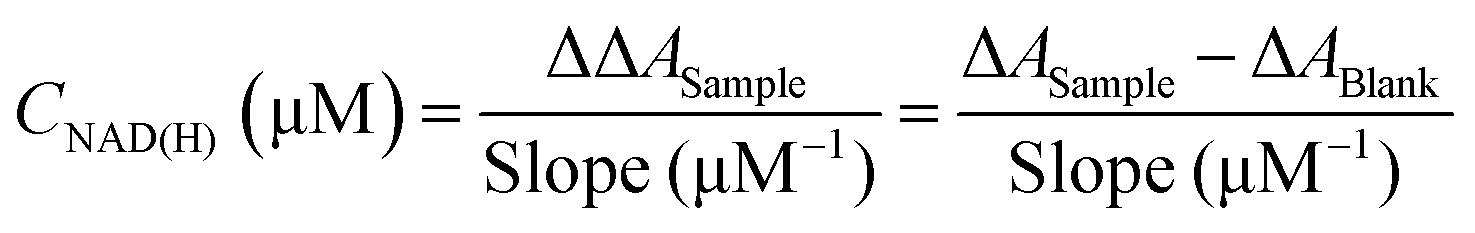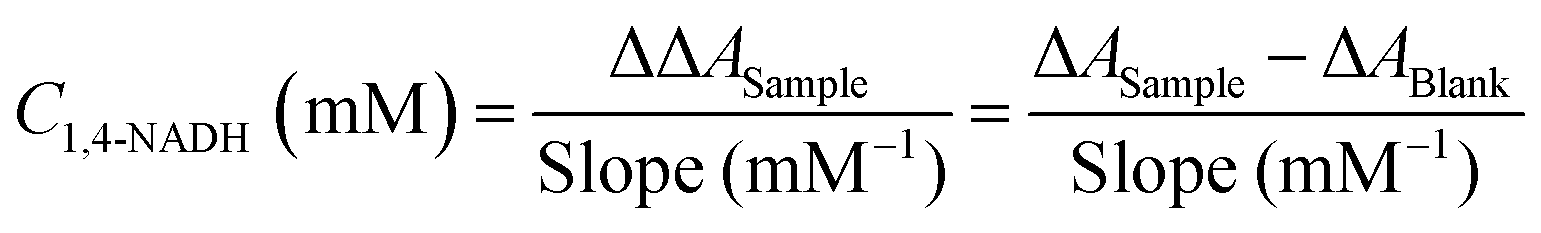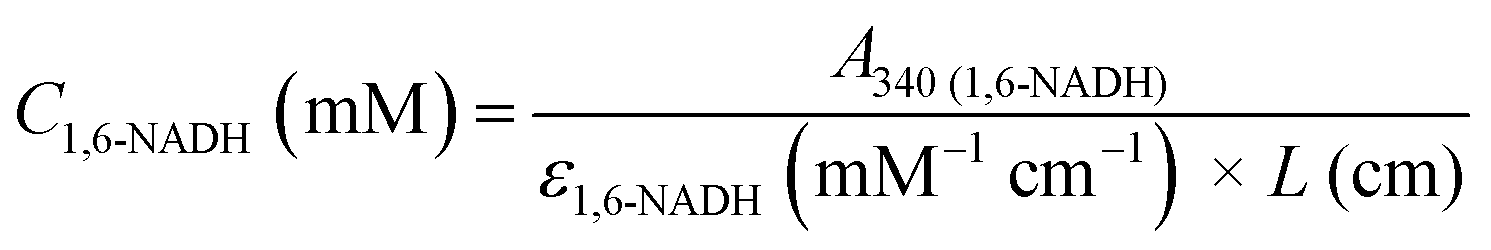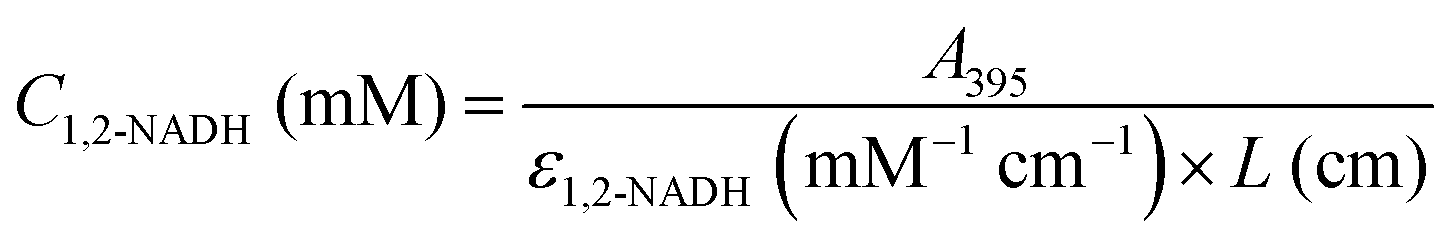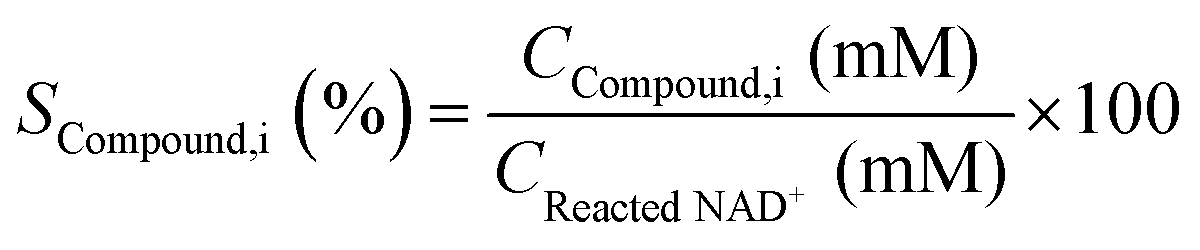DOI:
10.1039/C9CC07805C
(Communication)
Chem. Commun., 2020,
56, 1231-1234
A facile analytical method for reliable selectivity examination in cofactor NADH regeneration†
Received
4th October 2019
, Accepted 18th December 2019
First published on 18th December 2019
Abstract
This study demonstrates a novel method to quantify selective (1,4-NADH) and unselective products (1,2- and 1,6-NADH) in NADH regeneration using combined UV-Vis spectroscopy and biological assays. The validity of the proposed method was tested in the Pt/C promoted NAD+ hydrogenation using hydrogen as reducing agent.
1,4-NADH is a valuable cofactor that is used in tandem with enzymes (i.e. oxidoreductases) in the chemical and pharmaceutical industries for the manufacture of intermediates and drugs. Notable examples of these products include the Lipitor (atorvastatin), the antilipemic agent ZETIA (ezetimibe), the HIV protease inhibitor REYATAZ (atazanavir) and L-tert-leucine building block, among others.1 Cofactor 1,4-NADH is very expensive ($ 2600 per mol) making its stoichiometric supply in a bioreaction not economically viable and necessitating its in situ regeneration.2 1,4-NADH regeneration has been the area of extensive research over the past ∼40 years with various methods having been investigated, including biocatalytic, electrochemical, chemical, photocatalytic, homogeneous catalytic, and heterogeneous catalytic.3
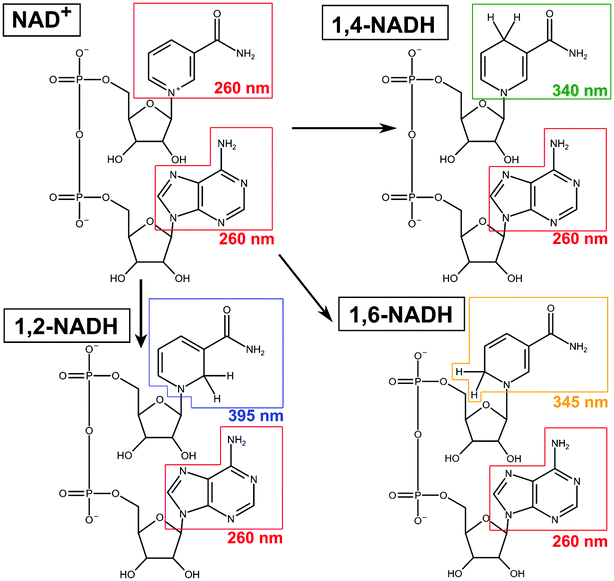
Besides the commercialised enzymatic method, other regeneration strategies, currently under development, may suffer from non-selective reduction of NAD+ to a mixture of the enzymatically active 1,4-NADH, and its inactive 1,2-NADH and 1,6-NADH isomers4 (see Scheme 1). These molecules absorb light at 340, 395 and 345 nm, respectively.5 It is furthermore noteworthy that 1,2-NADH exhibits high instability in phosphate buffer solutions (PBS) at pH ≤ 7 with a life time of only ∼30 min.6 Considering these facts, it is surprising to note that a significant number of literature has been relying solely on UV-Vis at 340 nm to quantify 1,4-NADH concentration.7 Moreover, Morrison et al.8 stated that unusual situations (e.g. discrepancies in HPLC analyses of NADH isomers) were observed in isomers preparation. This may lead to inaccurate or misleading conclusions, as other isomers were potentially not considered or distinguished. Another fact in NADH regeneration research is that the yield of 1,4-NADH (calculated based on the initial concentration of NAD+) is often reported, without determining NAD+ conversion (i.e. with no evidence of 100% selectivity or not including a process materials balance). These limitations impose an extreme challenge in understanding the reaction mechanism for further optimisation. Given the significant recent interest and effort in this field (see Table S1, ESI†),3,7 it is crucial that the concentrations of the products and unreacted reactant are unambiguously quantified.
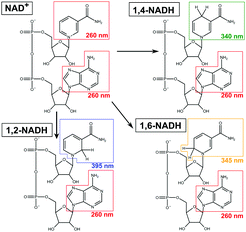 |
| | Scheme 1 Representative scheme for a non-selective NADH regeneration process in which NAD+ reacts into 1,2-, 1,4- and 1,6-NADH. | |
We report here, for the first time, an analytical method that can track NAD+ and both the desired and undesired forms of NADH using a combination of UV-Vis spectrophotometry and biological assays. The developed approach has been validated in a representative regeneration process.
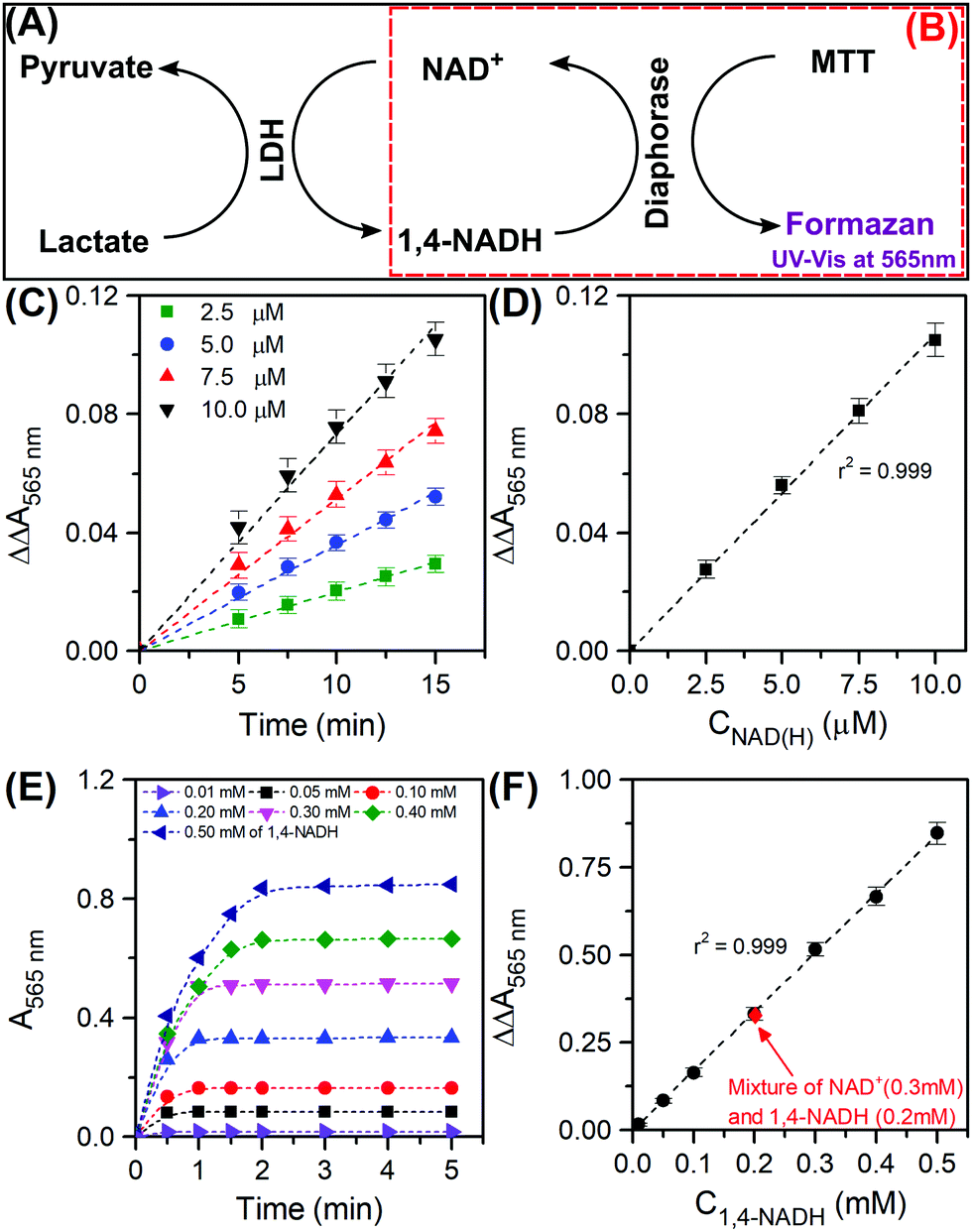
In the course of a reaction, the reaction medium may consist of all four molecules as shown in Scheme 1. The most important indicator for catalytic performance and kinetic analysis is the conversion of NAD+ and the selectivity to 1,4-NADH. We have accordingly customised an enzymatic assay kit to determine both 1,4-NADH and NAD+. The kit works as an enzymatic cyclic assay (Fig. 1A) in which lactate dehydrogenase (LDH) promotes the oxidation of lactate to pyruvate and NAD+ plays the role of the hydride acceptor, getting reduced to 1,4-NADH. The latter in turn releases the hydride to 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (known as MTT, yellow colour) to be reduced by diaphorase to (E,Z)-5-(4,5-dimethylthiazol-2-yl)-1,3-diphenylformazan (known as formazan, purple colour) that has a characteristic UV-Vis absorbance at 565 nm (Fig. 1B). The procedure presented here was specifically developed to be easily applicable to the various types of spectrometers available, not requiring a plate reader or other expensive analytical equipment. For this purpose, a working reagent was prepared by mixing 60 μL of the assay buffer, 1 μL of LDH, 1 μL of diaphorase, 14 μL of Lactate and 14 μL of MTT. 80 μL of the working reagent were added to 240 μL of the NAD(H) (referring to the total amount of 1,4-NADH and NAD+, assay buffer was used as a diluent when needed). The mixture was thoroughly stirred very briefly, then transferred to a quartz cuvette (2 mm path length, see ESI,† Part III for the detailed procedure and Fig. S1 and S2 for representative scans). The difference in absorbance at 565 nm between the sample (i.e. with cofactors) and blank (i.e. with no cofactors) was recorded with time using eqn (1) below:
| | | ΔΔASample = ΔASample − ΔABlank | (1) |
where Δ
ASample =
ASample at time t min −
ASample at 0 min; Δ
ABlank =
ABlank at time t min −
ABlank at 0 min. The difference in absorbance at 565 nm is directly proportional to the amount of NAD(H) in the system provided it stays linear with time.
9Fig. 1C shows the results of the kinetic measurements using our customised procedure which shows an excellent linear fit for the range of NAD(H) concentrations considered.
Fig. 1D shows the calibration curve at
t = 15 min for the total cycle and the resulting slope can be used for the quantification of the total amount of NAD(H) as follows:
| | 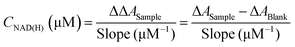 | (2) |
With the concentration of NAD(H) known, if either of the two components can be quantified, the other can be directly determined. To determine the concentration of 1,4-NADH, we have innovatively used the reduction side (right half) of the kit as depicted in
Fig. 1B, where 1,4-NADH reacts with MTT to form both NAD
+ and formazan. The working reagent was prepared by mixing 60 μL of the assay buffer, 1 μL of diaphorase and 14 μL of the MTT. The whole working reagent (75 μL) was then added to 240 μL of 1,4-NADH sample (in the range of 0.01 to 0.5 mM) and the absorbance was recorded with respect to time (see ESI,
† Part IV for the detailed procedure). The kinetic results are shown in
Fig. 1E. The absorbance increases rapidly and then stabilises. The absorbance at the plateau is an indication of the concentration of 1,4-NADH in solution and it was plotted against the actual concentration as seen in the calibration curve (
Fig. 1F).
Eqn (3) is used for the quantification of 1,4-NADH:
| | 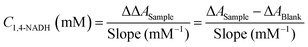 | (3) |
The validity of the half cycle's novel use was tested by preparing a mixture of both NAD
+ (0.3 mM) and 1,4-NADH (0.2 mM). The absorbance measurement fit exceptionally well the calibration curve (see
Fig. 1F), confirming that the existence of NAD
+ in solution has no practical effect on the 1,4-NADH analysis. The amount of NAD
+ is then deduced by subtracting the concentration of 1,4-NADH from the total NAD(H) using
eqn (4):
| | | CNAD+ (mM) = CNAD(H) (mM) − C1,4-NADH (mM) | (4) |
To elucidate the reaction pathway and mechanism, knowing only the concentrations of NAD
+ and 1,4-NADH is not sufficient, unless the amount of reacted NAD
+ is equal to that of 1,4-NADH produced,
i.e. the reaction is 100% selective. When this is not the case, the key information to obtain is the concentrations of the NADH isomers produced, which is hampered by the fact that 1,4- and 1,6-NADH exhibit similar characteristic absorbance peak (∼340 nm) and 1,2-NADH decays quickly in PBS with pH ≤ 7 (which is commonly used in NADH regeneration). We have therefore developed a systematic approach to distinguish and quantify the three isomers. In order to quantify 1,2- and 1,6-NADH in a reaction mixture, their molar absorptivities must be determined. Unfortunately, there is no supplier providing commercial isomer products (apart from 1,4-NADH). For this reason, we have synthesised a mixture of 1,2-, 1,4- and 1,6-NADH with known concentrations according to an established experimental procedure,
10i.e. sodium borohydride reduction of NAD
+ (see ESI,
† Part V). We conducted first the synthesis in PBS at pH = 7 as the high instability of 1,2-NADH at this condition (exhibits rapid decay) leaves only 1,4- and 1,6-NADH in the reaction medium.
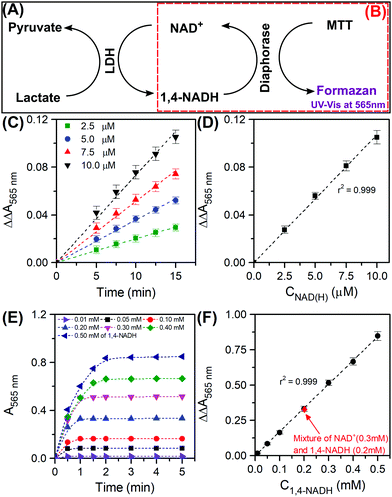 |
| | Fig. 1 Representative scheme of the reactions involved in the total enzymatic cyclic assay using both NAD+ and 1,4-NADH (in black borders (A)) and the reactions involved in the enzymatic half cycle using 1,4-NADH only (in dashed red borders (B)); absorbance as function of time for the total cyclic assay for different NAD(H) concentrations (2.5 to 10 μM) (C) and the corresponding calibration curve (D); absorbance as function of time for the half enzymatic cycle for different 1,4-NADH concentrations (0.01 to 0.5 mM) (E) and the corresponding calibration curve (F). | |
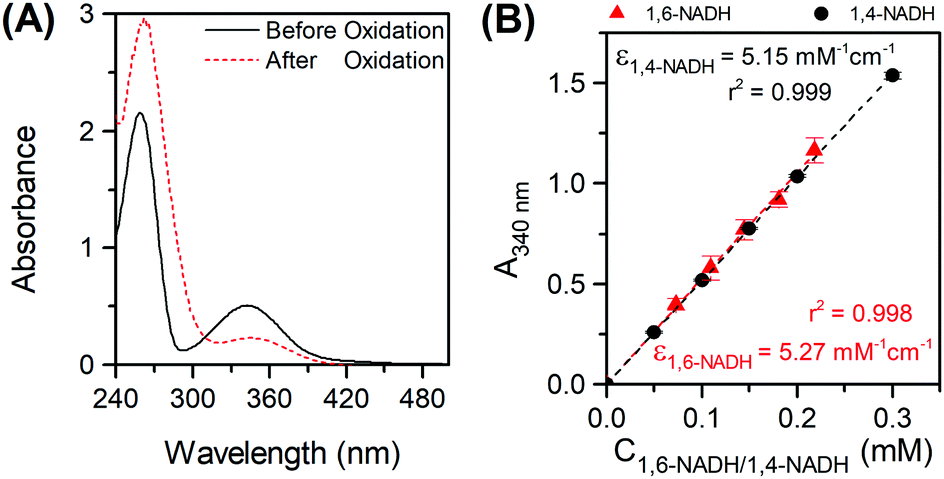
In order to distinguish between the two isomers at 340 nm, active 1,4-NADH was reoxidised to NAD+ by alcohol dehydrogenase (ADH) catalysed acetaldehyde reduction11 (see the ESI,† Part VI). A reduction of almost half of the initial 340 absorbance was observed (Fig. 2A), in line with previous studies.10c,12 Upon 1,4-NADH oxidation, the absorbance at 260 nm increased dramatically indicating the formation of NAD+ that has a high molar absorptivity at 260 nm compared to all isomers.5 The residual absorbance at 340 nm (due to 1,6-NADH) at five different initial concentrations of NAD+ was then translated into a calibration curve of 1,6-NADH, which is depicted in Fig. 2B. A standard calibration curve for commercial 1,4-NADH at 340 nm was generated in the range of 0.05 to 0.3 mM. The slope of the linear curve corresponds to the molar absorptivity of the 1,4-NADH in mM−1 cm−1 (Fig. 2B).
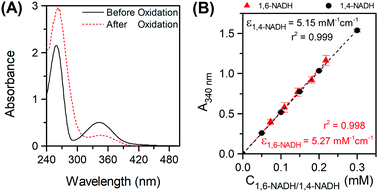 |
| | Fig. 2 Oxidation of a NAD+ reduction mixture in phosphate buffer pH 7 with acetaldehyde and ADH with Ci(NAD+) = 0.25 mM (A) and 1,6- and 1,4-NADH calibration curves at 340 nm (B). | |
In a course of a reaction, the absorbance of a sample at time t at 340 nm, A340 (total), is a combination of both 1,6-NADH and 1,4-NADH absorptions. The latter can be determined by converting 1,4-NADH concentration (determined from the enzymatic half cycle) using ε(1,4-NADH) according to eqn (5):
| | | A340(1,4-NADH) = ε1,4-NADH (mM−1 cm−1) × L (cm) × C1,4-NADH (mM) | (5) |
The absorbance and concentration of 1,6-NADH are then calculated using its molar absorptivity at 340 nm and the difference between the
A340(total) and
A340(1,4-NADH) (see
eqn (6) and (7)):
| | | A340(1,6-NADH) = A340(total) − A340(1,4-NADH) | (6) |
| | 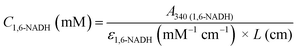 | (7) |
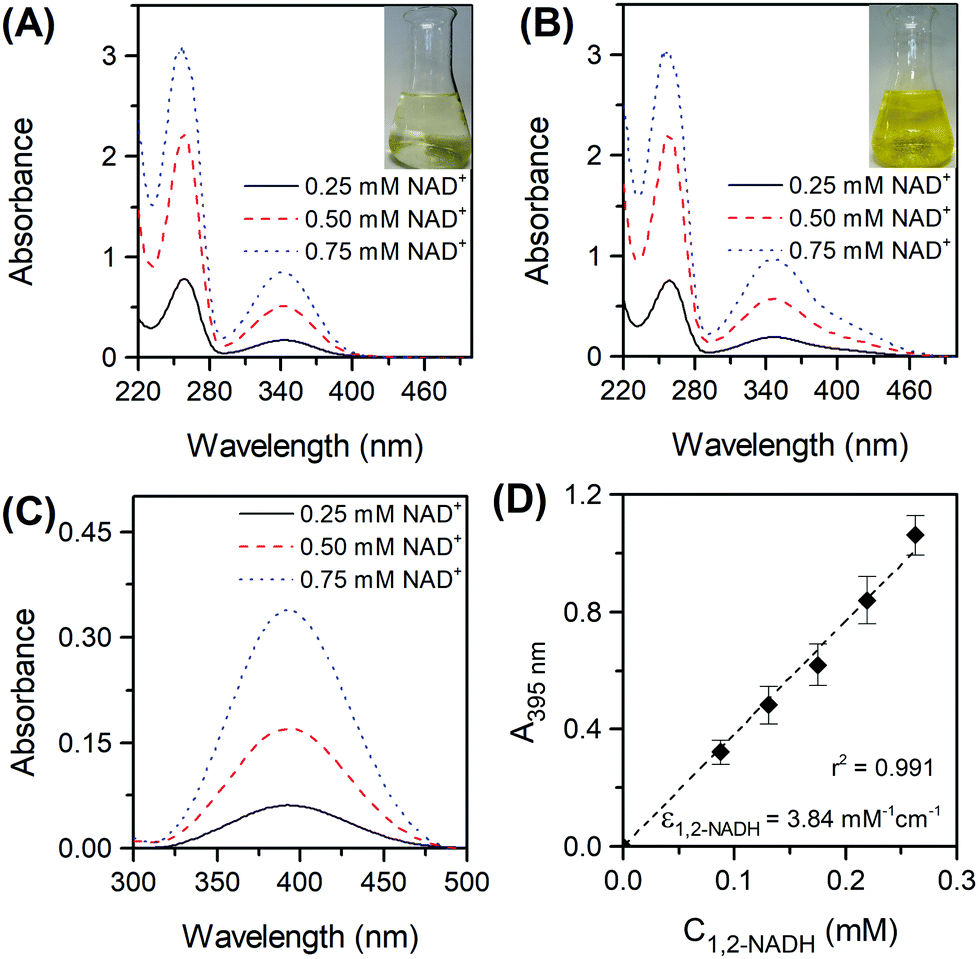
Fig. 3A presents the UV-Vis spectra of the products formed in the borohydride reduction of NAD
+ in PBS at pH 7. The reaction produced in the first few seconds a bright yellow mixture, which disappeared 1 minute later forming a very pale-yellow colour product (
Fig. 3A inset). There is also no clear peak at 395 nm, characteristic of 1,2-NADH, suggesting 1,2-NADH has decayed.
10b In contrast, when NAD
+ was reduced in Tris buffer at pH 8.5, it generated a persistent bright yellow mixture with a wide spectrum between 300 and 480 nm (
Fig. 3B). This vivid colour is associated with an absorbance band at 395 nm,
i.e. presence of 1,2-NADH. The difference spectrum between the two sets of reduction data (
Fig. 3C) clearly shows the presence of the 395 nm absorbing species in Tris buffer. These results prove the impossibility to track the formation of 1,2-NADH in a PBS system at pH 7. The difference in absorbance at 395 nm was then used for a calibration curve of 1,2-NADH (
Fig. 3D). It is important to mention that the
A395nm values in
Fig. 3C are not identical to those in
Fig. 3D because different dilution factors were used. The concentration of 1,2-NADH can be calculated using the absorbance of the sample at 395 nm following
eqn (8):
| | 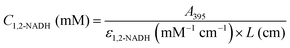 | (8) |
The validity of the proposed method was tested in the regeneration of NADH
via the hydrogenation of NAD
+ (see ESI,
† Part VII) using a commercial Pt/C catalyst, as previously reported.
13 The concentration of each species was determined using the method described before, and the selectivity of each component was calculated using
eqn (9):
| |  | (9) |
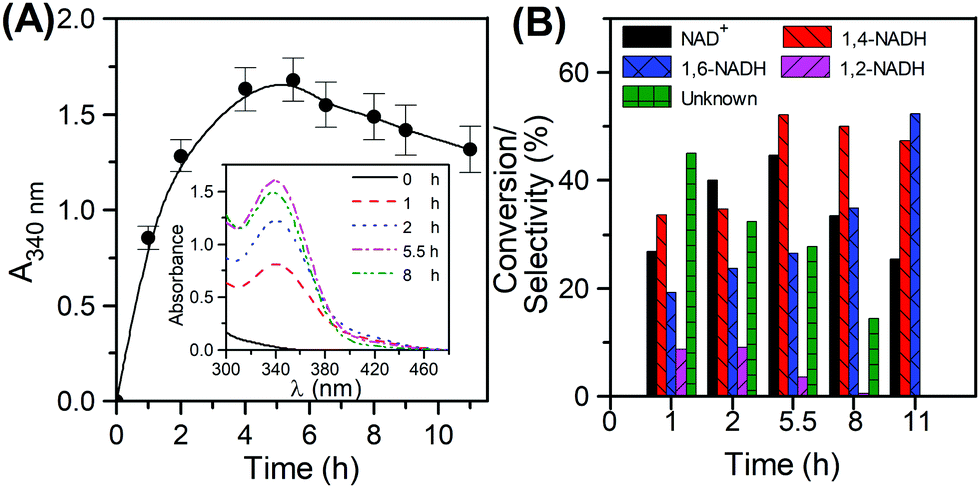
The reaction profile in the hydrogenation of NAD
+ was first tracked by measuring 340 nm absorbance with time (
Fig. 4A). The scanned reaction mixtures (
Fig. 4A inset) clearly show wide absorbance spectra, indicating the formation of all isomers. Based on
Fig. 4B, the catalyst exhibits its maximum conversion at 5.5 h. The analysis of the reaction mixture using the reductive cycle of the kit after 1 h reaction time, showed that only 60% of the total 340 absorbance amounted to the enzymatically active 1,4-NADH, with the remaining 40% being a contribution from 1,6-NADH. The colour of the sample taken at 1 h was slightly yellowish, but persistent, suggesting the formation of 1,2-NADH as well. This was confirmed by UV-Vis analysis at 395 nm. The amount of reacted NAD
+ was determined using the total cycle of the kit by subtracting the concentration of NAD
+ left from the initial NAD
+. Subtracting the concentration of all isomers (1,2-, 1,4-, and 1,6-NADH) from the reacted NAD
+ resulted in a mole balance closure of 89% with the rest being attributed to unknowns. In order to understand the origin of these unknowns, it was important to perform a selectivity measurement of all components throughout the reaction. The results are shown in
Fig. 4B. At the initial stages of the reaction (up to the first hour), high selectivity towards the unknown products is observed, followed by those of 1,4, 1,6 and 1,2-NADH in decreasing order. As the reaction progresses and the conversion increases, the selectivity to the unknowns drops dramatically from 45% reaching eventually a zero value, while that of 1,4-NADH continues to rise reaching 52% at the maximum conversion). These results suggest that the unknown product formed may be an intermediate to the isomers rather than an ultimate by-product from NAD
+ hydrogenation. At the maximum conversion (45%), the yields of 1,4-NADH, 1,6-NADH and 1,2-NADH were 23%, 7%, 2%, respectively. Using the 340 nm UV absorbance only (
i.e. the method widely employed in the literature), the yield of 1,4-NADH would have been reported as 31%, with an error of 35% with respect to the actual yield. This demonstrates the inaccuracy of using the 340 nm only method for 1,4-NADH quantification. It should be noted that the reported analytical method can also be used in other more complicated regeneration systems relying on sacrificial organic electron donors or hole scavengers (
e.g. TEOA, see Fig. S3, ESI
†).
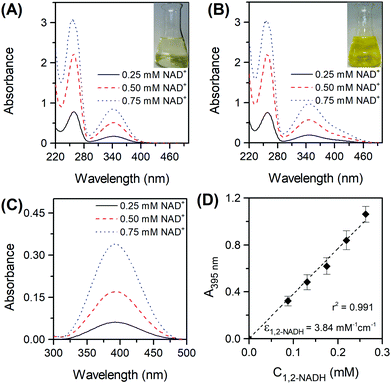 |
| | Fig. 3 Spectra of NAD+ reduced by sodium borohydride in phosphate buffer pH 7 (A) and in Tris buffer pH 8.5 (B) at three different initial concentrations of NAD+ (reaction conditions: Ci(NAD+) = 0.25–0.75 mM, Ci(NaBH4) = 70Ci(NAD+), 22 °C, 300 rpm, atmospheric pressure). Spectra difference obtained when spectra of the Tris-buffered reaction mixture was measured against the phosphate-buffered reaction mixture (C) and the corresponding 1,2-NADH calibration curve (D). | |
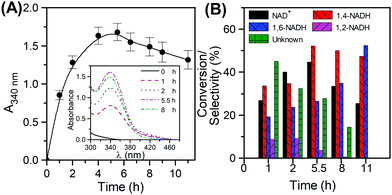 |
| | Fig. 4 NADH regeneration using Pt/C. Evolution of the 340 absorbance with time (A), conversion of NAD+ and selectivity of reaction products over time (B) (reaction conditions: 22 °C, 8 atm, Tris-buffer pH 8.5, 900 rpm and Ci(NAD+) = 1 mM. | |
In conclusion, we reported a method that uses UV-Vis spectrophotometry alongside biological assays for the determination of the concentrations of NAD+ and both selective and non-selective compounds in NADH regeneration. The developed method, validated in a Pt/C catalysed regeneration reaction, can be applied to other NADH regeneration systems, providing critical information for mechanism/pathway analysis and further process optimisation.
This work was supported by The Royal Society (ICA\R1\180317 and IES\R3\170162).
Conflicts of interest
There are no conflicts to declare.
Notes and references
- C. Morrison, E. Heitmann, W. Armiger, D. Dodds and M. Koffas, Adv. Appl. Microbiol., 2018, 105, 51–86 CrossRef PubMed.
-
K. Faber, Biotransformations in Organic Chemistry, Springer, 2018 Search PubMed.
-
(a)
R. Wichmann and D. Vasic-Racki, Technology transfer in biotechnology, Springer, 2005, pp. 225–260 Search PubMed;
(b) H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Green Chem., 2013, 15, 1773–1789 RSC;
(c) X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Chem, 2017, 2, 621–654 CrossRef CAS.
- H. K. Chenault, E. S. Simon and G. M. Whitesides, Biotechnol. Genet. Eng. Rev., 1988, 6, 221–270 CrossRef CAS PubMed.
- H. Jaegfeldt, J. Electroanal. Chem. Interfacial Electrochem., 1981, 128, 355–370 CrossRef.
- B. A. Beaupre, M. R. Hoag, J. Roman, F. H. Försterling and G. R. Moran, Biochemistry, 2015, 54, 795–806 CrossRef CAS PubMed.
-
(a) H. Song, S. H. Lee, K. Won, J. H. Park, J. K. Kim, H. Lee, S. Moon, D. K. Kim and C. B. Park, Angew. Chem., Int. Ed., 2008, 47, 1749–1752 CrossRef CAS PubMed;
(b) S. Kim, G. Y. Lee, J. Lee, E. Rajkumar, J. Baeg and J. Kim, Electrochim. Acta, 2013, 96, 141–146 CrossRef CAS;
(c) X. Ji, C. Liu, J. Wang, Z. Su, G. Ma and S. Zhang, J. Mater. Chem. A, 2017, 5, 5511–5522 RSC;
(d) D. Chen, D. Yang, Q. Wang and Z. Jiang, Ind. Eng. Chem. Res., 2006, 45, 4110–4116 CrossRef CAS;
(e) S. Kim, G. Y. Lee, J. Baeg, Y. Kim, S. Kim and J. Kim, J. Phys. Chem. C, 2014, 118, 25844–25852 CrossRef CAS;
(f) K. E. Taylor and J. B. Jones, J. Am. Chem. Soc., 1976, 98, 5689–5694 CrossRef CAS PubMed;
(g) L. Zhang, N. Vila, G. Kohring, A. Walcarius and M. Etienne, ACS Catal., 2017, 7, 4386–4394 CrossRef CAS;
(h) V. Ganesan, D. Sivanesan and S. Yoon, Inorg. Chem., 2017, 56, 1366–1374 CrossRef CAS PubMed;
(i) J. Canivet, G. Süss-Fink and P. Štěpnička, Eur. J. Inorg. Chem., 2007, 4736–4742 CrossRef CAS;
(j) X. Huang, J. Liu, Q. Yang, Y. Liu, Y. Zhu, T. Li, Y. H. Tsang and X. Zhang, RSC Adv., 2016, 6, 101974–101980 RSC;
(k) S. K. Kuk, R. K. Singh, D. H. Nam, R. Singh, J. K. Lee and C. B. Park, Angew. Chem., Int. Ed., 2017, 56, 3827–3832 CrossRef CAS PubMed;
(l) D. H. Nam, S. K. Kuk, H. Choe, S. Lee, J. W. Ko, E. J. Son, E. G. Choi, Y. H. Kim and C. B. Park, Green Chem., 2016, 18, 5989–5993 RSC;
(m) S. Wang, M. Li, A. J. Patil, S. Sun, L. Tian, D. Zhang, M. Cao and S. Mann, J. Mater. Chem. A, 2017, 5, 24612–24616 RSC.
- C. S. Morrison, W. B. Armiger, D. R. Dodds, J. S. Dordick and M. A. Koffas, Biotechnol. Adv., 2017, 36, 120–131 CrossRef PubMed.
-
(a) H. Li, K. E. Worley and S. Calabrese Barton, ACS Catal., 2012, 2, 2572–2576 CrossRef CAS;
(b) Y. Gibon and F. Larher, Anal. Biochem., 1997, 251, 153–157 CrossRef CAS PubMed;
(c) C. Vilcheze, T. R. Weisbrod, B. Chen, L. Kremer, M. H. Hazbon, F. Wang, D. Alland, J. C. Sacchettini and W. R. Jacobs Jr, Antimicrob. Agents Chemother., 2005, 49, 708–720 CrossRef CAS PubMed.
-
(a) M. B. Mathews, J. Biol. Chem., 1948, 176, 229–232 CAS;
(b) S. Chaykin and L. Meissner, Biochem. Biophys. Res. Commun., 1964, 14, 233–240 CrossRef CAS PubMed;
(c) S. Chaykin, L. King and J. G. Watson, Biochim. Biophys. Acta, 1966, 124, 13–25 CrossRef CAS PubMed.
- S. Chaykin, K. Chakraverty, L. King and J. G. Watson, Biochim. Biophys., 1966, 124, 1–12 CrossRef CAS.
- M. B. Mathews and E. E. Conn, J. Am. Chem. Soc., 1953, 75, 5428–5430 CrossRef CAS.
- X. Wang and H. H. P. Yiu, ACS Catal., 2016, 6, 1880–1886 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc07805c |
| ‡ Present address: Department of Engineering, Lancaster University, Lancaster LA1 4YW, UK. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Xiaodong
Wang‡
and
Xiaodong
Wang‡