. D. C. Dietzel, S. Bordiga and A. Zecchina, J. Am. Chem. Soc., 2008, 130, 8386–8396 CrossRef CAS.
- Z. Hu and D. Zhao, CrystEngComm, 2017, 19, 4066–4081 RSC.
- D. S. Sholl and R. P. Lively, J. Phys. Chem. Lett., 2015, 6, 3437–3444 CrossRef CAS.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- S. Dissegna, K. Epp, W. R. Heinz, G. Kieslich and R. A. Fischer, Adv. Mater., 2018, 30, 1704501 CrossRef.
- C. Perego and R. Millini, Chem. Soc. Rev., 2013, 42, 3956–3976 RSC.
- X. S. Zhao, X. Y. Bao, W. Guo and F. Y. Lee, Mater. Today, 2006, 9, 32–39 CrossRef CAS.
- J. Liang, Z. Liang, R. Zou and Y. Zhao, Adv. Mater., 2017, 29, 1701139 CrossRef.
- M. Dusselier and M. E. Davis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS.
- S. M. Csicsery, Zeolites, 1984, 4, 202–213 CrossRef CAS.
- C.-D. Wu and M. Zhao, Adv. Mater., 2017, 29, 1605446 CrossRef.
- Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- B. Li, H.-M. Wen, W. Zhou, J. Q. Xu and B. Chen, Chem, 2016, 1, 557–580 CAS.
- M. C. Das, S. Xiang, Z. Zhang and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 10510–10520 CrossRef CAS.
- H.-Y. Guan, R. J. LeBlanc, S.-Y. Xie and Y. Yue, Coord. Chem. Rev., 2018, 369, 76–90 CrossRef CAS.
- L. M. Aguirre-Díaz, D. Reinares-Fisac, M. Iglesias, E. Gutiérrez-Puebla, F. Gándara, N. Snejko and M. Ángeles Monge, Coord. Chem. Rev., 2017, 335, 1–27 CrossRef.
- C. K. Brozek and M. Dincă, Chem. Sci., 2012, 3, 2110–2113 RSC.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- M. Zhang, M. Bosch, T. Gentle III and H.-C. Zhou, CrystEngComm, 2014, 16, 4069–4083 RSC.
- M. Eddaoudi, H. Li and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 1391–1397 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- J. C. van der Waal, P. J. Kunkelera, K. Tan and H. van Bekkum, J. Catal., 1998, 173, 74–83 CrossRef CAS.
- J. F. Haw, Phys. Chem. Chem. Phys., 2002, 4, 5431–5441 RSC.
- B. Yan, L.-Z. Tao, A. Mahmood, Y. Liang and B.-Q. Xu, ACS Catal., 2017, 7, 538–550 CrossRef CAS.
- A. Herbst and C. Janiak, CrystEngComm, 2017, 19, 4092–4117 RSC.
- A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Catal. Sci. Technol., 2013, 3, 2509–2540 RSC.
- A. Dhakshinamoorthy, M. Alvaro, A. Corma and H. Garcia, Dalton Trans., 2011, 40, 6344–6360 RSC.
- J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y.-G. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642–5650 RSC.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- K. Nita, S. Nakai, S. Hidaka, T. Mibuchi, H. Shimakawa, K.-I. Ii and K. Inamura, Stud. Surf. Sci. Catal., 1987, 34, 501–511 CrossRef CAS.
- A. R. Sujan, D.-Y. Koh, G. Zhu, V. P. Babu, N. Stephenson, A. Rosinski, H. Du, Y. Luo, W. J. Koros and R. P. Lively, Ind. Eng. Chem. Res., 2018, 57, 11757–11766 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro, P. Concepcion and H. Garcia, Catal. Commun., 2011, 12, 1018–1021 CrossRef CAS.
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS PubMed.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144 CrossRef CAS PubMed.
- B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. Santiago-Portillo, A. M. Asiri and H. Garcia, ChemCatChem, 2019, 11, 899–923 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- F. Vermoortele, B. Bueken, G. Le Bars, B. van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier and V. van Speybroeck, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- M. Taddei, R. J. Wakeham, A. Koutsianos, E. Andreoli and A. R. Barron, Angew. Chem., Int. Ed., 2018, 57, 11706–11710 CrossRef CAS PubMed.
- M. Taddei, Coord. Chem. Rev., 2017, 343, 1–24 CrossRef CAS.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- F. X. Llabrés i Xamena, A. Abad, A. Corma and H. Garcia, J. Catal., 2007, 250, 294–298 CrossRef.
- Z. Liu, J. Zhu, C. Peng, T. Wakihara and T. Okubo, React. Chem. Eng., 2019, 4, 1699–1720 RSC.
- B. G. Rao, P. Sudarsanam, B. Mallesham and B. M. Reddy, RSC Adv., 2016, 6, 95252–95262 RSC.
- G.-Y. Jeong, A. K. Singh, M.-G. Kim, K.-W. Gyak, U. Ryu, K. M. Choi and D.-P. Kim, Nat. Commun., 2018, 9, 3968–3976 CrossRef PubMed.
- M. Rubio-Martinez, M. P. Batten, A. Polyzos, K.-C. Carey, J. I. Mardel, K.-S. Lim and M. R. Hill, Sci. Rep., 2014, 4, 5443–5447 CrossRef CAS PubMed.
- X. Liu, B. Ünal and K. F. Jensen, Catal. Sci. Technol., 2012, 2, 2134–2138 RSC.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- P. Plouffe, A. Macchi and D. M. Roberge, Org. Process Res. Dev., 2014, 18, 1286–1294 CrossRef CAS.
- S. Souzanchia, L. Nazaria, K. T. Venkateswara Rao, Z. Yuana, Z. Tanb and C. Xu, Catal. Today, 2019, 319, 76–83 CrossRef.
- J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC.
- M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS PubMed.
- C. Moreno-Marrodan, F. Liguori and P. Barbaro, Beilstein J. Org. Chem., 2017, 13, 734–754 CrossRef CAS PubMed.
- U. Hintermair, G. Francio and W. Leitner, Chem. Commun., 2011, 47, 3691–3701 RSC.
- J.-I. Yoshida, H. Kim and A. Nagaki, ChemSusChem, 2011, 4, 331–340 CrossRef CAS PubMed.
- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC.
- S. Marre and K. F. Jensen, Chem. Soc. Rev., 2010, 39, 1183–1202 RSC.
- C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300–2318 CrossRef CAS PubMed.
- B. K. Singh, N. Kaval, S. Tomar, E. Van der Eycken and V. S. Parmar, Org. Process Res. Dev., 2008, 12, 468–474 CrossRef CAS.
- D. Mark, S. Haeberle, G. Roth, F. von Stetten and R. Zengerle, Chem. Soc. Rev., 2010, 39, 1153–1182 RSC.
- T. Noel and S. L. Buchwald, Chem. Soc. Rev., 2011, 40, 5010–5029 RSC.
- Y. Monguchi, T. Ichikawa, T. Yamada, Y. Sawama and H. Sajiki, Chem. Rec., 2019, 19, 3–14 CrossRef CAS PubMed.
- C. A. Hone and C. O. Kappe, Top. Curr. Chem., 2019, 377, 2 CrossRef PubMed.
- V. Pascanu, P. R. Hansen, A. B. Gomez, C. Ayats, A. E. Platero-Prats, M. J. Johansson, M. A. Pericas and B. Martin-Matute, ChemSusChem, 2015, 8, 123–130 CrossRef CAS PubMed.
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC.
- J. Zhang, J. Chen, S. Peng, S. Peng, Z. Zhang, Y. Tong, P. W. Miller and X.-P. Yan, Chem. Soc. Rev., 2019, 48, 2566–2595 RSC.
- H. G. Jolliffe and D. I. Gerogiorgis, Chem. Eng. Res. Des., 2016, 112, 310–325 CrossRef CAS.
- P. D. Morse, R. L. Beingessner and T. F. Jamison, Isr. J. Chem., 2017, 57, 218–227 CrossRef CAS.
- I. Rossetti, Catal. Today, 2018, 308, 20–31 CrossRef CAS.
- D. K. B. Mohamed, X. Yu, J. Li and J. Wu, Tetrahedron Lett., 2016, 57, 3965–3977 CrossRef CAS.
- L. Shi, X. Wang, C. A. Sandoval, Z. Wang, H. Li, J. Wu, L. Yu and K. Ding, Chem. – Eur. J., 2009, 15, 9855–9867 CrossRef CAS PubMed.
- C. Wang, B. An and W. Lin, ACS Catal., 2019, 9, 130–146 CrossRef CAS.
- X. Chen, H. Jiang, B. Hou, W. Gong, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2017, 139, 13476–13482 CrossRef CAS PubMed.
- Y. Chen, S. Zhang, F. Chen, S. Cao, Y. Cai, S. Li, H. Ma, X. Ma, P. Li, X. Huang and B. Wang, J. Mater. Chem. A, 2018, 6, 342–348 RSC.
- P. Ji, X. Feng, P. Oliveres, Z. Li, A. Murakami, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 14878–14888 CrossRef CAS PubMed.
- J. Hou, Y. Luan, X. Huang, H. Gao, M. Yang and Y. Lu, New J. Chem., 2017, 41, 9123–9129 RSC.
- C. G. Piscopo, M. Schwarzer, M. Herrmann, A. Affini, P. Pelagatti, G. Maestri, R. Maggi and S. Loebbecke, ChemCatChem, 2016, 8, 1293–1297 CrossRef CAS.
- H. Gao, Y. Luan, K. Chaikittikul, W. Dong, J. Li, X. Zhang, D. Jia, M. Yang and G. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4667–4674 CrossRef CAS PubMed.
- B. R. James, J. A. Boissonnault, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, RSC Adv., 2018, 8, 2132–2137 RSC.
- H. D. Park, M. Dinca and Y. Roman-Leshkov, J. Am. Chem. Soc., 2018, 140, 10669–10672 CrossRef CAS PubMed.
- W. Ouyang, D. Zhao, Y. Wang, A. M. Balu, C. Len and R. Luque, ACS Sustainable Chem. Eng., 2018, 6, 6746–6752 CrossRef CAS.
- A. Sachse, R. Ameloot, B. Coq, F. Fajula, B. Coasne, D. De Vos and A. Galarneau, Chem. Commun., 2012, 48, 4749–4751 RSC.
- V. Pascanu, A. B. Gomez, C. Ayats, A. E. Platero-Prats, F. Carson, J. Su, Q. Yao, M. A. Pericas, X. Zou and B. Martín-Matute, ACS Catal., 2015, 5, 472–479 CrossRef CAS.
- C. G. Piscopo, L. Voellinger, M. Schwarzer, A. Polyzoidis, D. Bošković and S. Loebbecke, ChemistrySelect, 2019, 4, 2806–2809 CrossRef CAS.
- H. Li, L. Ma, L. Zhou, J. Gao, Z. Huang, Y. He and Y. Jiang, Chem. Commun., 2018, 54, 10754–10757 RSC.
- D. S. Kundu, J. Schmidt, C. Bleschke, A. Thomas and S. Blechert, Angew. Chem., Int. Ed., 2012, 51, 5456 CrossRef CAS PubMed.
- H. S. Ozdemir, E. Sahin and M. Cakici, Tetrahedron, 2015, 71, 2882 CrossRef CAS.
- F. Guo, D. Chang, G. Lai, T. Zhu, S. Xiong, S. Wang and Z. Wang, Chem. – Eur. J., 2011, 17, 11127 CrossRef CAS PubMed.
- Y.-F. Sheng, Q. Gu, A.-J. Zhang and S.-L. You, J. Org. Chem., 2009, 74, 6899 CrossRef CAS PubMed.
- I. Fechete, Y. Wang and J. C. Vedrine, Catal. Today, 2012, 189, 2 CrossRef CAS.
- T. Tsuruoka, S. Furukawa, Y. Takashima, K. Yoshida, S. Isoda and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4739–4743 CrossRef CAS PubMed.
- J. Cravillon, R. Nayuk, S. Springer, A. Feldhoff, K. Huber and M. Wiebcke, Chem. Mater., 2011, 23, 2130–2141 CrossRef CAS.
- E. Angeletti, C. Canepa, G. Martinetti and P. Venturello, J. Chem. Soc., Perkin Trans. 1, 1989, 105–107 RSC.
- A. Sachse, A. Galarneau, F. Di Renzo, F. Fajula and B. Coq, Chem. Mater., 2010, 22, 4123–4125 CrossRef CAS.
- D.-Z. Xu, S. Shi and Y. Wang, RSC Adv., 2013, 3, 23075–23079 RSC.
- T. Jackson, J. H. Clark, D. J. Macquarrie and J. H. Brophy, Green Chem., 2004, 6, 193–195 RSC.
- E. A. Feijani, H. Mandavi and A. Tavasoli, Chem. Eng. Res. Des., 2015, 96, 87–102 CrossRef CAS.
- S. L. Qiu, M. Xue and G. S. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- B. Seoane, J. Coronas, I. Gascon, M. E. Benavides, O. Karvan, J. Caro, F. Kapteijn and J. Gascon, Chem. Soc. Rev., 2015, 44, 2421–2454 RSC.
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- L. Wu, T. Moteki, A. A. Gokhale, D. W. Flaherty and F. D. Toste, Chem, 2016, 1, 32–58 CrossRef CAS PubMed.
- N. Alonso-Fagúndez, V. Laserna, A. Alba-Rubio, M. Mengibar, A. Heras, R. Mariscal and M. L. Granados, Catal. Today, 2014, 234, 285–294 CrossRef.
- J. D. Lewis, S. Van de Vyver, A. J. Crisci, W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. Román-Leshkov, ChemSusChem, 2014, 7, 2255–2265 CrossRef CAS PubMed.
- K.-i. Shimizu, E. Hayashi, T. Hatamachi, T. Kodama, T. Higuchi, A. Satsuma and Y. Kitayama, J. Catal., 2005, 231, 131–138 CrossRef CAS.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC.
- K. Kasuga, T. Kato, N. Kabata and M. Handa, Bull. Chem. Soc. Jpn., 1996, 69, 2885–2888 CrossRef CAS.
- D. Y. Hong, Y. K. Hwang, C. Serre, G. Ferey and J. S. Chang, Adv. Funct. Mater., 2009, 19, 1537 CrossRef CAS.
- Y. D. Y. L. Getzler, V. Kundnani, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 6842 CrossRef CAS PubMed.
- M. L. A. Jansen and W. M. van Gulik, Curr. Opin. Biotechnol., 2014, 30, 190 CrossRef CAS PubMed.
- H. Li, Z. Fang and S. Yang, ACS Sustainable Chem. Eng., 2016, 4, 236–246 CrossRef CAS.
- A. H. Valekar, K.-H. Cho, S. K. Chitale, D.-Y. Hong, G.-Y. Cha, U.-H. Lee, D. W. Hwang, C. Serre, J.-S. Chang and Y. K. Hwang, Green Chem., 2016, 18, 4542–4552 RSC.
- Y. Kuwahara, H. Kango and H. Yamashita, ACS Sustainable Chem. Eng., 2017, 5, 1141–1152 CrossRef CAS.
- J. He, H. Li, Y. Lu, Y. Liu, Z. Wu, D. Hu and S. Yang, Appl. Catal., A, 2016, 510, 11–19 CrossRef CAS.
- X. Tang, H. Chen, L. Hu, W. Hao, Y. Sun, X. Zeng, L. Lin and S. Liu, Appl. Catal., B, 2014, 147, 827–834 CrossRef CAS.
- J. Gascon and F. Kapteijn, Angew. Chem., Int. Ed., 2010, 49, 1530 CrossRef CAS PubMed.
- S. Aguado, J. Canivet and D. Farrusseng, J. Mater. Chem., 2011, 21, 7582 RSC.
- E. V. Ramos-Fernandez, M. Garcia-Domingos, J. Juan-Alcaniz, J. Gascon and F. Kapteijn, Appl. Catal., A, 2011, 391, 261 CrossRef CAS.
- E. Perez-Mayoral and J. Cejka, ChemCatChem, 2011, 3, 157 CrossRef CAS.
- E. Pérez-Mayoral, Z. Musilová, B. Gil, B. Marszalek, M. Položij, P. Nachtigall and J. Čejka, Dalton Trans., 2012, 41, 4036–4044 RSC.
- N. Zotova, K. Hellgardt, G. H. Kelsall, A. S. Jessiman and K. K. M. Hii, Green Chem., 2010, 12, 2157–2163 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Chem. Commun., 2017, 53, 10851–10869 RSC.
- H. Liu, Y. Liu, Y. Li, Z. Tang and H. Jiang, J. Phys. Chem. C, 2010, 114, 13362–13369 CrossRef CAS.
- A. Santiago-Portillo, M. Cabrero-Antonino, M. Alvaro, S. Navalon and H. Garcia, Chem. – Eur. J., 2019, 25, 9280–9286 CrossRef CAS PubMed.
- C. W. Yoon, K. F. Hirsekorn, M. L. Neidig, X. Yang and T. Don Tilley, ACS Catal., 2011, 1, 1665–1678 CrossRef CAS.
- C. Hammond and G. Tarantino, Catalysts, 2015, 5, 2309–2323 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |