
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclic peptide scaffold with ability to stabilize and deliver a helical cell-impermeable cargo across membranes of cultured cancer cells†
Nicole Lawrence‡
 *a,
Grégoire J.-B. Philippe‡a,
Peta J. Harveya,
Nicholas D. Condona,
Aurélie H. Benfieldab,
Olivier Chenevala,
David J. Craik*a and
Sónia Troeira Henriques*ab
*a,
Grégoire J.-B. Philippe‡a,
Peta J. Harveya,
Nicholas D. Condona,
Aurélie H. Benfieldab,
Olivier Chenevala,
David J. Craik*a and
Sónia Troeira Henriques*ab
aInstitute for Molecular Bioscience, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: sonia.henriques@qut.edu.au; n.lawrence@imb.uq.edu.au; d.craik@imb.uq.edu.au; Tel: +61 7 34437342 Tel: +61 7 33462014 Tel: +61 7 33462019
bQueensland University of Technology, School of Biomedical Sciences, Institute of Health & Biomedical Innovation and Translational Research Institute, Brisbane, Queensland 4102, Australia
First published on 20th October 2020
Abstract
Cell penetrating peptides (CPPs) are valuable tools for developing anticancer therapies due to their ability to access intracellular targets, including protein–protein interactions. cPF4PD is a newly described CPP designed from a transduction domain of the human defense protein platelet factor 4 (PF4), that also has antimalarial activity. The cPF4PD peptide recapitulates the helical structure of the PF4 domain and maintains activity against intracellular malaria parasites via a selective membrane-active mechanism. We hypothesized that cPF4PD and PF4-derived peptide analogues would enter cancer cells and have utility as scaffolds for delivering a peptide dual inhibitor (pDI) sequence with ability to inhibit p53:MDM2/X interactions and reactivate the p53 pathway. Here we designed and produced PF4 peptide and PF4 peptide-pDI grafted analogues with low micromolar activity toward melanoma and leukemia. Two grafted analogues achieved a stable helical structure and inhibited interaction with MDM2 and MDMX. These peptides reached the cytoplasm of cells but were unable to reactivate the p53 pathway. Instead, the cytotoxic mechanism was attributed to peptide binding to mitochondrial membranes that perturbed function within two hours of treatment. These studies of PF4-derived CPPs suggest their potential as scaffolds for delivering cell-impermeable cargoes into the cytoplasm of cells and highlight the importance of characterizing the internalization and cell death mechanism of designer peptide-based drugs.
Introduction
Cancer is a serious global health burden with 18.1 million new cases and 9.6 million deaths in 2018. Due to increases in population growth and ageing, and lifestyle changes associated with social and economic development, cancer incidence is expected to increase to 29.5 million new cases in 2040.1 New approaches to developing cancer therapies are required to stay ahead of the disease progression, and to improve on conventional treatments that include surgery, radiotherapy and chemotherapy. Targeted therapies that can distinguish between cancerous and noncancerous cells based on their different characteristics provide an advantage over chemotherapeutic drugs that target rapidly dividing cells, and successful therapeutic molecules that employ antibody-based2 or peptide-based3 targeting approaches are emerging.A distinction between cancer cells and healthy mammalian cells is the presence of phospholipids containing anionic phosphatidylserine (PS)-headgroups exposed at the cell surface that results in cancer cell membranes being more negatively charged.4,5 In healthy cells, PS-phospholipids are predominantly maintained in the inner leaflet through ATP-dependent flippase enzyme activity.6 Thus, surface exposure of phospholipids with anionic PS-headgroups is a useful biomarker for selectively targeting cancer cells.7
Peptides with desirable properties for developing targeted anticancer therapies have been described, including antimicrobial peptides that can distinguish between anionic and neutral lipids;8–11 cell penetrating peptides (CPPs) that can cross cell membranes without disruption;12–15 and peptide macrocycles that are amenable to modification and substitution with desirable target sequences through a process known as molecular grafting.16,17
Recently we engineered an antimalarial peptide that is a structural mimic of an amphipathic helical transduction domain from the human defense protein platelet factor 4 (PF4). The peptide, cyclic PF4 peptide dimer (cPF4PD), recapitulated the ability of the parent PF4 protein18 to directly cross the plasma membrane of infected red blood cells (RBCs) and kill malaria parasites by disrupting their anionic digestive vacuole membrane.19 The direct entry of cPF4PD, and the larger PF4 protein, into the cytoplasm of malaria parasites inside RBCs suggests that endocytic pathways are not required for cell entry.19 This entry mechanism is more desirable than endocytic mechanisms employed by other CPPs, for example the Tat peptide, that was also developed from a transduction domain of a larger protein.12,20
The ability of PF4 and cPF4PD to enter infected RBCs, but not uninfected cells, is probably due to interaction between cationic residues within the amphipathic helices and anionic PS-headgroups on the outer leaflet of infected RBC membranes.21,22 This membrane selectivity led us to hypothesize that the peptide would enter cancer cells, which also have an increased proportion of PS-headgroups on their outer leaflet.
We proposed that the helical structure of cPF4PD would be useful for stabilizing peptides that require helix-formation for bioactivity, including those designed to inhibit protein–protein interactions (PPIs), and chose the peptide dual inhibitor (pDI)23 as a model sequence to graft into the peptide scaffold (Fig. 1). Stabilization of pDI in an alpha helix displays residues Phe3, Trp7 and Leu10 on the same face of the peptide, and improves insertion into clefts on the surface of p53 inhibitory proteins MDM2 and MDMX24 (e.g. see Fig. 1A). Inhibition of p53:MDM2 and p53:MDMX interactions in the cytoplasm of cells promotes activation of the p53 pathway to trigger targeted cell death in cancers with wild-type p53.25
 | ||
| Fig. 1 Stabilizing pDI helical structure using interaction between PF4P and pDI helices. (A) High affinity interaction between pDI and MDM2 requires pDI residues F3, W7 and L10 to align and insert deeply into a cleft on the MDM2 surface (PDB 3G03). (B) Proposed interaction surface between PF4P and [Q9L]pDI. Amino acid side chains involved in helix–helix stabilization are shown within the grey box. The Q9L mutation (*) improves the interaction. Lys residues (K5, 6, 9, 10) from PF4P that are involved in selective membrane binding, and Phe3, Trp7 and Leu10 involved in binding to MDM2, are on opposite sides of the helices. | ||
Peptide mimics of the p53 interaction surface, including Hp53-C,26 pDI23 and lactam-stabilized analogue KD3,27 are unable to enter cells. One approach used to overcome this limitation is to incorporate these peptides into cyclized helix-loop-helix scaffolds with cell-penetrating properties. For example, cHLH-p53-R,26 cHLH-pDI-R analogues and cHLH-KD3-R28 all achieve a helical structure that inhibits p53:MDM2 and p53:MDMX interactions. Among these peptides, cHLH-p53-R enters cells with the highest efficacy and is the most potent at killing cancer cells, but its cytotoxic activity depends on membrane disruption, and not on reactivation of the p53 pathway.28
Here we explore these hypotheses by characterizing target-specific versus membrane-specific activity of PF4-derived scaffold peptides and grafted peptide analogues. We demonstrate selective binding to model membranes with PS-headgroups and ability to cross cancer cell membranes at nanomolar concentrations. Two helical PF4-pDI grafted peptides inhibited protein–protein interactions in vitro, but did not induce intracellular activation of the p53 pathway. Instead, the cytotoxic mechanism of PF4 scaffold and grafted peptide analogues from this study relates to the ability of the peptide scaffold to target and disrupt mitochondrial membrane function.
Results and discussion
Peptide design
The sequences and schematic representations of the peptides included in this study are shown in Table 1. Peptides were synthesized and oxidized to form disulfide bonds (cyclic, c), or ligated via thioether linkage (cyclic thioether, ct). Purity and correct mass were confirmed with analytical HPLC (Fig. S1, ESI†) and MS (Fig. S2, ESI†), and the retention time (RT) from analytical HPLC was determined to compare relative overall hydrophobicity (Table 1).| a Primary peptide sequence where * is C-terminal amide; cyclized peptides are indicated with a bracket between C–C for disulfide and C–G for thioether bond cyclization; pDI sequence is indicated in blue; X is azidoalanine; Alexa Fluor 488 (A488) dye was incorporated onto acetone-linked peptides using oxime ligation (cPF4PD-A488) or attached at azidoalanine using copper catalyzed cycloaddition chemistry (PF4P_pDI-A488, cPF4Pext_pDI-A488). b Expected average mass was calculated from the amino acid sequence. c Observed mass was confirmed from m/z using MS. d Coefficient of extinction at 280 nm (ε280) calculated based on contributions from Tyr and Trp residues, and disulfide bonds. e Retention time (RT) was determined using analytical HPLC with a 2% min−1 gradient of solvent B 90% acetonitrile (ACN), 0.1% trifluoroacetic acid (TFA) in solvent A (0.1% TFA). f Charge at pH 7.4 (Chg) was calculated from contributions of charged amino acid side chains. g Predicted structure of the peptides, where cylinders represent α-helices. For cyclized peptides disulfide bonds are shown in yellow, and thioether bonds in black. Location of the A488 dye is indicated with green ovals. |
|---|
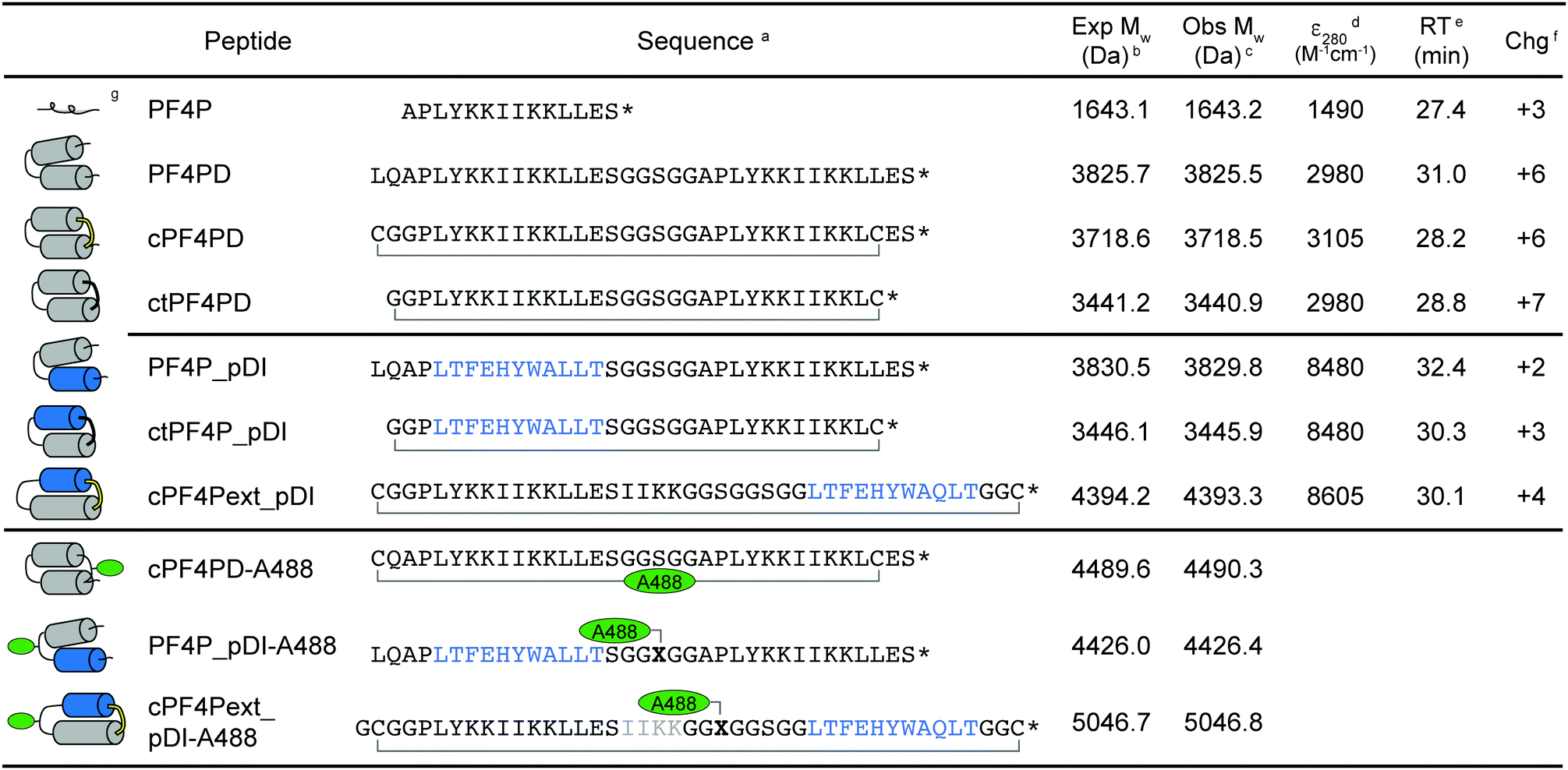 |
Three variations of PF4 peptide scaffolds were produced to examine structure activity relationships and identify the most promising format for developing anticancer drug leads. cPF4PD is the disulfide-cyclized head-to-tail homodimer that is helical in both aqueous and hydrophobic environments; PF4PD is an open head-to-tail homodimer that becomes helical in hydrophobic environments;19 and ctPF4PD is a new analogue cyclized via a thioether bond that was included to determine whether thioether cyclization would improve stability and/or cytotoxic activity. The PF4P monomer was included in this study as an unstructured control peptide. PF4PD had a longer RT from HPLC, compared to cPF4PD and ctPF4PD (see RT in Table 1), suggesting that the open dimer had a more hydrophobic surface than the cyclized analogues. Removal of the anionic Glu residue from ctPF4PD increased its overall charge to +7, but did not affect its overall hydrophobicity, as suggested by its similar RT to cPF4PD.
To investigate whether the PF4 scaffolds could be re-engineered as anticancer drugs able to inhibit intracellular PPIs, we replaced one of the PF4P helices with pDI,23 with the expectation that helix-stabilizing interactions between PF4P and pDI would drive helix formation, and that PF4P residues involved in selective cell entry and pDI residues Phe3, Trp7 and Leu10, would be presented on opposing faces (see Fig. 1B). The variant [Q9L]pDI sequence (LTFEHYWALLT) was grafted into PF4PD and ctPF4PD to produce PF4PD_pDI and ctPF4P_pDI. The Leu substitution was included to promote the formation of hydrophobic bonds between the heterogeneous helices, similar to those present for the parent PF4P helices. An extended PF4P helix was included to improve the likelihood of helix formation in cPF4Pext_pDI, a disulfide cyclized heterodimer with the original pDI sequence. Grafting the pDI sequence into the PF4 scaffold resulted in peptides with increased hydrophobicity and decreased charge compared to the parent peptides (see for instance the longer RT obtained for PF4P_pDI, Table 1).
Peptide structure and stability
The secondary structure of the peptides in aqueous solution (100 mM NaF, 10 mM KH2PO4, pH 7.5) was examined using CD spectroscopy. The cyclic homodimer peptides cPF4PD and ctPF4PD had spectra with minima at 208 and 222 nm suggesting helical structure, whereas the monomer peptide PF4P and open homodimer peptide PF4PD did not display the minima that are typical for a helical conformation and are probably unstructured in aqueous solution (Fig. 2A). The relative helicity was highest for cPF4PD (45%), which corresponds to 16 out of 22 amino acid residues predicted to be helical (see Fig. S3, ESI†).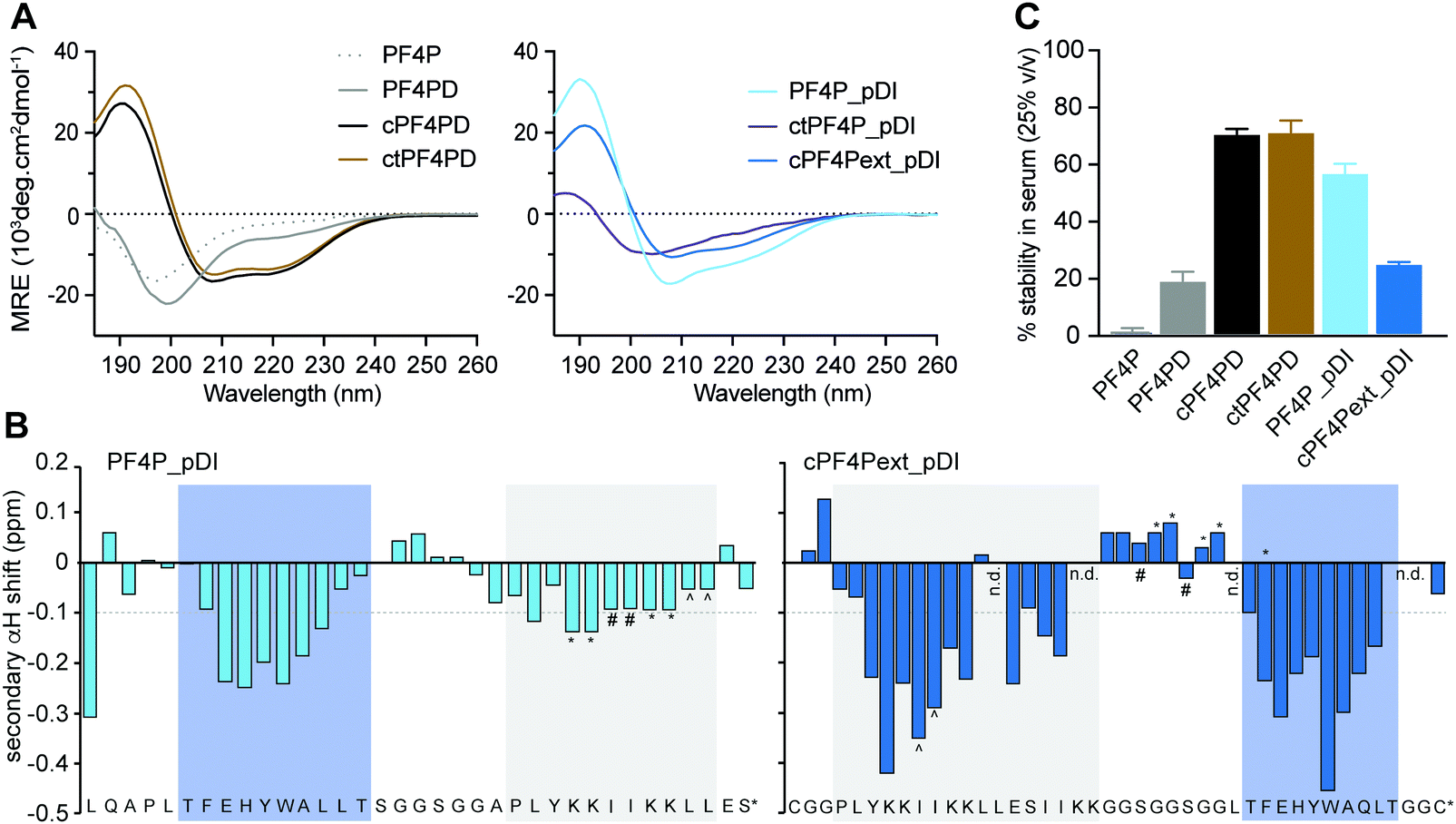 | ||
| Fig. 2 Structure and stability of PF4 scaffold and grafted peptides. (A) CD spectra of 50 μM peptide in aqueous solution (100 mM NaF, 10 mM KH2PO4 [pH 7.5]), where the presence of α-helical structure is indicated by mean residual ellipticity (MRE) minima at 208 and 222 nm. (B) Two-dimensional NMR secondary αH shifts determined for ∼1 mM peptide in aqueous solution (10% D2O/90% H2O v/v [pH ∼ 3]) where α-helical structure is indicated for residues with shifts ≤−0.1 ppm. Symbols (*, #, ^) denote ambiguity between these residues due to peak overlap. The label n.d. refers to those residues whose shifts could not be determined due to peak overlap. Shaded boxes show predicted α-helical regions; blue, pDI; grey, PF4P. (C) Stability of 50 μM peptide following 24 h incubation in human serum (25% v/v in PBS [pH 7.5]) as a percentage of peptide recovered at 0 h. Values have been calculated from area under the curve for recovered peptide peaks at 215 nm using analytical HPLC. Data points are mean ± SD from three technical replicates from the same experiment. | ||
Helicity of the pDI-grafted peptides followed the trend PF4P_pDI > cPF4Pext_pDI > ctPF4P_pDI. The open peptide PF4P_pDI had the strongest tendency to form helices in aqueous solution (39% of helical content, 14/21 of predicted amino acid residues) suggesting that the heterogeneous helices were stabilized by inter-helix molecular interactions. The reduced helicity of ctPF4P_pDI (18% or 6/21 predicted residues) suggested that the rotational freedom and ability to form favorable inter-helix interactions may have been restricted in the cyclic heterodimer.
Open-format peptides PF4PD and PF4P_pDI had improved helicity in a less polar solution (50% trifluoroethanol, see Fig. S4, ESI†), suggesting that these peptides might become helical when they interact with membranes and/or with hydrophobic protein binding pockets. NMR spectroscopy and analysis of the secondary α-proton shifts of PF4P_pDI and cPF4Pext_pDI was used to confirm that both pDI and PF4P components of the heterodimeric peptides formed helices (Fig. 2B, see shaded regions). A higher proportion of helical residues was observed for cPF4Pext_pDI from the NMR analysis (19/27 predicted residues) compared to the CD spectra analysis (11/27 predicted residues), most likely due to differences in sampling conditions, including higher peptide concentration and lower pH (less representative of physiological conditions) in the NMR samples.
To investigate the relationship between the degree of helicity achieved by the peptides and their ability to resist degradation by serum proteases, we incubated the PF4 scaffold and grafted peptides with 25% human serum for 24 h at 37 °C (Fig. 2C). cPF4PD and ctPF4PD were the most stable peptides with ∼75% of molecules remaining intact at 24 h. Open format PF4P_pDI was more resistant than the parent PF4PD (60% vs. 20% of intact peptide after 24 h), but disulfide cyclized cPF4PDext_pDI was less stable, with only 26% of intact peptide remaining at 24 h. Even though the format of the peptide scaffold (open, disulfide cyclic, thioether cyclic) was not predictive of stability for the grafted peptides, a strong positive correlation (r2 = 94%) was observed between the percentage of peptide with helical conformation and stability in human serum (Fig. S3, ESI†).
Selective binding and disruption of anionic membranes
We previously demonstrated that PF4-derived peptides selectively bind and disrupt membranes that contain a high proportion of anionic phospholipids.19 To determine whether this selectivity was maintained for the designer grafted peptides, we investigated their ability to bind and disrupt neutral versus anionic model membranes using surface plasmon resonance (SPR) and vesicle leakage assays. Membranes composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) were used to mimic the properties of the outer leaflet of healthy mammalian cell membranes, which are fluid and rich in zwitterionic phospholipids containing phosphatidylcholine (PC)-headgroups; and membranes composed of a mixture of POPC with negatively charged 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS; POPC/POPS (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio)) were used to mimic the outer leaflet of cancer cell membranes.
:1) membranes with higher affinity than to neutral POPC membranes, as quantified by peptide-to-lipid ratio (P/L, mol/mol) obtained at equilibrium with 16 μM peptide, and by the P/L at saturation (P/Lmax) obtained from dose–response curves (Table S1, ESI†). The open heterodimer PF4P_pDI had increased binding affinity for both neutral POPC and POPC/POPS (4:1) membranes compared to the parent homodimer peptides, and removed the lipid from the SPR chip at 16 μM peptide (see Fig. S5, ESI†), suggesting detergent-like properties at higher peptide concentrations. The increased affinity of PF4P_pDI for both lipid systems, and loss of selectivity for negatively charged membranes are probably due to its higher overall hydrophobicity and reduced charge. It is important to take these biophysical characteristics in account when describing the activity of peptide-based drugs, as they can greatly affect membrane lysis and cell toxicity.28,29
:1 molar ratio)) were used to mimic the outer leaflet of cancer cell membranes.
:1) membranes with higher affinity than to neutral POPC membranes, as quantified by peptide-to-lipid ratio (P/L, mol/mol) obtained at equilibrium with 16 μM peptide, and by the P/L at saturation (P/Lmax) obtained from dose–response curves (Table S1, ESI†). The open heterodimer PF4P_pDI had increased binding affinity for both neutral POPC and POPC/POPS (4:1) membranes compared to the parent homodimer peptides, and removed the lipid from the SPR chip at 16 μM peptide (see Fig. S5, ESI†), suggesting detergent-like properties at higher peptide concentrations. The increased affinity of PF4P_pDI for both lipid systems, and loss of selectivity for negatively charged membranes are probably due to its higher overall hydrophobicity and reduced charge. It is important to take these biophysical characteristics in account when describing the activity of peptide-based drugs, as they can greatly affect membrane lysis and cell toxicity.28,29
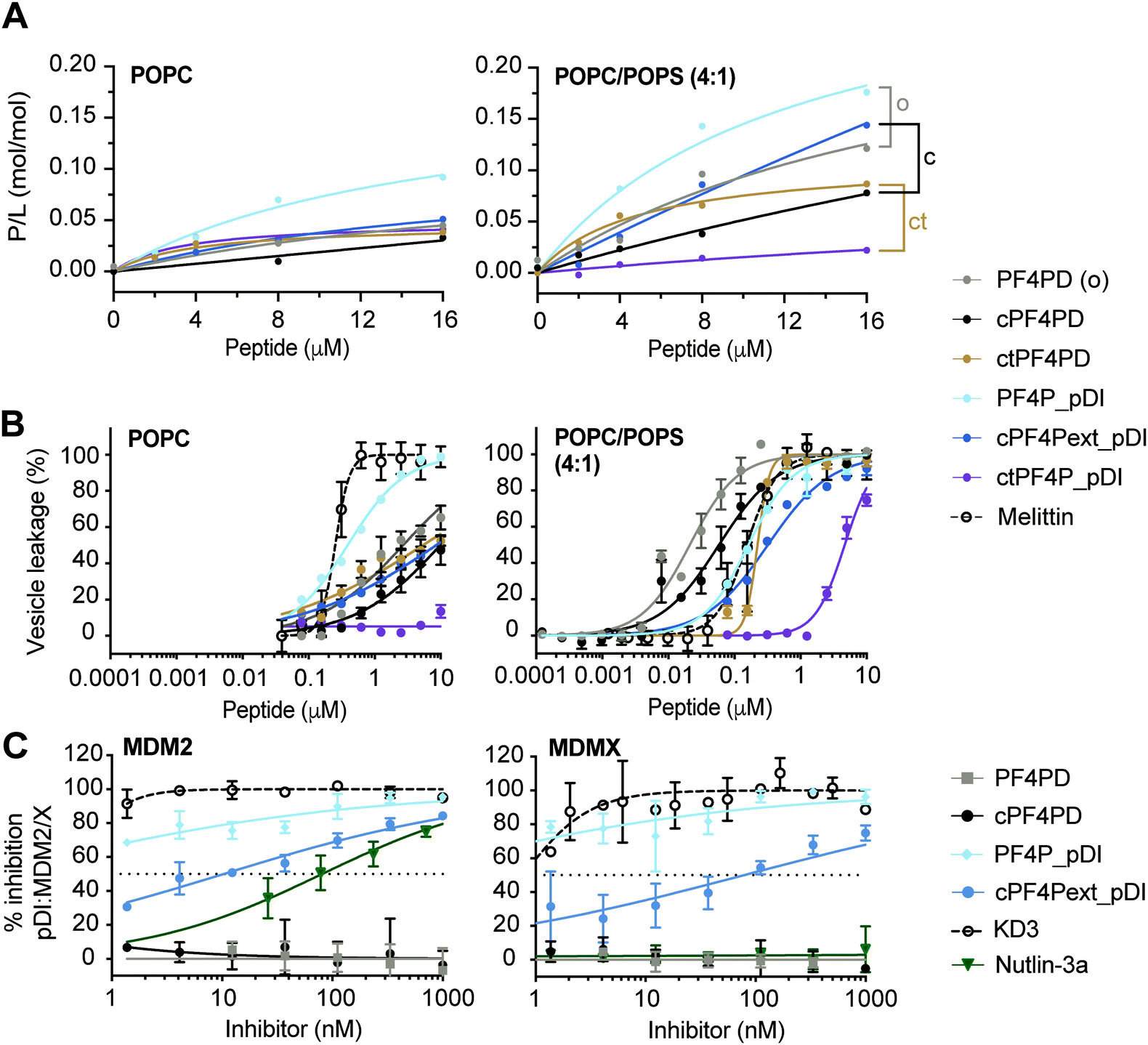 | ||
| Fig. 3 Grafted peptides bind and lyse model membranes with PS-headgroups and also inhibit pDI:MDM2/X interactions. (A) Binding of peptide to neutral (POPC) and anionic (POPC/POPS, 4:1) phospholipid bilayers. Peptides were injected over lipid bilayers on an L1 chip at varying concentrations and peptide-to-lipid ratio (P/L) was determined at the end of the association phase (170 s) when peptide–lipid binding reached a plateau (see Fig. S5, ESI†). Grafted peptides were compared to the parent peptide according to their format: o, open; c, disulfide cyclized; ct, thioether cyclized, by considering relative differences in P/L for 2–16 μM peptide. (B) Membrane leakage induced by peptides. Peptides at varying concentrations were incubated with vesicles (lipid concentration at 5 μM) loaded with self-quenching concentrations of carboxyfluorescein (CF); the percentage of vesicles with their membrane disrupted was determined by measuring the fluorescence emission intensity of CF released (λexc = 485 nm/λem = 520 nm). 100% of vesicles with leaky membranes was established using 0.1% (v/v) Triton X-100. Melittin, a membrane-disruptive peptide, was included as a positive control. Data points and LC50 values are mean ± SD from at least two technical replicates from the same experiment. (C) Inhibition pDI:MDM2 and pDI:MDMX interactions by cPF4PD and analogues. FITC-labeled pDI (F-pDI) was used as a p53 mimetic and inhibition of biding to MDM2 and MDMX was determined by measuring the variation in fluorescence polarization of F-pDI. The fluorescence polarization of free F-pDI was used to define 100% inhibition of pDI:MDM2/X interactions, and the fluorescence polarization obtained with pDI:MDM2/X was used to define 0% of inhibition. Controls included: KD3, a lactam-stabilized pDI peptide with high affinity for MDM2 and MDMX; and Nutlin-3a, a small molecule with high affinity for MDM2 but not MDMX. The inhibitor concentration required to inhibit 50% of F-pDI:MDM2/X interactions (IC50) was determined from the dose response curves. Data points are mean ± SD from three technical replicates from the same experiment. | ||
The extended disulfide cyclized heterodimer cPF4Pext_pDI bound to POPC/POPS (4:1) membranes with higher affinity than the parent cPF4PD peptide (see Fig. 3A, Table S1 P/Lmax and Fig. S5 SPR sensorgrams, ESI†). Even though the overall charge of this heterodimer (+4) was reduced compared to cPF4PD (+6), presentation of the six Lys residues on the same face of the extended α-helix may have increased the ability of cPF4Pext_pDI to interact with negatively charged PS-headgroups. Thioether cyclized ctPF4PD had increased binding affinity for POPC/POPS (4:1) at the lower peptide concentrations tested (2–8 μM), but both ctPF4PD and ctPF4P_pDI had a reduced P/Lmax, compared to disulfide cyclized and open format peptides (see Table S1, ESI†).
:1) membranes was investigated by measuring leakage of CF from large unilamellar vesicles (LUVs).19,30 The dose response curves for peptide-induced membrane lysis are shown in Fig. 3B; the peptide concentration required to induce leakage for 50% of the vesicles (LC50; final lipid concentration of 5 μM) and the relative selectivity for lysing LUVs composed of POPC/POPS (4:1) compared to POPC are shown in Table S1 (ESI†). All of the grafted peptides, with the exception of PF4P_pDI, disrupted POPC/POPS (4:1) with a higher efficacy than POPC LUVs, but with reduced selectivity (25 to 75-fold selectivity) compared to the parent PF4PD and cPF4PD peptides, which had 135-fold and 165-fold higher affinity for POPC/POPS (4:1) compared to POPC. PF4P_pDI lost selectivity for negatively charged membranes and lysed POPC and POPC/POPS (4:1) LUVs with similar efficacy as the control membranolytic peptide Melittin. ctPF4P_pDI induced minimal lysis of POPC LUVs up to the highest concentration tested (10 μM), and ∼5 μM was required to lyse 50% of LUVs composed of POPC/POPS (4:1). The reduction in membrane lytic capacity of ctPF4P_pDI agrees with its weak binding affinity to POPC and POPC/POPS (4:1) membranes (see Fig. 1A) and correlates with the inability to acquire helical conformation, even when in a hydrophobic environment (see Fig. S4B, ESI†).Inhibition of p53:MDM2/X interactions
We evaluated the ability of PF4P_pDI and cPF4Pext_pDI, and their respective parent peptides PF4PD and cPF4PD, to inhibit interaction between fluorescein-labeled pDI (F-pDI),28 a peptide mimic of p53, and MDM2 or MDMX. The peptide concentrations required to inhibit binding of 50% of F-pDI (IC50) to MDM2 and MDMX were determined from dose response curves (Fig. 3C) and used to compare the inhibitory activity of the peptides with KD3, a lactam-stabilized pDI peptide with high affinity for MDM2 and MDMX;27 and Nutlin-3a, a small molecule inhibitor with high affinity for MDM2 but not MDMX.25PF4P_pDI was the best inhibitor of pDI:MDM2 and pDI:MDMX interactions, and inhibited binding of >50% of F-pDI at the lowest tested concentration of 1 nM. cPF4Pext_pDI bound with lower affinity, requiring 10 nM and 80 nM peptide to inhibit 50% of pDI binding to MDM2 and MDMX, respectively. Neither of the grafted peptides were able to completely reproduce the inhibitory activity of KD3 (IC50 ≪ 1 nM), suggesting that optimal access to MDM2 and MDMX binding surfaces were not achieved by the larger peptides. However, both PF4P_pDI and cPF4Pext_pDI inhibit binding to MDM2 with higher efficacy than Nutlin-3a (IC50 80 nM), an inhibitory potency that is within the range previously reported for Nutlin-3a binding to MDM2 (Ki 52 nM)25 and inhibition of p53:MDM2 (∼110 nM).31 The parent peptides PF4PD and cPF4PD did not inhibit the pDI:MDM2/X interactions, confirming that the grafted pDI sequence within PF4P_pDI and cPF4Pext_pDI was responsible for the inhibitory activity of these peptides.
The ability of PF4P_pDI and cPF4Pext_pDI to inhibit pDI:MDM2 and pDI:MDMX interactions confirms that, in the grafted peptides, pDI is present in a more energetically favorable α-helical conformation than F-pDI. This structure promotes the display of key binding residues Phe, Trp and Leu on the same face of the grafted peptides and results in the increased affinity for the p53-binding pocket of MDM2 and MDMX24 (see Fig. 3C). Compared to previously reported cHLH-p53-R26 and analogues,28 PF4P_pDI and cPF4Pext_pDI are several hundred times more potent at inhibiting pDI:MDM2 and pDI:MDMX interactions.
Activity of PF4 peptide scaffolds and grafted peptides
The cytotoxic activity of the peptides was tested against a panel of cancerous and noncancerous cell lines, and also peripheral blood mononuclear cells (PBMCs) and RBCs isolated from healthy adult donors. The panel of cancer cell lines was selected to represent different types of cancer, including melanoma (MM96L), epithelial breast cancer (MCF7 and MDA-MB-231), cervical cancer (HeLa), colorectal cancer (HCT116), and leukemia (K562). Cell lines with wild-type or mutant p53 were selected to provide a comparison for determining whether pDI-containing peptides could induce p53 reactivation, which is only expected for wild-type p53 cells. Cell morphology and origin was considered when selecting control cells. The immortalized, but noncancerous keratinocyte (HaCaT) cell line was included as a skin fibroblast cell control for MM96L; the immortalized noncancerous kidney (HEK293) cell line as an epithelial cell control for MCF7, MDA-MB-231, HeLa and HCT116; and PBMCs as a mononuclear blood cell control for K562. RBCs were included to represent healthy cells with neutral membranes. The peptide concentrations required to kill 50% of cells after 24 h incubation (CC50) with the various peptides are compared in Table 2.| Peptide | CC50a (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Skin fibroblast cells | Epithelial cells | Blood cells | ||||||||
| Breast | Breast | Cervix | Colorectal | Kidney | ||||||
| MM96L | HaCaT noncancer | MCF7 | MDA MB-231 | HeLa | HCT116 | HEK293 noncancer | K562 | PBMC noncancer | RBC noncancer | |
| p53: WT | mut | WT | mut | WT (rep) | WT | WT | mut | WT | WT | |
| a CC50 were obtained from dose response curves from 0.25–32 μM peptide. Human cancer cell lines include MM96L, melanoma; K562, chronic myeloid leukemia; HeLa, cervical; MCF7 and MDA-MB-231, breast; and HCT116, colorectal epithelial. Noncancerous immortalized human cell lines include HaCaT, keratinocyte; and HEK293, embryonic kidney. Cells isolated from human blood include PBMCs and RBCs. p53 status is wild-type (WT), mutant (mut), repressed (rep). Cells (except RBCs) were incubated with peptide for 24 h and cell death was measured using resazurin. RBC toxicity was measured following 1 h incubation with peptide by detecting released hemoglobin. Data represent mean ± SD from three independent experiments conducted on different days, except PBMCs and RBCs where data represent mean ± SD from a single experiment using cells from three individual donors. | ||||||||||
| PF4P | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >64 | |
| PF4PD | 1.6 ± 0.1 | 4.3 ± 0.2 | 17.6 ± 2.0 | >32 | 17.2 ± 2.2 | 4.8 ± 0.8 | 13.7 ± 2.2 | 4.4 ± 0.1 | 14.0 ± 0.9 | >64 |
| cPF4PD | 4.1 ± 0.2 | 9.1 ± 0.7 | ∼32 ± 5 | ∼32 ± 2 | >32 | 23.3 ± 2.2 | >32 | 5.7 ± 0.4 | 9.1 ± 1.3 | >64 |
| ctPF4PD | 3.5 ± 0.2 | 16.9 ± 0.8 | 20.7 ± 2.2 | 2.8 ± 0.2 | 5.1 ± 0.6 | >64 | ||||
| PF4P_pDI | 13.2 ± 0.7 | 17.1 ± 1.6 | >32 | >32 | >32 | 25.5 ± 2.1 | 27.5 ± 2.3 | 16.4 ± 3.3 | >32 | >64 |
| ctPF4P_pDI | >32 | >32 | >32 | >32 | 20.5 ± 0.8 | >64 | ||||
| cPF4Pext_pDI | 2.5 ± 0.1 | 7.2 ± 0.3 | 18.6 ± 2.0 | 29.0 ± 1.9 | ∼32 ± 5 | 11.7 ± 0.9 | 19.1 ± 0.5 | 5.1 ± 0.2 | 4.3 ± 0.4 | >64 |
These cytotoxicity data suggest that cPF4Pext_pDI was more toxic for cancer cells with wild-type p53. However, western blot analysis of MM96L and HCT116 cells that were treated with PF4PD, PF4P_pDI, cPF4PD and cPF4Pext_pDI under similar conditions to the cytotoxicity assays, did not reveal any increase in p53 expression (Fig. S6, ESI†). From the peptide–lipid binding studies above, we showed that the pDI-grafted peptides; including open PF4P_pDI, and cyclized cPF4Pext_pDI and ctPF4P_pDI, bound with higher affinity to more negatively charged membranes compared to their respective parent peptides (see Fig. 3A). From these combined observations, differences in cytotoxic activity of PF4 scaffold and pDI-grafted peptides most likely relate to differences in binding to, and/or lysis of, anionic membranes, and not variations in expression levels of p53.
The expected selectivity of the PF4 scaffolds and grafted peptides for all cancer cells compared to all noncancerous cells was not observed. Toxicity toward noncancer cells followed the trend RBC < HEK293 < PBMC < HaCaT, which is consistent with overall differences in activity observed for cancerous epithelial, blood, and skin fibroblast cell lines. Therefore, it is likely that that differences between the membrane composition of different cell types might also modulate susceptibility to treatment with PF4 peptides.
Cellular internalization of PF4 peptide scaffolds and grafted peptides
To further examine our hypothesis that PF4-derived peptides can deliver a therapeutic cargo sequence to the cytoplasm of cells without becoming trapped in endosomes, we conducted internalization studies with cPF4PD, PF4P_pDI and cPF4Pext_pDI labelled with Alexa Fluor 488 (A488). We used flow cytometry to compare relative amounts of peptide uptake into cells, and confocal microscopy of live cells to characterize intracellular location of the peptides over time. We have previously demonstrated the value of these methods for describing peptide entry without cell membrane leakage or residual binding of peptides to the cell surface; and for distinguishing peptide entry into the cell cytoplasm via endocytosis (with and without release from vesicles), or direct entry.28,32,33Labeled peptides were detected inside >95% of all tested cells following a one-hour treatment with 2 μM cPF4PD-A488, PF4P_pDI-A488 or cPF4Pext_pDI-A488 (Fig. 4). Notably, these peptides entered cells at non-toxic concentrations. (Table 2). For example, 0.5 μM cPF4PD or cPF4ext_pDI internalized into >95% of MM96L and MDA-MB-231 cells, but CC50 values (see Table 2) were 5–8 fold higher for MM96L and ∼16 fold higher for MDA-MB-231 cells. MM96L and HaCaT cells were additionally treated with Trypan Blue, a fluorescence quenching compound that cannot penetrate cells with intact membranes. Dose response curves showed identical fluorescence emission intensities and percentage of fluorescent cells in the presence and absence of Trypan Blue, suggesting that the labeled peptides locate inside cells and without damaging cell membranes (Fig. S7, ESI†).
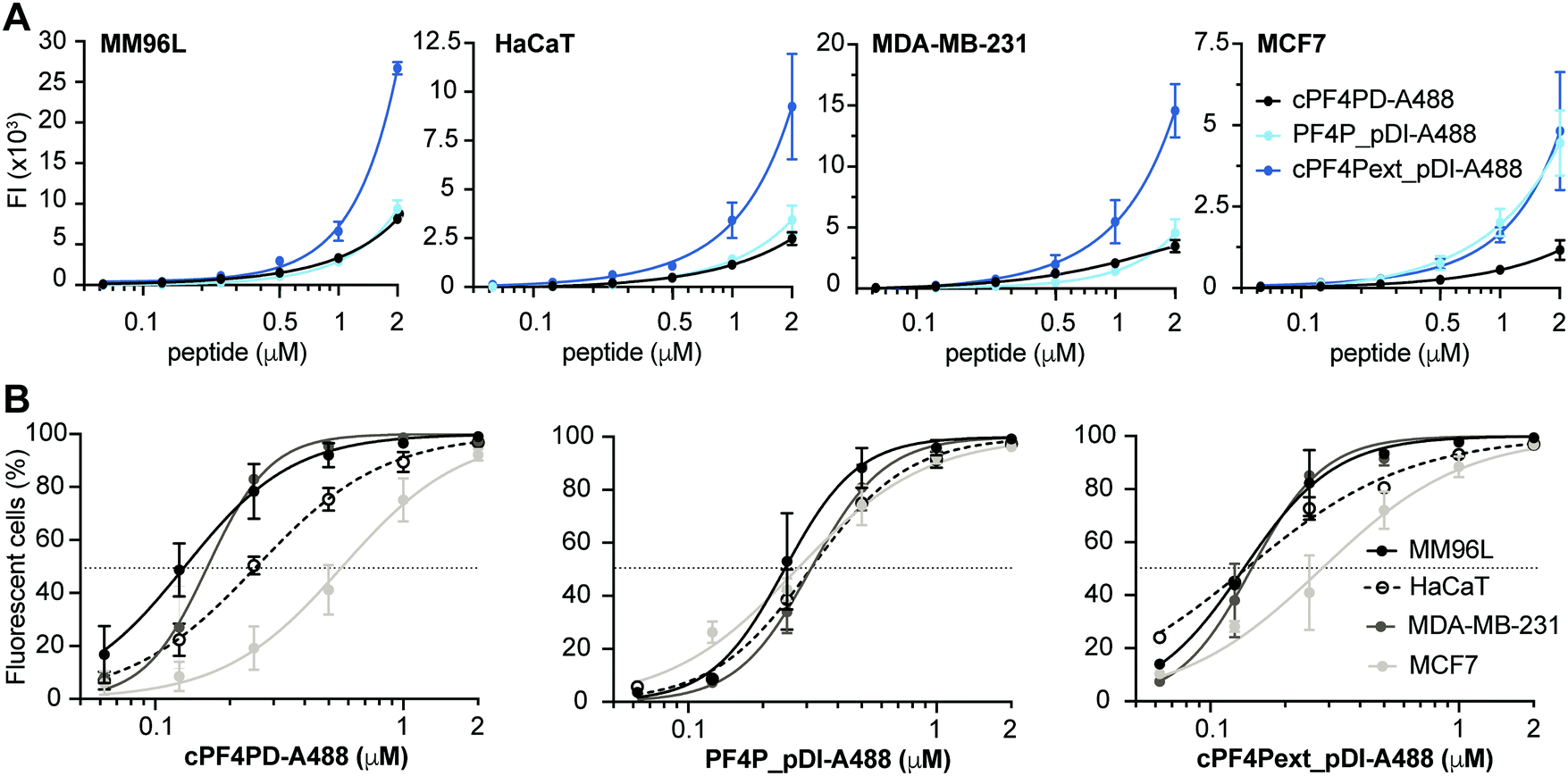 | ||
| Fig. 4 Internalization of peptides into cancer cells. Cells were treated with 0.06–2 μM labeled peptide. Mean fluorescence emission intensity (FI) was recorded for 10000 cells per sample (excitation at 488 nm and emission at 530/30 nm) at 330 V (except MM96L which was recorded at 370 V). Background FI for untreated cells was subtracted. (A) Dose response curves show the relative amounts of cPF4PD-A488, PF4P_pDI-A488 and cPF4Pext_pDI-A488 internalized into MM96L, HaCaT, MDA-MB-231 and MCF7 cells. The amount of internalized peptide was not compared between cell lines because differences in voltage settings affect FI signal amplification. (B) Dose response curves show the percentage (%) of MM96L, MDA-MB-231, MCF7 and HaCaT cells that were fluorescent following treatment with cPF4PD-A88, PF4P_pDI-A488 or cPF4Pext_pDI-A488. Peptide concentrations to achieve 50% fluorescent cells (dotted line; FC50) were used to compare internalization efficacy between the different cell lines. Data points represent the mean ± SD from at least two independent experiments conducted on different days. | ||
Mean fluorescence emission intensity (FI) was used to compare the relative amounts of peptide internalized within a cell line (Fig. 4A). cPF4Pext_pDI-A488 entered MM96L, MDA-MB-231 and HaCaT cells with higher efficacy than cPF4PD-A488 and PF4P_pDI-A488. This observation agrees with cPF4Pext_pDI being the most toxic peptide across all tested cell lines. However, differences in relative uptake of the peptides (up to 2 μM) did not explain relative differences in CC50 between peptides within each of the cell lines (Table 2). Likewise, cPF4Pext_pDI-A488 and cPF4Pext_pDI both entered MCF7 cells with higher efficacy than cPF4PD, but all of these peptides had similar poor toxicity against MCF7.
The percentage of fluorescent cells was recorded for each cell sample treated with peptide. Peptide concentrations required to detect fluorescence in 50% of cells (FC50) were determined from the dose response curves and used to compare internalization efficacy between the cell lines (Fig. 4B). The trend in internalization efficacy was similar for cPF4PD-A488 and cPF4Pext_pDI-A488 with MM96L ∼ MDA-MB-231 > HaCaT > MCF7. In contrast, PF4P_pDI appeared to enter each of the different cell types with similar efficacy. The differences in internalization efficacy that were observed between the different cell lines for cPF4PD-A488 and cPF4Pext_pDI-A488 did not correlate with relative differences in toxicity (see Table 2). For example, MM96L cells had the lowest FC50 for cPF4PD-A488 (0.13 μM) and also the lowest CC50 for cPF4PD (∼4 μM), whereas MDA-MB-231 cells were resistant to treatment with cPF4PD at concentrations up to 32 μM, but the labeled peptide (up to 2 μM) was able to enter these cells with similar efficacy (FC50 0.16 μM) to MM96L cells. Similarly, cPF4PD was not toxic to MCF7, but fluorescence was detected in 50% of cells following treatment with 0.65 μM cPF4PD-A488 (Fig. 4B).
Internalization of cPF4PD-A488, PF4P_pDI-A488 and cPF4Pext_pDI-A488 into melanoma (MM96L) cells was visualized using spinning disc confocal microscopy of live cells, and fluorescence micrographs from representative time points are shown in Fig. 5A for each of the peptides. Intracellular localization was examined for 1 μM (sub-lethal) and 4 μM (∼CC50) cPF4PD-A488; 6 μM (half-CC50) PF4P_pDI-A488; and 1 μM (half-CC50) cPF4Pext_pDI-A488. At these concentrations, the peptides appeared to localize at the cell surface (outlined arrowhead), then enter directly into the cell cytoplasm (closed arrowhead). Colocalization with structures that were identified as mitochondria by staining with MitoTracker Red was also observed (double-headed arrow Fig. 5A). The plasma membrane appeared to remain intact, although some cells increased in size suggesting that membrane permeation was affected. Representative time intervals showing the internalization of PF4P_pDI-A488 into MM96L cells are shown in Fig. 5B (see Movie S1 for the complete time series, ESI†).
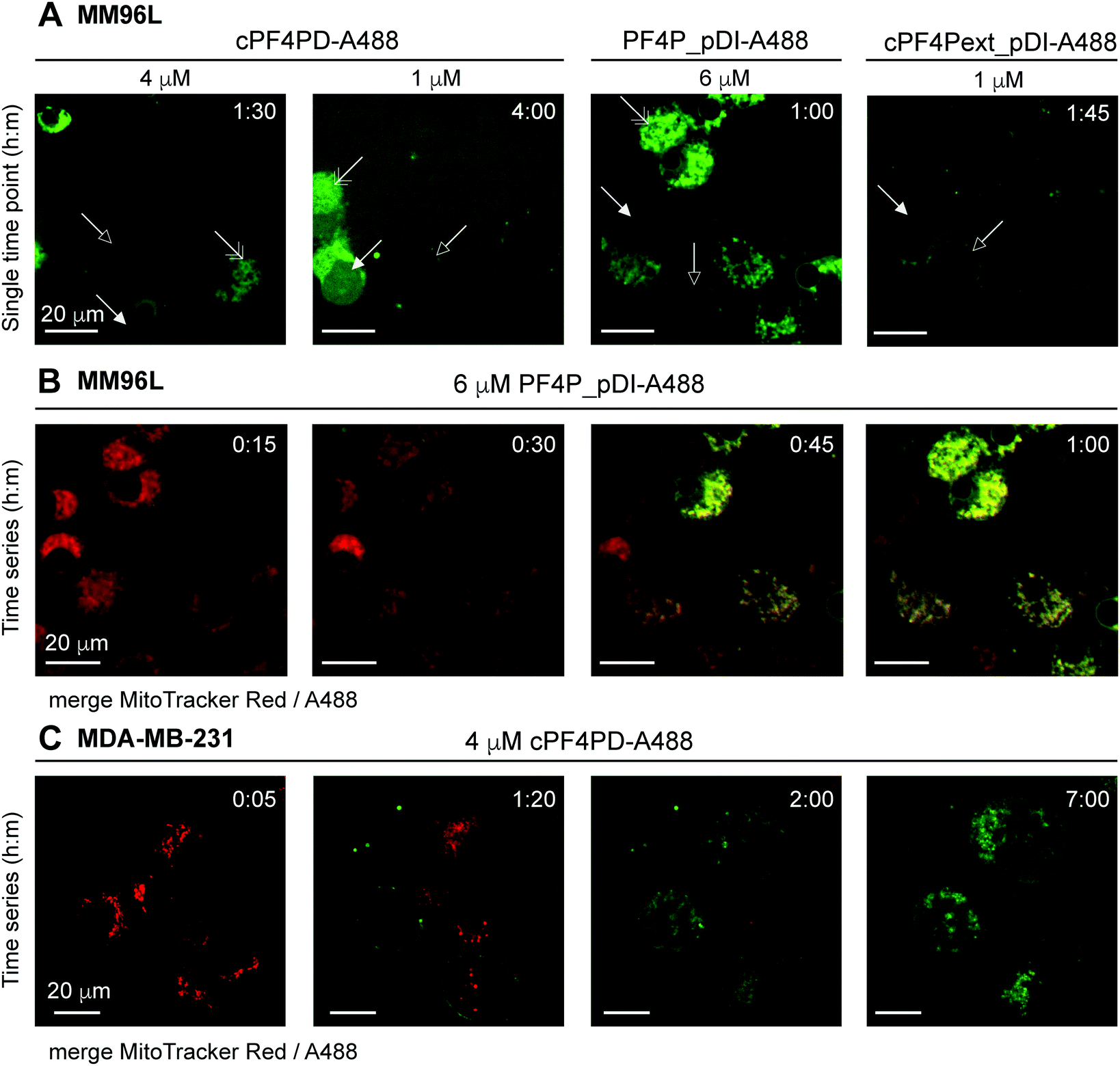 | ||
| Fig. 5 Fluorescence micrographs showing intracellular location of A488-labeled peptides and colocalization with mitochondrial membranes. (A) MM96L cells were monitored for up to four hours after adding cPF4PD-A488, PF4P_pDI-A488 or cPF4Pext_pDI-A488. Each panel represents a single time point. Peptide identity and concentration are shown above each panel. Arrows indicate: outlined, peptide binding to the cell surface; closed, peptide located in the cell cytoplasm; double-headed, peptide colocalization with mitochondria. (B) MM96L cells were labeled with MitoTracker Red for 5 min and the dye was removed prior to treatment with 6 μM PF4P_pDI-A488. Time intervals of 15 min are shown for merged MitoTracker Red and A488 channels (see Movie S1 for the complete time course for the A488 channel, 1 frame per min, ESI†). (C) MDA-MB-231 cells were stained with MitoTracker Red (5 min pulse) and treated with 4 μM cPF4PD-A488. Cells were monitored for seven hours and representative time intervals are shown here. The scale bars on all panels represent 20 μm. Colocalization of cPF4PD-A488 with mitochondrial membranes is shown in more detail in Fig. S8 (ESI†) for the seven-hour time interval. | ||
The location of 4 μM (sub-lethal) cPF4PD-A488 inside MDA-MB-231 cells was also examined and entry of the labeled peptide into the cytoplasm was observed within two hours. Intracellular accumulation of cPF4PD-A488 increased over the next five hours (Fig. 5C). During this time, peptide accumulation was observed both within the cell cytoplasm, and also colocalized with mitochondrial membranes (see Fig. S8 for overlay of A488 and MitoTracker Red fluorescence, ESI†). An increase in the size of some cells was also observed over this time period. Notably, these observed events occurred at a concentration of cPF4PD-A488 that was eight-fold lower than the CC50 for MDA-MB-231 cells (∼30 μM, see Table 2).
Together these observations of peptide internalization into MM96L and MDA-MB-231 cells support the hypothesis that cPF4PD, and pDI-grafted peptides PF4P_pDI and cPF4Pext_pDI, can directly translocate plasma membranes to access the cell cytoplasm without becoming trapped in endosomes. Differences in peptide concentrations required to detect intracellular accumulation within (MM96L) and between (MM96L vs. MDA-MB-231) cell types were consistent with differences in cytotoxic concentrations of cPF4PD, PF4P_pDI and cPF4ext_pDI (Table 2). Colocalization of the peptides with mitochondrial membranes is consistent with their high affinity and preference for binding to anionic membranes (see Fig. 2). Mitochondrial membranes are more negatively charged than the cell plasma membrane, as they are rich in negatively charged lipids including cardiolipin and phospholipids with phosphatidylglycerol headgroups.34
Peptide effects on mitochondrial transmembrane potential
Mitochondrial membranes can be described as polarized, and maintenance of a high transmembrane potential (more negative inside) is important for mitochondrial respiration and production of ATP. We have previously demonstrated that cationic peptides can depolarize biological membranes by neutralizing the surface charge;35 and here we were interested in determining whether PF4 scaffold and grafted peptides affect the polarization of mitochondrial membranes, thereby affecting mitochondrial function. We treated MM96L cells with PF4PD, cPF4PD, PF4P_pDI and cPF4Pext_pDI, then stained the cells with tetramethylrhodamine ethyl ester (TMRE). This cell permeant dye was used to report on whether peptide treatment affected mitochondrial transmembrane potential because it only accumulates on the inner membrane of active mitochondria (high transmembrane potential, negative inside).Treatment of MM96L cells with the peptides at their respective CC50 (see Table 2) induced depolarization of mitochondrial membranes, as suggested by a drop in the FI of TMRE, by at least as much as cells treated with the control proton ionophore carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) that depolarizes membranes by decoupling oxidative phosphorylation in the inner mitochondrial membrane.36 Transmembrane potential continued to decrease for up to eight hours after treatment (Fig. 6A). Notably, the TMRE FI of peptide-treated cells was lower than that of FCCP-treated cells, suggesting that the mitochondrial transmembrane potential had been greatly affected.
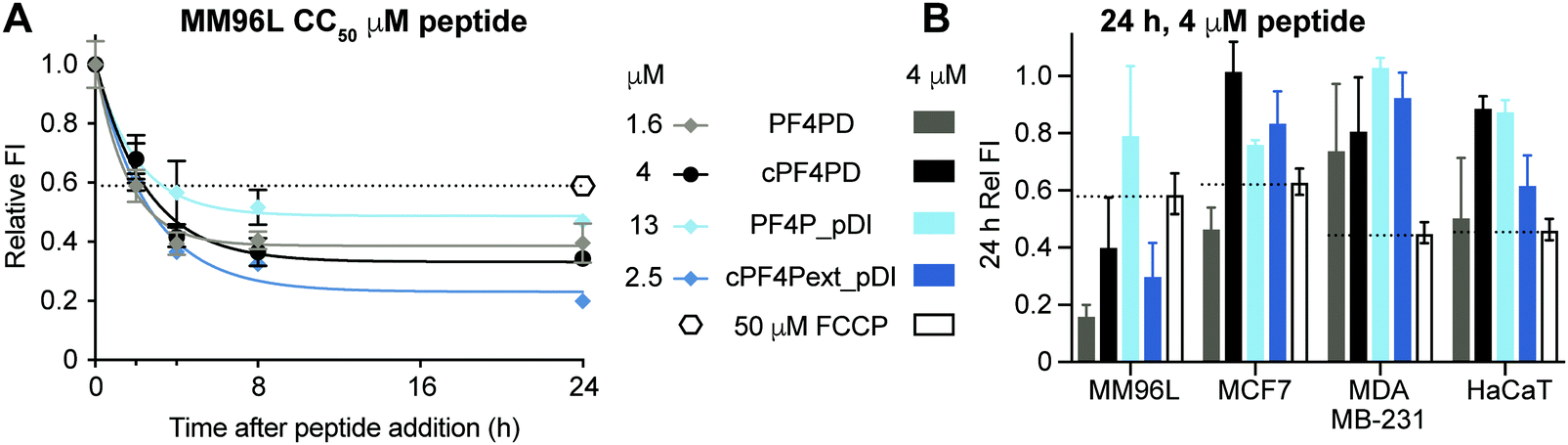 | ||
| Fig. 6 Depolarization of mitochondrial membranes following treatment with PF4 scaffold, or grafted peptides, measured with TMRE fluorescence. (A) MM96L cells treated with peptides at CC50 concentrations for two, four, eight and 24 h end-time points. TMRE was added to each sample and incubated for 30 min before measuring fluorescence emission intensity (λexc = 549 nm/λem = 575 nm). Fluorescence emission intensity (FI) values are relative to the fluorescence obtained with TMRE when bound to mitochondria of untreated cells (FI = 1). FCCP decouples oxidative phosphorylation in the inner membrane of mitochondria and was used as a positive control for membrane depolarization. (B) Relative TMRE FI in four different cell lines treated with peptides at 4 μM for 24 h is shown compared to cells treated with FCCP for 24 h. Data points are mean ± SD from three technical replicates from the same experiment. | ||
The effect of peptide treatment on mitochondrial membrane potential was compared between representative cell lines MM96L, MCF7, MDA-MB-231 and HaCaT following treatment with 4 μM PF4PD, cPF4PD, PF4P_pDI and cPF4Pext_pDI for 24 h. The relative TMRE FI of the treated cells are shown in Fig. 6B. Peptides that produced a relative FI below the FCCP positive control were considered to have caused depolarization of mitochondrial membranes. The greatest perturbation of mitochondrial membrane potential was observed for MM96L cells treated with 4 μM PF4PD, cPF4PD and cPF4ext_pDI. The open dimer PF4PD similarly depolarized mitochondrial membranes of MM96L and MCF7 cells, at the same concentration. cPF4PD and cPF4ext_pDI did not appear to affect the mitochondrial transmembrane potential of MDA-MB-231 cells, despite the ability of both these peptides (4 μM peptide labeled with A488) to enter MM96L and MDA-MB-231 cells (Fig. 3 panels A and C). Higher concentrations of cPF4PD-A488 appeared to colocalize with mitochondria of MM96L cells (Fig. 4, panel A vs. C), and these higher localized peptide concentrations likely explain the higher degree of mitochondrial membrane perturbation caused by cPF4PD and also cPF4Pext_pDI in MM96L.
The relative levels of mitochondrial membrane depolarization observed from the TMRE fluorescence data agree with differences in cytotoxicity observed between the different cell types, and also between peptides within the same cell type. For example, MM96L cells had a similar trend for relative mitochondrial membrane depolarization and cytotoxicity (CC50); PF4PD > cPF4Pext_pDI > cPF4PD ≫ PF4P_pDI (see Fig. 6B and Table 2), which suggests that perturbation of mitochondrial membrane function contributes strongly to the cytotoxic mechanism of action of these peptides. This mechanism is similar to that reported for a designer peptide with a helical KLAK motif and a tumor-homing and cell-penetrating RGD motif (RGD-4C)-GG-D(KLAKLAK)2, that was able to reduce both tumor and metastatic burden in mice with tumor xenografts.37
The ability of PF4 scaffold and grafted peptides to target and enter cancer cells to neutralize more negatively charged mitochondrial membranes represents an interesting parallel with cPF4PD's ability to target and enter RBCs infected with Plasmodium parasites to lyse the more negatively charged digestive vacuole membranes.19 This crossover highlights the utility of selective membrane-active peptides for developing targeted therapies to a range of diseases.
Conclusion
CPPs that can reach the cytoplasm of cancer cells are valuable tools for developing peptide-based drugs; especially those with selective activity toward negatively charged membranes that correspond to diseased cells, including cancer. Newly characterised peptide cPF4PD recapitulates the intrinsic selective and cell-penetrating properties of a transduction domain from the human defense protein PF4, and here we demonstrate the ability of cPF4PD and analogues PF4PD and ctPF4PD to enter and kill cancer cells, especially melanoma and leukemia, at low micromolar concentrations.cPF4PD analogues were redesigned to incorporate pDI, a peptide that inhibits interactions between p53:MDM2 and p53:MDMX in cancer cells with wild-type p53. Grafted peptides PF4P_pDI and cPF4Pext_pDI successfully acquired a helical structure, which correlated with their resistance to degradation by serum proteases and ability to inhibit p53:MDM2/X interactions at low nanomolar concentrations. cPF4Pext_pDI also maintained the selectivity of the parent PF4 scaffold peptides for binding and lysing model membranes that contained phospholipids with negatively charged PS-headgroups.
Internalization studies showed that cPF4PD, PF4P_pDI and cPF4Pext_pDI (labeled with A488) enter cancer cells at non-toxic concentrations (nM to low μM) without disrupting the plasma membrane or becoming trapped in endosomes. The ability to directly translocate across cell membranes to access the cytoplasm is tremendously important as it improves the ability to access and disrupt intracellular protein–protein interactions. Therefore, PF4 scaffold and grafted peptides have an advantage compared to other CPPs that become trapped in endosomes, including gold standard CPPs Tat20 and Arg9,15,38 and previously reported grafted cyclic helix-loop-helix peptide (cHLH-p53-R).28
PF4 scaffold and pDI-grafted peptide analogues entered susceptible MM96L cells and less susceptible MDA-MBA-231 cells at similarly low nanomolar concentrations. Peptides accumulated in the cytoplasm and also on mitochondria. The extent of mitochondrial accumulation was more pronounced for MM96L cells, and this was consistent with a greater reduction in mitochondrial membrane potential. Furthermore, the extent of mitochondrial membrane depolarization correlated with differences in peptide toxicity both between and within cell lines. The reasons for differences in the intracellular accumulation of and localization of peptides within the tested cell lines remain unclear but might relate to differences in cell membrane composition and/or biophysical properties (e.g. overall charge and fluidity, exposure of proteoglycans).
Despite PF4P_pDI and cPF4Pext_pDI reaching the cytoplasm of cancer cells, selective toxicity was not observed for cancer cells with wild-type p53, as these peptides did not increase p53 levels in MM96L or HCT116 cells. Instead, the mechanism of action for the PF4 scaffold and grafted peptides was related to their ability to perturb mitochondrial membrane function, which occurred within the first two hours of peptide treatment. This rapid mechanism of cell death likely precludes the cells ability to accumulate p53 and enter an apoptotic cell death pathway. We have also reported a predominantly membrane-active mechanism of action in other p53-targeted designer peptides, including the previously reported cHLH-p53-R peptide,26 where cytotoxic activity is mediated by selective cell membrane disruption, and not activation of the p53 pathway.28 Together, these studies highlight the importance of characterizing peptide–membrane interactions during the development of peptide-based drug leads. This is especially relevant for hydrophobic p53 mimic peptides, including pDI, that can increase affinity for biological membranes.
In conclusion, cPF4PD and redesigned analogues kill melanoma and leukemia cells at low micromolar concentrations, with two-fold selectivity over noncancer cells of the same cell type. cPF4PD analogues are also promising CPP scaffolds that can stabilize peptide sequences in a bioactive conformation to inhibit protein–protein interactions. The redesigned peptides maintain selectivity for negatively charged membranes and can enter cells at nanomolar to low micromolar concentrations via direct membrane translocation. cPF4PD analogues have application for cytosolic delivery of cell-impermeable cargoes using grafting or conjugation strategies. This approach is best suited to fast-acting agents with sub-micromolar activity and is applicable to a range of disease applications, including intracellular protein–protein interactions in cancer, and intracellular pathogens such as malaria parasites.
Experimental
Peptide synthesis, oxidation and cyclization
All peptide analogues used in this study were synthesized on rink amide resin using solid phase peptide synthesis (symphony® Protein technologies) and standard Fmoc chemistry. Amino acid coupling was achieved using 2 molar equivalents of Fmoc-protected amino-acid, 2 molar equivalents of O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluroniumhexafluorophosphate (HCTU) and 2.5 molar equivalents of N,N-diisopropylethylamine (DIPEA). For peptides that required thioether cyclization (ctPF4PD, ctPF4P_pDI), Cys was incorporated on the C-terminus and a final coupling was used to include chloroacetic acid on the N-terminus.Final N-terminus Fmoc deprotection was conducted on all peptides that did not have an N-terminal chloroacetic acid by treating the resin with 30% piperidine in DMF. Peptides were deprotected and cleaved from the resin using H2O:TIPS:TFA (2:2:96, v/v/v), then filtered and precipitated with ice–cold diethyl ether and extracted with 50% acetonitrile (ACN): 0.05% TFA. The crude deprotected (linear) peptides were purified by RP-HPLC using Shimadzu preparative system and Phenomenex Jupiter C18 column with a 1% min−1 gradient of solvent B (90% ACN, 0.05% TFA v/v) against solvent A (0.05% TFA v/v) and a flow rate of 8 mL min−1.
cPF4PD and cPF4Pext_pDI were cyclized by disulfide bond formation. Purified and lyophilized peptide was dissolved at 0.5 mg mL−1 in 50% ACN, 0.05% TFA (v/v), flushed with nitrogen for 5 minutes and oxidized by adding iodine solution (peptide:iodine, 1:20) then stirring for 30 min at room temperature in the dark. The oxidizing buffer was quenched with ascorbic acid and purified by RP-HPLC as above. ctPF4PD and ctPF4P_pDI were cyclized by thioether bond formation in DMSO with trimethylamine.
All peptides were further purified by RP-HPLC. Purity >95% was confirmed using analytical RP-HPLC (C18 column 2.1 × 150 mm), with a flow rate of 0.3 mL min−1 and a 2% min−1 gradient of solvent B (starting with 1% solvent B in solvent A); or a 2% min−1 gradient of ACN, 0.1% formic acid (v/v) (starting with 1% of ACN, 0.1% formic acid (v/v) in 0.1% formic acid (v/v)). Correct sequence and cyclization were confirmed from the mass as determined using ESI-MS or LC-MS.
The overall hydrophobicity of the peptides was compared in a single experiment with consecutive runs using analytical RP-HPLC (C18 column 2.1 × 150 mm), with a flow rate of 0.3 mL min−1 and a 2% min−1 gradient of solvent B (starting with 1% solvent B in solvent A). RT of the peptides is shown in Table 1. Peptide sample concentrations were determined by absorbance at 280 nm (see ε280 in Table 1).
Peptide labeling
cPF4PD-A488 was prepared using oxime ligation.39 Peptide (5 mg) was reacted with 1.1 molar equivalents of tris-(2-carboxyethyl)phosphine (TCEP) and 1.5 molar equivalents of dichloroacetone in 30 mL of NH4HCO3 pH 8.1 for 2 h at room temperature. The peptide was purified by RP-HPLC. To add the label, 0.5 mg of purified acetone-linked peptide was reacted with 2 molar equivalents of Alexa Fluor 488 hydroxylamine dye (ThermoFisher Scientific) in 200 μL of 50 mM NaOAc, 150 mM NaCl pH 5 at 37 °C overnight. The labeled peptide was purified by RP-HPLC and the correct mass confirmed by MS.PF4P_pDI-A488 and cPF4Pext_pDI-A488 were prepared by first producing peptide analogues with an azidoalanine replacing a Ser residue in the loop between PF4P helices (see Table 1). Copper-catalyzed azide–alkyne cycloaddition was used to incorporate the A488 label onto the azide-containing peptides.40 Azide-containing peptide (0.5 mg) was combined with 2 molar equivalents of Alexa Fluor 488 Alkyne dye (AF 488 Alkyne, Click Chemistry Tools) with a total reaction volume of 500 μL in 100 mM sodium phosphate buffer pH 7, 20% dimethylsulfoxide. Copper was introduced by adding a 1:5 mixture of CuSO4:tris(3-hydroxypropyltriazolylmethyl)amine (final 0.1 mM Cu). The reaction was initiated by adding sodium ascorbate (final 5 mM) then allowed to proceed for one hour at ∼22 °C. Labeled peptides were purified by RP-HPLC and the correct mass confirmed by MS.
CD spectroscopy
The overall secondary structure of peptides was estimated using CD spectroscopy.41 The peptides were prepared at 50 μM in an aqueous solution (100 mM NaF, 10 mM KH2PO4 pH 7.5) or at 25 μM in a 1:1 mixture of aqueous solution with helix inducing trifluoroethanolamine (TFE). CD spectra were collected and averaged from 5 scans, at 1 nm intervals between wavelengths of 185 nm to 260 nm, using a Jasco J810 spectropolarimeter. Blanks were subtracted, and the mean residue ellipticity (θ) was calculated from the CD output response unit in millidegrees, and the percentage of helicity of each peptide was determined using the Luo-Baldwin formula: Hα = (θ222nm − θC)/(θ∞222nm − θC), with θ222nm the lowest value between 218 and 222 nm, θC = 2220–53T and θ∞222nm = (−44000 + 250T)(1 − k/Nres) with T in °C and k = 3.0.42
NMR spectroscopy
Samples of PF4P_pDI and cPF4Pext_pDI were prepared by dissolving peptide in 10% D2O/90% H2O (v/v) at a concentration of ∼1 mM and pH of 3.5. Spectra were recorded on a Bruker Avance III 600 MHz NMR spectrometer at 298 K, and using excitation sculpting as solvent suppression. Chemical shifts of backbone resonances were assigned by analysis of 2D TOCSY (with an 80 ms MLEV-17 spin lock) and NOESY (mixing time of 200 ms). Spectra were processed with Topspin 3.5 (Bruker Biospin) and analyzed with CcpNMR analysis.43 Secondary αH chemical shifts were calculated as the difference between the observed αH chemical shifts and that of the corresponding residues in a random coil peptide.44Stability of peptides in human serum
The ability of peptides to resist serum degradation was quantified by following the amount of peptide that remained intact at different time points using analytical RP-HPLC. Peptides (final concentration 50 μM) were incubated for 0, 1, 2, 4, 8 and 24 h in the presence of 25% (v/v) human serum in phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). Remaining peptide was recovered by precipitating and removing serum proteins following the addition of ACN with 2% (v/v) TFA. Recovered peptides were analyzed using RP-HPLC (1% min−1 gradient of solvent B, starting with 1% solvent A), and the area of the peak corresponding to full-length peptide (as confirmed by MS) in the chromatogram obtained with absorbance at 215 nm. The amount of peptide remaining was calculated as a percentage of the amount of peptide recovered following 0 h incubation with 25% (v/v) serum. No appreciable peptide degradation was detected for the peptides in a control incubation in PBS over 24 h. Data were collected from a single experiment performed with three technical replicates.Liposome preparation
Synthetic lipids POPC and POPS (Avanti Polar Lipids) were solubilized in chloroform. Lipid films of POPC and POPC/POPS (4:1 molar ratio), were prepared by drying the chloroform under a nitrogen flow then in a vacuum desiccator for >2 h. Lipid vesicles were prepared using repeated freeze/thaw cycles followed by extrusion of 1 mM lipid mixtures in HEPES buffer (10 mM HEPES, 150 mM NaCl, pH 7.4).45 Small unilamellar vesicles (SUVs) were prepared by extrusion through membranes with 50 nm pores; and LUVs with 100 nm.
SPR
The affinity of peptides for lipid membranes was monitored using SPR at 25 °C (Biacore 3000 instrument: GE healthcare). SUVs composed of POPC and POPC/POPS (4:1) were deposited onto a L1 chip for 40 min at a flow rate of 2 μL min−1, reaching a steady-state plateau and confirming chip surface coverage of deposited lipid bilayers. After the plateau of deposition of the lipid was reached, serial dilution of peptides (in HEPES running buffer) were injected over the lipid bilayer (flow rate 5 μL min−1, 180 s), then peptide–lipid dissociation was followed for 600 s. The response units (RU) at the end of the association phase (t = 170 s) of the sensorgrams were converted to peptide to lipid ratio (P/L, mol/mol) for each tested condition by assuming 1 RU = 1 pg mm−2. P/Lmax was determined from fitting P/L dose response curves.
Vesicle leakage
POPC and POPC/POPS (4:1) LUVs were prepared as above, except with 50 mM CF in HEPES buffer. CF-loaded vesicles were purified using a 10 mL gel filtration column prepared from pre-swollen Sephadex G-50 beads in HEPES buffer. The lipid concentration of the purified vesicles was determined against a standard curve prepared with ferrothiocyanate reagent (100 mM FeCl3·6H2O, 400 mM NH4SCN; Stewart assay46). Serial dilutions of peptide (prepared in HEPES buffer) were incubated with LUVs containing 5 μM lipid in black 96-well plates. The fluorescence intensity (FI; λex = 489 nm, λem = 515 nm) was measured using a Tecan fluorescence spectrometer plate-reader after a 10 min incubation. Triton X-100 (0.1% v/v) was included to measure 100% leakage, and HEPES buffer to measure 0% leakage. The percentage of leakage induced by the peptides was calculated using the formula: (FI sample − FI HEPES)/(FI TX-100 − FI HEPES) × 100. Data were collected from a single experiment with three technical replicates.
Inhibition of p53:MDM2/X interactions
The ability of peptides to inhibit interactions of p53 with MDM2 or MDMX (Abcam) was measured in a competition assay using the F-pDI peptide (N-terminal FITC label) as a p53 mimetic.27 The concentration of F-pDI was determined using the Pulcon method.47 The assay was conducted with 10 nM F-pDI, and 8 nM MDM2 or 45 nM MDMX, which are the previously determined protein concentrations required to bind 80% of F-pDI.27 Serial dilution of the peptides were incubated with F-pDI:MDM2/X in Tris buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA). The fluorescence polarization signal obtained for F-pDI:MDM2 or F-pDI:MDMX (λexc = 489 nm/λem = 515 nm) was used to establish 0% inhibition of pDI:MDM2/X interactions, and F-pDI in Tris buffer was used to establish 100% inhibition. The percent inhibition of F-pDI binding in the presence of inhibitor was determined by measuring the variation in fluorescence polarization compared to F-pDI. KD3 (a lactam-stabilized pDI peptide with PEG linker) was included to compare binding affinity of the larger peptides to a smaller stabilized small peptide with the same sequence. Nutlin-3a was included as positive control with high affinity for MDM2, and as negative control for MDMX. The inhibitor concentration required to have 50% of F-pDI in solution (IC50) was determined from the dose response curves. Data were collected with three technical replicates.Cell culture
MM96L, HaCaT and K562 cell lines were grown in RPMI medium supplemented with 2 mM L-glutamine and 10 mM sodium pyruvate. HeLa, MCF7, MDA-MB-231 and HEK293 cell lines were grown in DMEM medium. Media were supplemented with 10% (v/v) fetal bovine serum, 100 units mL−1 of penicillin and 100 μg mL−1 of streptomycin. Flasks of cultured cells were maintained in a humidified incubator at 37 °C, 5% CO2 and passaged every 2–3 days to maintain cultures between 20–90% confluence. Cell line identity was verified by comparing STR profiles of the cell lines against reported database entries. ATCC profiles were used for all cell lines except MM96L, compared to QIMR database (Brisbane, Australia); and HaCaT, compared to Huang et al., 2017.48PBMCs isolation
Blood was donated from healthy adults with informed consent for all the experiments and following protocols approved by the Human Research Ethics Committees (University of Queensland approval number 2013000582). 20 mL of blood from each donor was collected into heparin tubes on the day of the experiment. Heparinized blood was diluted with an equal volume of PBS and then gently layered onto Ficoll-Paque Premium solution (GE Healthcare). PBMCs were collected following centrifugation at 1400 rpm for 30 min by collecting the cell layer between Ficoll-Paque and plasma layers. The cells were washed three times with PBS prior to use in toxicity assays.Cell toxicity
Toxicity to the cell lines and to PBMCs was tested following peptide treatment by measuring the metabolism of resazurin into fluorescent rezofurin. The assays were performed with 5000 cells per well (96-well plate) with incubation of serially diluted peptides for 24 h at 37 °C, 5% CO2. 100% of toxicity was established using 0.1% Triton-X 100 and 0% using PBS. Fluorescence (λex = 560 nm, λem = 585 nm) was measured using a Tecan infinite M1000Pro multiplate reader and CC50 values were determined from dose response curves. Data were collected from three independent experiments conducted on different days, except PBMCs where data were collected from a single experiment using cells from three individual donors.RBC hemolysis
Blood was collected from healthy adult donors following protocols approved by the Human Research Ethics Committees (University of Queensland approval number 2013000582). RBCs were isolated by centrifugation at 4000 rpm for 1 min and washing three times with PBS. Peptides were diluted in PBS and incubated with RBC (final concentration 0.25% v/v) for one hour at 37 °C. Triton X-100 (0.1% v/v final) was included as a control to measure 100% hemolysis, and PBS to measure 0% hemolysis. Plates were centrifuged 1000 rpm for 5 min to pellet RBCs. The supernatant was transferred to a flat bottom 96-well plate and the absorbance at 415 nm was recorded to detect released hemoglobin. The percentage hemolysis was calculated using the following formula: (A415 sample − A415 PBS)/(A415 TX-100 − A415 PBS) × 100. Data were collected from a single experiment using RBCs from three individual donors.Western blots
Cells were plated at 10000 cells per well (24-well plate) and incubated overnight at 37 °C, 5% CO2. Overnight media was replaced with serum-free media, supplemented with 0.3% BSA, and peptides were added to a final concentration of 5 μM or 10 μM. Nutlin-3a was added to a final concentration of 10 μM, as a positive control for reactivation of p53. Cells were incubated as above for 24 h, media was removed before addition of 120 μL of lysis buffer (150 mM NaCl, 1% Triton X-100, 0.1% SDS, 50 mM Tris pH 8, with protease inhibitors [Roche]), with incubation for 30 min on ice. Cell debris was pelleted by centrifugation at 12000 rpm for 20 min. The supernatant was collected, and total protein concentration was estimated from a BSA standard curve using a BCA assay (Pierce). Cell extracts (20 μg) were separated with SDS-PAGE and transferred to nitrocellulose membrane using standard procedures. Blots were blocked with 3% BSA in Tris buffered saline with Tween 20 (TBST; 20 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween 20). Blots were incubated with p53 antibody (D-07, mouse; Thermo Fisher) diluted 1:250 in 0.5% BSA in TBST overnight at 4 °C. Blots were washed three times in TBST, then incubated with 1:5000 goat-anti-mouse-HRP conjugate (Thermo Fisher) and 1:10000 rhodamine-conjugated Tubulin antibody (BioRad) in 0.5% TBST for 1 h at room temperature. The blots were washed three more times prior to development with Clarity Western ECL substrate (BioRad). Western blot images were recorded using a BioRad Chemiluminescent imaging system and densitometry measurements (intensity) were determined for area under the curve for each of the bands using ImageJ.49
Flow cytometry
Internalization of labeled peptides was measured using a flow cytometer.32 Cells were seeded at 105 cells per wells (24-well plate) and incubated at 37 °C, 5% CO2 overnight. Serial dilutions of labeled peptides in serum free medium were added to the cells and incubated for 1 h. Cells were harvested, and the fluorescence detected using the flow cytometry (BD FACS Canto II; excitation at 488 nm and emission with a 530/30 nm filter). Fluorescence readings were repeated after addition of the non-permeable quenching reagent Trypan Blue (0.02% (w/v) final, Sigma Aldrich). The percentage of cells with fluorescence above a background threshold for cells without peptide was determined (% fluorescent cells). Data were collected from at least two independent experiments conducted on different days.Fluorescence microscopy
Localization of labeled peptides inside MM96L and MDA-MB-231 cells was investigated using Zeiss spinning disc confocal microscope. Cells were plated with 16000 cells per well (8-well chamber slides) in complete media and incubated overnight at 37 °C, 5% CO2. Before the assay, cells were washed, and the media was replaced with serum-free media. Cells were stained with MitoTracker Red (final 100 nM, Invitrogen) for 5 minutes to label the membranes of viable mitochondrial. The medium was removed and replaced with serum-free media with the final concentration of labeled peptide. Cells were incubated at 37 °C with 5% CO2 during imaging. Images were post processed using Fiji software.49
Mitochondrial membrane potential
Cells were plated at 5000 cells per well (96-well plate) and incubated overnight at 37 °C, 5% CO2. Serial dilutions of peptides were added to the cells and incubated for up to 24 h. Trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP, Abcam) was included on each plate as a control compound for establishing cell fluorescence when mitochondrial membranes are depolarized. FCCP was added 10 min prior to staining to a final concentration of 50 μM. Mitochondrial membrane staining was achieved by adding tetramethylrhodamine, ethyl ester, perchlorate (TMRE, Invitrogen) to a final concentration of 0.5 μM. Control wells with PBS ± TMRE were included on each plate. The plates were incubated for 30 min at 37 °C, 5% CO2 prior to removing the media and washing the cells two times with 0.2% BSA in PBS. Fluorescence was measured using a Tecan infinite M1000Pro multiplate reader (λex = 549 nm, λem = 575 nm). Relative fluorescence was determined: (sample − PBS−TMRE)/(PBS+TMRE − PBS−TMRE). Replicate plates were prepared where multiple time points were measured, and each plate contained at least three technical replicates for every peptide and control treatment.Non-linear regression
Dose response curves of percentage of vesicle leakage (Fig. 3B), pDI:MDM2/MDMX inhibition (Fig. 3C), cell toxicity (CC50 from Table 2), or fluorescent cells (Fig. 4B) were fitted using saturation binding with Hill slope, (%) = 100 × [peptide]H/(CH50 + [peptide]H), C50 is the peptide concentration required to reach 50% of leakage (LC50), inhibition (IC50), cell death (CC50), or fluorescent cells (FC50), and H is the Hill slope; SPR dose–response curves (Fig. 3A) were fitted using saturation binding with Hill slope (P/L) = P/Lmax[peptide]H/(KHD + [peptide]H), where P/Lmax is the peptide-to-lipid ratio at binding saturation, and KD is the peptide concentration required to reach half of P/Lmax; TMRE FI time-course curves (Fig. 6A) were fitted using one phase decay (FI = (FI0 − FI∞) × e−Kt + FI∞, where FI is the fluorescence emission signal of TMRE, FI0 is FI at time 0, FI∞ is FI at infinite time, K is the rate constant, and t is time). All curves were fitted using a least squares regression without weighting using Prism, GraphPad software, Inc.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council (FL150100146 to D. J. C., FT150100398 to S. T. H.) and the National Health and Medical Research Council, Australia (APP1084965 to D. J. C. and S. T. H.). G. J.-B. P. was supported by a University of Queensland Postgraduate Scholarship. Live cell microscopy (Spinning disc confocal microscopy) was performed at the Australian Cancer Research Foundation (ACRF)/Institute for Molecular Bioscience Cancer Biology Imaging Facility UQ, which was established with the support of the ACRF. The Translational Research Institute is supported by a grant from the Australian Government. The authors thank Dr R. Kapetanovic (Institute for Molecular Bioscience) for providing technical support and reagents for the western blot experiments.References
- IARC and WHO, International Agency for Research on Cancer. World Health Organisation, 2019.
- C. H. Chau, P. S. Steeg and W. D. Figg, The Lancet, 2019, 394, 793–804 CrossRef CAS.
- F. Araste, K. Abnous, M. Hashemi, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2018, 292, 141–162 CrossRef CAS.
- S. D. Vallabhapurapu, V. M. Blanco, M. K. Sulaiman, S. L. Vallabhapurapu, Z. Chu, R. S. Franco and X. Qi, Oncotarget, 2015, 6, 34375–34388 CrossRef.
- V. A. Fadok, D. R. Voelker, P. A. Campbell, J. J. Cohen, D. L. Bratton and P. M. Henson, J. Immunol., 1992, 148, 2207 CAS.
- M. Seigneuret and P. F. Devaux, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 3751–3755 CrossRef CAS.
- B. Sharma and S. S. Kanwar, Semin. Cancer Biol., 2018, 52, 17–25 CrossRef CAS.
- M. R. Yeaman and N. Y. Yount, Pharmacol. Rev., 2003, 55, 27–55 CrossRef CAS.
- H. Sato and J. B. Feix, Biochim. Biophys. Acta, 2006, 1758, 1245–1256 CrossRef CAS.
- S. T. Henriques, N. Lawrence, S. Chaousis, A. S. Ravipati, O. Cheneval, A. H. Benfield, A. G. Elliott, A. M. Kavanagh, M. A. Cooper, L. Y. Chan, Y. H. Huang and D. J. Craik, ACS Chem. Biol., 2017, 12, 2324–2334 CrossRef.
- F. Vernen, J. P. Harvey, A. S. Dias, S. A. Veiga, Y.-H. Huang, J. D. Craik, N. Lawrence and S. Troeira Henriques, Int. J. Mol. Sci., 2019, 20, 4184 CrossRef.
- M. Green and P. M. Loewenstein, Cell, 1988, 55, 1179–1188 CrossRef CAS.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, J. Biol. Chem., 1994, 269, 10444–10450 CAS.
- D. J. Mitchell, D. T. Kim, L. Steinman, C. G. Fathman and J. B. Rothbard, J. Pept. Res., 2000, 56, 318–325 CrossRef CAS.
- W. B. Kauffman, T. Fuselier, J. He and W. C. Wimley, Trends Biochem. Sci., 2015, 40, 749–764 CrossRef CAS.
- T. K. Sawyer, A. W. Partridge, H. Y. K. Kaan, Y.-C. Juang, S. Lim, C. Johannes, T. Y. Yuen, C. Verma, S. Kannan, P. Aronica, Y. S. Tan, B. Sherborne, S. Ha, J. Hochman, S. Chen, L. Surdi, A. Peier, B. Sauvagnat, P. J. Dandliker, C. J. Brown, S. Ng, F. Ferrer and D. P. Lane, Bioorg. Med. Chem., 2018, 26, 2807–2815 CrossRef CAS.
- D. J. Craik and J. Du, Curr. Opin. Chem. Biol., 2017, 38, 8–16 CrossRef CAS.
- B. J. McMorran, L. Wieczorski, K. E. Drysdale, J. A. Chan, H. M. Huang, C. Smith, C. Mitiku, J. G. Beeson, G. Burgio and S. J. Foote, Science, 2012, 338, 1348–1351 CrossRef CAS.
- N. Lawrence, A. S. M. Dennis, A. M. Lehane, A. Ehmann, P. J. Harvey, A. H. Benfield, O. Cheneval, S. T. Henriques, D. J. Craik and B. J. McMorran, Cell Chem. Biol., 2018, 25, 1140–1150 CrossRef CAS.
- J. P. Richard, K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik and B. Lebleu, J. Biol. Chem., 2003, 278, 585–590 CrossRef CAS.
- S. Eda and I. W. Sherman, Cell. Physiol. Biochem., 2002, 12, 373–384 CrossRef CAS.
- D. Engelbrecht and T. L. Coetzer, Parasitol. Int., 2016, 65, 715–727 CrossRef CAS.
- B. Hu, D. M. Gilkes and J. Chen, Cancer Res., 2007, 67, 8810 CrossRef CAS.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948 CrossRef CAS.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K.-H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445 CrossRef CAS.
- D. Fujiwara, H. Kitada, M. Oguri, T. Nishihara, M. Michigami, K. Shiraishi, E. Yuba, I. Nakase, H. Im, S. Cho, J. Y. Joung, S. Kodama, K. Kono, S. Ham and I. Fujii, Angew. Chem., Int. Ed., 2016, 55, 10612–10615 CrossRef CAS.
- G. Philippe, Y. H. Huang, O. Cheneval, N. Lawrence, Z. Zhang, D. P. Fairlie, D. J. Craik, A. D. de Araujo and S. T. Henriques, Biopolymers, 2016, 106, 853–863 CrossRef CAS.
- G. J. B. Philippe, D. Gaspar, C. Sheng, Y. H. Huang, A. H. Benfield, J. Weidman, N. Lawrence, A. Lower, M. A. R. B. Castanho, D. J. Craik and S. T. Henriques, ACS Chem. Biol., 2019, 14(9), 2071–2087 CrossRef CAS.
- G. H. Bird, E. Mazzola, K. Opoku-Nsiah, M. A. Lammert, M. Godes, D. S. Neuberg and L. D. Walensky, Nat. Chem. Biol., 2016, 12, 845–852 CrossRef CAS.
- S. T. Henriques, Y.-H. Huang, K. J. Rosengren, H. G. Franquelim, F. A. Carvalho, A. Johnson, S. Sonza, G. Tachedjian, M. A. R. B. Castanho, N. L. Daly and D. J. Craik, J. Biol. Chem., 2011, 286, 24231–24241 CrossRef CAS.
- Y. Ji, S. Majumder, M. Millard, R. Borra, T. Bi, A. Y. Elnagar, N. Neamati, A. Shekhtman and J. A. Camarero, J. Am. Chem. Soc., 2013, 135, 11623–11633 CrossRef CAS.
- I. M. Torcato, Y.-H. Huang, H. G. Franquelim, D. D. Gaspar, D. J. Craik, M. A. R. B. Castanho and S. T. Henriques, ChemBioChem, 2013, 14, 2013–2022 CrossRef CAS.
- S. T. Henriques, Y. H. Huang, S. Chaousis, M. A. Sani, A. G. Poth, F. Separovic and D. J. Craik, Chem. Biol., 2015, 22, 1087–1097 CrossRef CAS.
- L. C. Schenkel and M. Bakovic, Int. J. Cell Biol., 2014, 2014, 709828 Search PubMed.
- S. T. Henriques, E. Deplazes, N. Lawrence, O. Cheneval, S. Chaousis, M. Inserra, P. Thongyoo, G. F. King, A. E. Mark, I. Vetter, D. J. Craik and C. I. Schroeder, J. Biol. Chem., 2016, 291, 17049–17065 CrossRef CAS.
- R. Benz and S. McLaughlin, Biophys. J., 1983, 41, 381–398 CrossRef CAS.
- H. M. Ellerby, W. Arap, L. M. Ellerby, R. Kain, R. Andrusiak, G. D. Rio, S. Krajewski, C. R. Lombardo, R. Rao, E. Ruoslahti, D. E. Bredesen and R. Pasqualini, Nat. Med., 1999, 5, 1032–1038 CrossRef CAS.
- J. He, W. B. Kauffman, T. Fuselier, S. K. Naveen, T. G. Voss, K. Hristova and W. C. Wimley, J. Biol. Chem., 2013, 288, 29974–29986 CrossRef CAS.
- N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew. Chem., Int. Ed., 2015, 54, 8665–8668 CrossRef CAS.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS.
- N. E. Shepherd, H. N. Hoang, G. Abbenante and D. P. Fairlie, J. Am. Chem. Soc., 2005, 127, 2974–2983 CrossRef CAS.
- W. F. Vranken, W. Boucher, T. J. Stevens, R. H. Fogh, A. Pajon, P. Llinas, E. L. Ulrich, J. L. Markley, J. Ionides and E. D. Laue, Proteins: Struct., Funct., Bioinf., 2005, 59, 687–696 CrossRef CAS.
- D. S. Wishart, C. G. Bigam, A. Holm, R. S. Hodges and B. D. Sykes, J. Biomol. NMR, 1995, 5, 332 CrossRef CAS.
- S. T. Henriques, L. K. Pattenden, M. I. Aguilar and M. A. Castanho, Biochemistry, 2009, 48, 4198–4208 CrossRef CAS.
- J. C. M. Stewart, Anal. Biochem., 1980, 104, 10–14 CrossRef CAS.
- A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS.
- Y. Huang, Y. Liu, C. Zheng and C. Shen, PLoS One, 2017, 12, e0170384 CrossRef.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1. Comparison of peptide binding and lysis of model membranes composed of POPC and POPC/POPS (4:1). Fig. S1. Analytical HPLC trace for peptides showing peptide purity. Fig. S2. Mass determination of peptides from MS trace and m/z peaks. Fig. S3. Comparison of the peptide helicity and resistance to degradation in human serum. Fig. S4. CD spectra of peptides in aqueous and hydrophobic solutions. Fig. S5. Binding of PF4 scaffold and grafted peptides to model membranes. Fig. S6. Expression of p53 in HCT116 and MM96L cells following treatment with peptides. Fig. S7. Internalization of A488-labeled peptides into the cytoplasm of MM96L and HaCaT cells without damaging the cell membrane. Fig. S8. Colocalization of cPF4PD-A488 with mitochondrial membranes of MDA-MB-231 cells. Movie S1: Internalization of 6 μM PF4_pDI in MM96L cells (full time course 1 frame per s). See DOI: 10.1039/d0cb00099j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |