
Versatile symport transporters based on cyclic peptide dimers†
Alberto
Fuertes
 ,
Manuel
Amorín
* and
Juan R.
Granja
*
,
Manuel
Amorín
* and
Juan R.
Granja
*
Singular Research Centre in Chemical Biology and Molecular Materials, (CIQUS), Organic Chemistry Department, University of Santiago de Compostela (USC), 15782, Santiago de Compostela, Spain. E-mail: manuel.amorin@usc.es; juanr.granja@usc.es
First published on 15th November 2019
Abstract
We present the synthesis and transmembrane transport properties of a new family of tris-pyridine-decorated cyclic peptides. These molecules are designed to self-assemble into dimeric shuttles in nonpolar media, which act as symport ionophores in which, apparently, the tris-pyridine scaffold complexes both cations and anions with high potency and efficacy.
The transmembrane transport of ions and other small polar molecules plays several crucial roles in cellular functions, such as cell communication, homeostasis or energy production. Generally, integral membrane proteins are responsible for carrying out these tasks in living organisms.1 Consequently, the malfunction of these natural transporters can give rise to a variety of diseases, generally known as channelopathies.2 Hence, the development of new synthetic molecules with the ability of transporting relevant ions across lipid bilayers is an appealing task in search for potential new therapeutic strategies.
Synthetic chemists often find inspiration in natural ionophores, which exhibit a lipophilic outer surface and a heteroatom-rich inner cavity, where the desolvated ion is accommodated masking its hydrophilicity.3 Artificial ionophores generally rely on a topologically prearranged scaffold (generally multipodal) to achieve a correct disposition of the analyte-stabilising groups in the transport-active conformation.4 Even though a wide variety of synthetic mobile carriers have been described in recent years, the number of transporters that are capable of translocating simultaneously the anion and cation (symport mechanism) is much scarcer. Furthermore, most of the examples in the literature rely on functionalized cavitands (i.e., calixpyrroles or calixarenes) with separated binding domains for cations and anions, whereas the discovery of transporters that utilise an ambivalent chemical motif for the complexation of both species remains elusive.5 The main advantage of the cooperative salt transport relies on the possibility of moving an ion against its gradient by coupling its transport with a counterion that presents a favorable gradient.6 In addition, symport transport should not dissipate the transmembrane pH gradients, an undesired side-effect for biomedical applications.
In this regard, we have designed a new class of transmembrane carriers based on a hydrophobic α,γ-cyclic peptide (CP) scaffold7 that was post-synthetically modified with three pyridine moieties that are projected perpendicularly from the plane of the ring (CP2/3, Scheme 1 and Schemes 2SI and 3SI, ESI†).8 Due to the coordinating capabilities of the pyridine rings and its correct topological disposition at the entrance of the CP dimeric cavity, we envision that these molecules could act as attractive ion shuttles across model lipid bilayers.
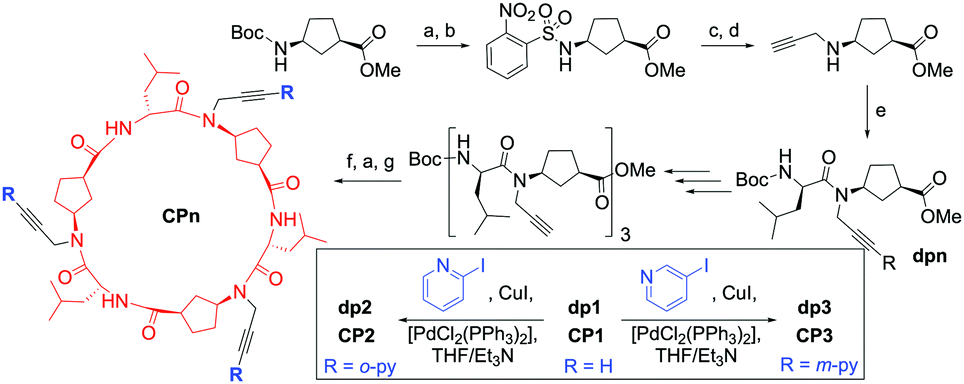 | ||
| Scheme 1 Synthetic route followed for the preparation of pyridine-decorated CP ionophores CP2 and CP3. (a) TFA/CH2Cl2; (b) NsCl, DIEA, CH2Cl2 (87%); (c) propargyl bromide, K2CO3, ACN; (d) PhSH, K2CO3, ACN (69%); (e) Boc-D-Leu-OH, N-HBTU, DIEA, CH2Cl2 (96%); (f) LiOH, MeOH/H2O; (g) N-TBTU, DIEA, CH2Cl2 (49%). | ||
The synthesis of the artificial transporters (Scheme 1) started with the preparation of a N-propargylated cyclic γ-amino acid by means of the Fukuyama strategy,9 which is then coupled with Boc-D-Leu-OH to generate the basic building block of the cyclic peptide. This dipeptide (dp1) was sequentially deprotected and coupled to afford a hexamer, whose protecting groups were removed to perform a cyclization that produced alkyne-functionalized cyclic α,γ-hexapeptide CP1 in 49% yield. CP1 was subsequently coupled with either 2- or 3-iodopyridine under Sonogashira conditions (PdCl2(PPh3)2/CuI/Et3N) to afford pyridine-decorated peptides CP2 and CP3, respectively.8
The resulting peptides, as their precursor CP1, assemble into the corresponding dimers (D2 and D3, Scheme 3SI, ESI†) in non-polar solvents, as confirmed by the down-field shift of amide protons involved in the dimerization process in the 1H NMR experiments. These signals at 8.11 (CP1), 8.26 (CP2) and 8.28 (CP3) ppm remain unchanged with peptide concentration, confirming, as previously observed for this type of peptides, the large association constant in this media (Ka > 106 M−1).7 FTIR, with bands at 3297, 1623 and 1532 cm−1 for CP1; 3400, 1655 and 1530 cm−1 for CP2 and 3302, 1628 and 1532 for CP3 also confirmed the antiparallel β-sheet type interactions characteristic of the dimeric structures.7
The ion transport capabilities across model lipid bilayers (EYPC) were examined using fluorescence assays.10,11 First, we evaluated the transport ability of these molecules to rectify a pH gradient across LUVs with entrapped 5(6)-carboxyfluorescein (CF⊂LUV, Fig. 1A), so the ionophore-facilitated vesicle basification would produce an increase in the fluorescence intensity, which is assumed to be associated with the co-transport of sodium ions.12 Remarkably, it was found that the intravesicular pH could be completely equilibrated in the case of CP2, whereas the meta-pyridine isomer CP3, although remarkable, only exhibited a Ymax value of 63%. Both ionophores presented EC50 values, determined by the Hill equation,10,13 in the sub-μM range (263 vs. 199 nM for CP2 and CP3, Fig. 2SI and 3SI, respectively, ESI†). On the other hand, no transport activity was observed for CP1 under similar conditions, confirming the need of pyridine moieties for the transport. The integrity of the vesicles in the presence of both transporters was confirmed by a self-quenched CF experiment (see Fig. 4SI, ESI†).
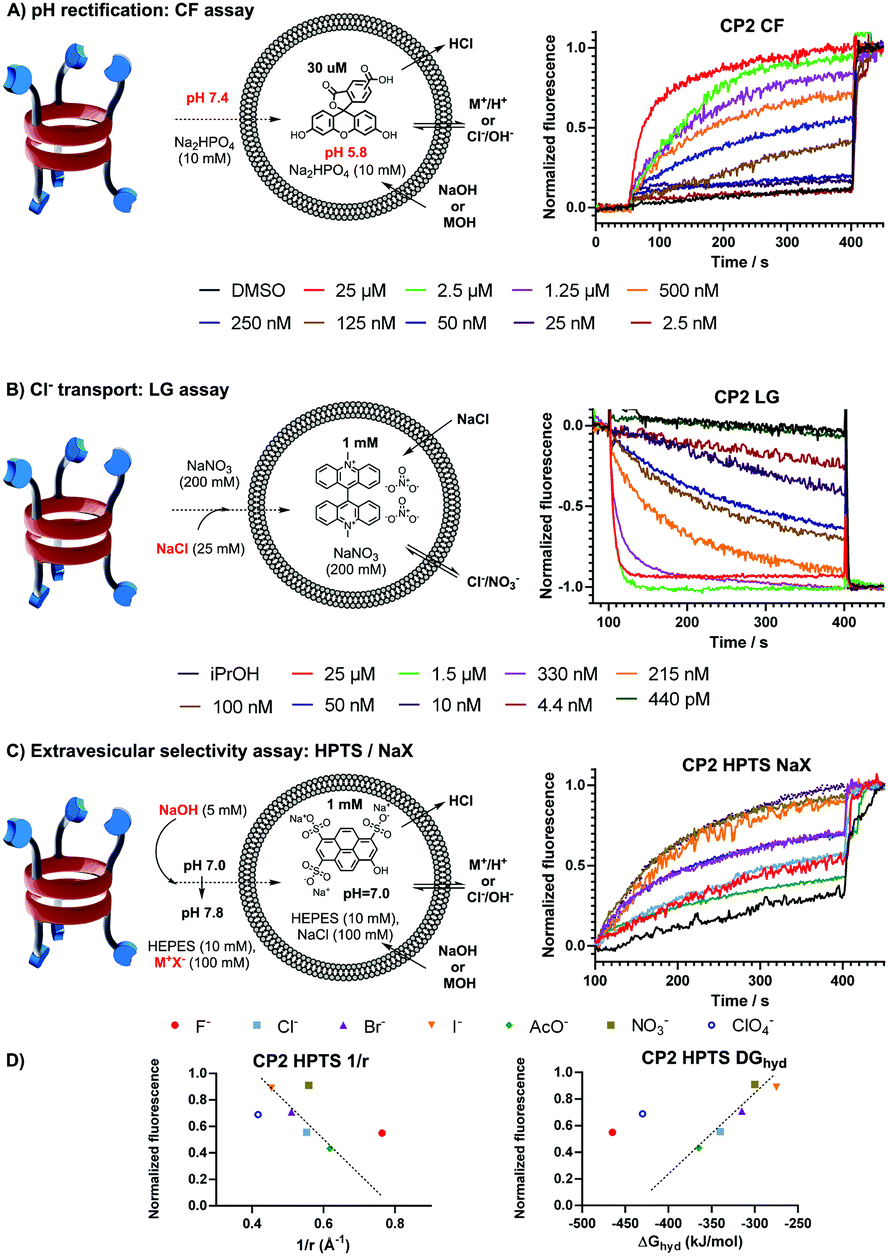 | ||
| Fig. 1 (A) pH dissipation assay on CF⊂LUV and (B) Cl− transport assay with CP2 at different concentrations. (C) HPTS⊂LUV experiment under different NaX extravesicular buffers. (D) Transport intensities plotted against 1/r and hydration energy (ΔGhyd) for each anion (the dashed line was incorporated to emphasize the linear relationship). | ||
Encouraged by these activities, we tested the selectivity of these transporters towards cations, expecting that the tris-pyridine caps should preferentially complex metal ions over anions due to their unique coordination properties. In this regard, the lucigenin assay was explored (LG⊂LUV, Fig. 1B), to rule out the possibility of transporting chloride ions.14 Surprisingly, it was found that the intravesicular delivery of chloride ions was carried out more efficiently than cations by both transporters. Again, CP2 outperformed CP3, showing a remarkable activity (EC50 = 45 nM), around four times more potent than CP3 (Fig. 6SI and 7SI, respectively, ESI†) and, also, five times more efficient in transporting chloride than proton/sodium exchange in CF⊂LUV experiments.
Next, we studied the ion selectivity; therefore, a new rectification assay was designed to analyse whether the basification of the intravesicular milieu exhibited a preference on the nature of the extravesicular buffer composition. With this idea, an HPTS⊂LUV (Fig. 1C) basification procedure was carried out under the presence of a variety of extravesicular salts to evaluate the importance of either anions or cations in the transport.15 Therefore, different experiments were carried out with a variety of sodium salts and, also, different alkaline and alkaline-earth chloride species. It was found that more accute variability was observed in the experiments where different anions were analysed compared to those carried out with chloride salts. Initially, it was hard to find any selectivity trend, however, the analysis of the efficacy versus the ionic radius and the hydration energy of different anions (Fig. 1D) showed two opposite transport trends for each transporter. For the ortho-pyridine substituted ionophore CP2, there was a preference for large and polarizable anions such as I− or NO3−, with fluoride performing better than expected. The meta-isomer CP3 exhibited an opposite selectivity where small and strongly hydrated anions, such as F−, were preferred (Fig. 11SI and 12SI, ESI†). These results clearly demonstrate that the position of the nitrogen atom either at the entrance or pointing towards the interior of the cavity (CP3 and CP2, respectively) plays a major role in the binding mode of different ionic species. Similar analysis was carried out with the cations, but no significant changes in transport activities were observed (see Fig. 10SI and 13SI, ESI†). Only transport of magnesium dichloride presented a different behaviour, perhaps because of its different dissociation and dehydration properties.
Intrigued by these results, the transport mechanism was studied. In this regard, vesicles made of DPPC phospholipids were prepared, given their higher phase transition temperature (41 °C)16 that allows the discrimination between mobile and static transporters.17 Carrier transporters should provide transport behaviour that depends on the phase of the membrane, where no transport should be observed with bilayers in the solid gel phase state (bellow Tm). Channel-forming molecules should not be affected by the lipid phase. Experiments carried out at higher (45 °C) and lower (25 °C) temperatures than Tm ruled out the pore-forming mechanism based on the observed differences in transport in HPTS⊂LUV(DPPC) assays induced by pH gradient (Fig. 2).
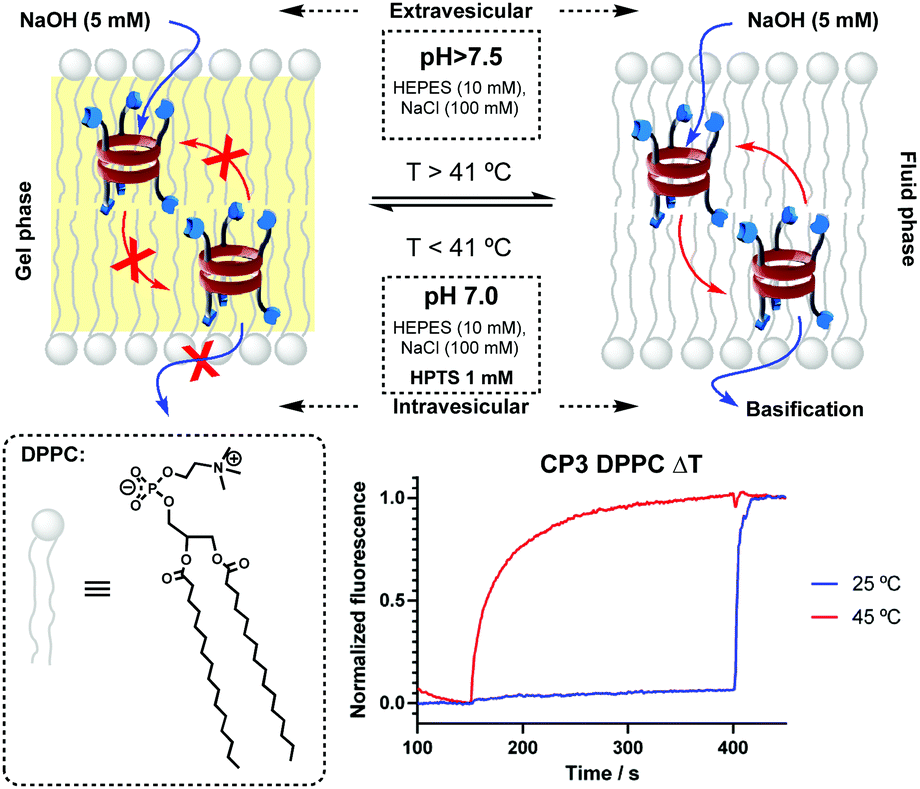 | ||
| Fig. 2 HPTS⊂LUV(DPPC) assay carried out at different temperatures was employed to reveal that membranes in the fluid state (T > Tm) were required for the transport to occur, confirming a carrier mechanism. | ||
Additionally, in an attempt to determine the sym- or antiport mechanism, additional Cl− transport experiments were carried out. It is proposed that in the chloride transport carried out in nitrate solutions, the halide transport is associated with the antiport transport of a nitrate ion.14 The substitution of nitrate for the more hydrophilic sulfate, whose high hydrophilicity18 makes its transport extremely difficult to achieve, can help to differentiate both mechanisms.19 Experiments with sulfate solutions provided a 50% reduction in transport activity (Fig. 8SI, ESI†), suggesting that the ionophores operate under a symporter mechanism, where both Na+ and Cl− migrate simultaneously until a balance in the osmotic pressure is reached. To confirm the ability to transport metal cations, we carried out some additional transport experiments. Sodium transport was confirmed by 23Na NMR experiments (Fig. 14SI, ESI†),20 in which liposomes containing LiCl (80 mM) were placed in a solution containing NaCl and dysprosium(III)tripolyphosphate (DyPPP). The chemical shift of sodium nuclei in the presence of DyPPP depends on the ratio between the ion and the dysprosium salt. A new signal (0.0 ppm) appears upon internalization of Na+ into the vesicles. Calcium transport was also analyzed using calcein⊂LUV(DPPC). In this case the increase of fluorescence emission upon peptide addition also confirmed the properties of CP2 to translocate Ca(II) into the vesicles (Fig. 15SI, ESI†).
Finally, a series of control experiments with other peptide scaffolds were also performed to determine the structural requirements of each carrier moiety for successful ion translocation activity. LG⊂LUV assays were accomplished with CP1, lacking pyridine rings, CP4,7a that incorporates Phe instead of Leu and also lacks pyridine moieties, and both protected dipeptides dp2 and dp3, which are the monomeric units repeated in CP2 and CP3, respectively. These experiments revealed that only dp3 exhibited Cl− transport activity at high concentrations. In any case, a detailed dose–response analysis compared with counterpart CP3 (Fig. 3, top) revealed that the cyclic scaffold was around ten times more active than dp3, a higher value than the one expected in statistical terms (i.e., 3-fold increase). This result clearly revealed that the incorporation of three pyridine rings onto the cyclic skeleton induced a cooperativity that promoted the ion translocation.
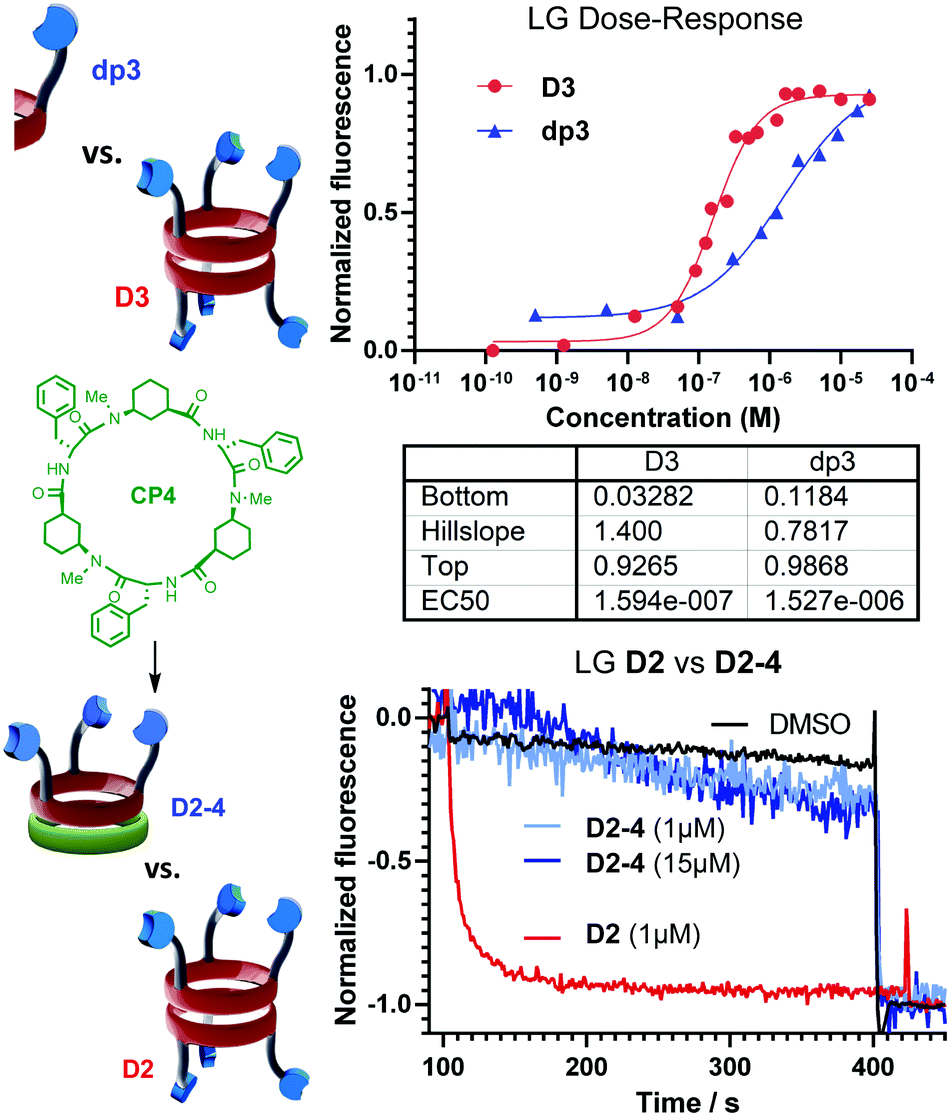 | ||
| Fig. 3 Top: Comparison between the cyclic scaffold D3 and its repeating unit dp3. Bottom: Effect of the heterodimerization on the transport efficiency after the addition of CP4 (in green). | ||
At this point, we also evaluated the influence of both tris-pyridine caps in the transport and, for this purpose, we carried out experiments with heterodimers in which only one of the CPs bears the three heterocyclic moieties. Previously, we have found that α,γ-CPs made of different types of γ-residues [3-aminocyclopentanecarboxylic acid (Acp) versus 3-aminocyclo-hexanecarboxylic acid (Ach) residues] form heterodimeric aggregates that are more stable than the corresponding homodimers.7b Therefore, CP4 (Fig. 3, green) with only N-methyl groups combined with CP2 gave rise to heterodimer D2-4. The formation of this heterodimer was confirmed by NMR, in which the appearance of proton signals that correspond to the heterodimer (D2-4) confirmed its formation (Fig. 14SI, ESI†). Consequently, transport experiments with equimolar amounts of CP4 and CP2 in LG⊂LUV assays were carried out (Fig. 3, bottom). None of these experiments showed ionophoric activity, suggesting that both tris-pyridine caps of the dimeric form D2/3 (Scheme 3SI, ESI†) are required for the transport to take place, perhaps with every single unit helping to stabilize each ion. With all these results in hand we propose a complexation mechanism in which the pyridine moieties not only act as coordinating ligands for metal ions but also of their ion-pair. Due to the electronic properties of the pyridine moieties and their close proximity of the tripodal arms, they might interact with anions through anion–π interactions or hydrogen bonds.21 The metal coordination ability was studied by NMR in which different salts (CuI, Zn(OTf)2, Fig. 17SI and 18SI, ESI†) were added to a solution of CP2. These additions provoke the characteristic shift of the signals of pyridine protons upon binding with metal ions. The sharp signals and C3 symmetry of the resulting spectra confirm the simultaneous coordination of the three pyridine moieties to the ion. Therefore, for CP2, we propose that the anion must lose the hydration waters in order for the pyridines to establish interactions with larger anions, because of the ortho-orientation of the heterocyclic moieties, fitting better in the tripodal cap (Fig. 4A). Alternatively, CP3 would interact with the hydrated anions and consequently pyridine moieties with meta-substitution would prefer the small ions (Fig. 4B). In both models, each CP would interact with a different ion. To the best of our knowledge, this example represents the first ion-pair transporter in which the same recognition motif can identify both anions and cations.5
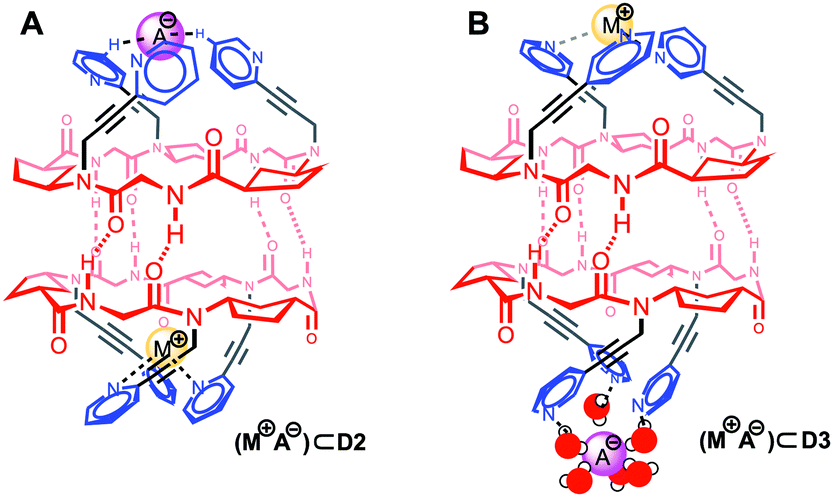 | ||
| Fig. 4 Proposed mechanism for the simultaneous complexation of cations and anions at both tri-pyridine entrances of the CP shuttle. | ||
In conclusion, we have developed a new family of symport ionophore molecules based on α,γ-cyclic peptide dimers decorated with pyridine rings, where the tris-pyridine decorated cap can act as the recognition unit for both cations and anions, although anion interaction is responsible for the observed selectivity. To do this, both pyridine caps of dimeric forms were required for the transmembrane activity. In general, ortho-decorated CP2 outperformed the meta-substituted isomer (CP3), especially regarding Cl− transport. Each transporter exhibited an inverse anion selectivity trend in the HPTS rectification assay, where the ionic radius and the hydration energy were key factors for the transport efficiencies. Finally, control experiments revealed that the cyclic scaffold plays a crucial role in the transport capability, showing a cooperative behaviour.
This work was supported by the Spanish Agencia Estatal de Investigación (AEI) and the ERDF (CTQ2016-78423-R), and by the Xunta de Galicia and the ERDF (ED431C 2017/25 and Centro singular de Investigación de Galicia accreditation 2016–2019, ED431G/09). We also thank the ORFEO-CINCA network and Mineco (CTQ2016-81797-REDC). A. F. thanks the Spanish Ministry of Education for his FPU fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Hesketh, J. Cell Sci., 2000, 113, 2925–2926 Search PubMed.
- J.-B. Kim, Korean J. Pediatr., 2014, 57, 1–18 CrossRef CAS PubMed; R. T. Moxley III and C. Heatwole, Channelopathies, Swaiman's Pediatric Neurology, 6th edn, 2017 CrossRef PubMed; D. M. Kullmann and S. G. Waxmann, J. Physiol., 2010, 588, 1823–1827 CrossRef PubMed; P. Imbrici, A. Liantonio, G. M. Camerino, M. De Bellis, C. Camerino, A. Mele, A. Giustino, S. Pierno, A. De Luca, D. Tricarico, J.-F. Desaphy and D. Conte, Front. Pharmacol., 2016, 7, 121 Search PubMed.
- N. Rodríguez-Vázquez, A. Fuertes, M. Amorín and J. R. Granja, Bioinspired Artificial Sodium and Potassium Ion Channels, The Alkali Metals Ions: Their Role for Life, Springer International, Cham, 2016 Search PubMed.
- A. Roy, D. Saha, A. Mukherjee and P. Talukdar, Org. Lett., 2016, 18, 5864–5867 CrossRef CAS PubMed; N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef PubMed.
- Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed.
- L. R. Forrest, R. Krämer and C. Ziegler, Biochim. Biophys. Acta, 2011, 1807, 167–188 CrossRef CAS PubMed.
- (a) M. Amorín, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2003, 125, 2844–2845 CrossRef; (b) R. J. Brea, M. Amorín, L. Castedo and J. R. Granja, Angew. Chem., Int. Ed., 2005, 44, 5710–5713 CrossRef CAS.
- A. Pizzi, H. L. Ozores, M. Calvelo, R. García-Fandiño, M. Amorín, N. Demitri, G. Terraneo, S. Bracco, A. Comotti, P. Sozzani, C. Bezuidenhout, P. Metrangolo and J. R. Granja, Angew. Chem., Int. Ed., 2019, 58, 14472–14476 CrossRef CAS.
- T. Kan and T. Fukuyama, Chem. Commun., 2004, 353–359 RSC; A. Fuertes, H. L. Ozores, M. Amorín and J. R. Granja, Nanoscale, 2017, 9, 748–753 RSC.
- S. Matile and N. Sakai, The Characterization of Synthetic Ion Channels and Pores, in Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Willey, Weinheim, 2007, pp. 391–418 Search PubMed.
- X. Wu, E. N. W. Howe and P. A. Gale, Acc. Chem. Res., 2018, 51, 1870–1879 CrossRef CAS.
- S. Massou, Biochem. Educ., 2000, 28, 171–173 CrossRef CAS; J. N. Weinstein, S. Yoshikami, P. Henkart, R. Blumenthal and W. A. Hagins, Science, 1977, 195, 489–492 CrossRef.
- A. V. Hill, Biochem. J., 1913, 7, 471–480 CrossRef CAS; S. Bhosale and S. Matile, Chirality, 2006, 18, 849–856 CrossRef.
- B. A. McNally, A. V. Koulov, B. D. Smith, J.-B. Joos and A. P. Davis, Chem. Commun., 2005, 1087–1089 RSC; J. A. Cooper, S. T. G. Street and A. P. Davis, Angew. Chem., Int. Ed., 2014, 53, 5609–5613 CrossRef CAS.
- V. Gorteau, G. Bollot, J. Mareda, A. Perez-Velasco and S. Matile, J. Am. Chem. Soc., 2006, 128, 14788–14789 CrossRef CAS; P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 6528–6529 CrossRef; S. V. Shinde and P. Talukdar, Org. Biomol. Chem., 2019, 17, 4483–4490 RSC.
- M. J. Pregel, L. Jullien, J. Canceill, L. Lacombe and J.-M. Lehn, J. Chem. Soc., Perkin Trans. 2, 1995, 417–426 RSC.
- C. M. Dias, H. Valkenier and A. P. Davies, Chem. – Eur. J., 2018, 24, 6262–6268 CrossRef CAS.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- C. C. Tong, R. Quesada, J. L. Sessler and P. A. Gale, Chem. Commun., 2008, 6321–6323 RSC.
- D. C. Buster, J. F. Hinton, F. S. Millett and D. C. Shungu, Biophys. J., 1988, 53, 145–152 CrossRef CAS.
- A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc06644f |
| This journal is © The Royal Society of Chemistry 2020 |