
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMet80 and Tyr67 affect the chemical unfolding of yeast cytochrome c: comparing the solution vs. immobilized state
Alessandro
Paradisi
 a,
Lidia
Lancellotti
b,
Marco
Borsari
b,
Marzia
Bellei
c,
Carlo Augusto
Bortolotti
c,
Giulia
Di Rocco
c,
Antonio
Ranieri
c,
Marco
Sola
c and
Gianantonio
Battistuzzi
*b
a,
Lidia
Lancellotti
b,
Marco
Borsari
b,
Marzia
Bellei
c,
Carlo Augusto
Bortolotti
c,
Giulia
Di Rocco
c,
Antonio
Ranieri
c,
Marco
Sola
c and
Gianantonio
Battistuzzi
*b
aDepartment of Chemistry, University of York, Heslington, YO10 5DD, York, UK
bDepartment of Chemistry and Geology, University of Modena and Reggio Emilia, via Campi 103, 41126 Modena, Italy. E-mail: gianantonio.battistuzzi@unimore.it; Tel: +39-0592058639
cDepartment of Life Sciences, University of Modena and Reggio Emilia, via Campi 103, 41126 Modena, Italy
First published on 9th September 2020
Abstract
Urea-induced denaturation of the Met80Ala and Met80Ala/Tyr67Ala variants of S. cerevisiae iso-1 cytochrome c (ycc) was studied through variable temperature diffusive cyclic voltammetry and electronic absorption, CD and MCD spectroscopies. The susceptibility to unfolding of both variants – represented by the free energy of unfolding at denaturant infinite dilution, 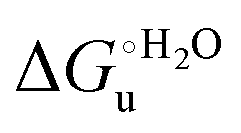 – is greater compared to the species showing an intact Met/His coordination, as observed previously for the same species immobilized onto a functionalized electrode. This is consistent with the role of the axial Fe–(S)Met bond and the H-bond network involving Tyr67 in stabilizing the polypeptide matrix in the heme crevice. Notably, we find that the unfolding propensity and axial heme iron coordination of the present Fe–(S)Met bond-deprived variants are affected by the motional regime of the protein. In particular, electrostatic adsorption onto a negatively charged SAM surface – which would mimic the phospholipidic inner mitochondrial membrane – facilitates unfolding compared to the solution state, especially at room temperature. This finding has physiological relevance related to the cytochrome c interaction with cardiolipin at the IMM in the early stages of apoptosis. Moreover, while both immobilized variants maintain the His/OH− axial heme iron coordination up to 7 M urea, the same species in solution are subjected to urea-induced replacement of the axial hydroxide ligand by a His ligand. The contributions of the enthalpic and entropic terms to
– is greater compared to the species showing an intact Met/His coordination, as observed previously for the same species immobilized onto a functionalized electrode. This is consistent with the role of the axial Fe–(S)Met bond and the H-bond network involving Tyr67 in stabilizing the polypeptide matrix in the heme crevice. Notably, we find that the unfolding propensity and axial heme iron coordination of the present Fe–(S)Met bond-deprived variants are affected by the motional regime of the protein. In particular, electrostatic adsorption onto a negatively charged SAM surface – which would mimic the phospholipidic inner mitochondrial membrane – facilitates unfolding compared to the solution state, especially at room temperature. This finding has physiological relevance related to the cytochrome c interaction with cardiolipin at the IMM in the early stages of apoptosis. Moreover, while both immobilized variants maintain the His/OH− axial heme iron coordination up to 7 M urea, the same species in solution are subjected to urea-induced replacement of the axial hydroxide ligand by a His ligand. The contributions of the enthalpic and entropic terms to 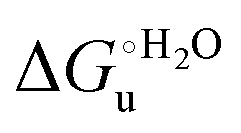 were found to be opposite (H–S compensation), indicating that the unfolding thermodynamics are strongly affected by changes in the hydrogen bonding network in the hydration sphere of the protein.
were found to be opposite (H–S compensation), indicating that the unfolding thermodynamics are strongly affected by changes in the hydrogen bonding network in the hydration sphere of the protein.
Introduction
Mitochondrial cytochrome c (cytc) is a small globular protein which shuttles electrons between cytochrome c reductase and cytochrome c oxidase in the mitochondrial intermembrane space.1–7 It contains a single six-coordinate heme c wedged into a hydrophobic environment, which is axially coordinated by His18 and Met80.1–7 The solution properties and binding events affect the heme environment and axial ligation,1,3–5,8–27 modifying the physiological role in vivo and the reactivity in vitro.1,5,9,13–16,18,19,28–40 Indeed, cytochrome c has proven to be a multi-tasking protein whose biological role is modulated by external stimuli and cell conditions.1,4,28,30,41 The paradigm of this tunable functionality in vivo is the interaction with cardiolipin (CL), a negatively charged glycerophospholipid found in the inner mitochondrial membrane (IMM),30,40,42 which induces the cleavage of the Fe–S(Met80) bond,1,5,9,19,21,28,30,32,39,42–72 imparting to cytc significant (lipo)peroxidase activity, which is crucial in triggering the apoptosis cascade.1,5,28,30,40,47,73In vitro, analogous heme axial ligand swapping from His/Met to His/His occurs upon chemical denaturation of both solution14 and surface-immobilized yeast cytochrome c (ycc).14,33,34,74–77 Under the latter conditions, ycc gains remarkable pseudo-peroxidase activity.32,34,75–77Because of their physiological relevance, the conformational equilibria leading to non-native states of cytochrome c have been studied in depth,1,6,7,14,18,22,24–27,41,43,78–80 making it a model system widely used to unravel the mechanistic details of protein folding and unfolding.5,8,9,22,23,25,38,41,78,81–84 In this work, we used a combination of spectroscopic and electrochemical techniques to analyze the urea-induced unfolding of the M80A and M80A/Y67A variants of ycc under freely diffusing conditions. The former mutation was selected to quantitatively assess the contribution of the distal axial heme ligation to the overall conformational stability of cytochrome c, whereas the evolutionary conserved Tyr67 was chosen because its OH group participates in the hydrogen bond (H-bond) network in the distal side of the heme connecting the Ω loops formed by residues 40–57 and 71–85 (Fig. 1),85–96 which are the least stable foldons.78,79 Therefore, Tyr67 plays a key role in stabilizing the three-dimensional structure of folded ycc and controlling the solvent accessibility to the heme crevice.85–96 Moreover, Tyr67 was indicated as a possible apoptotic trigger.85
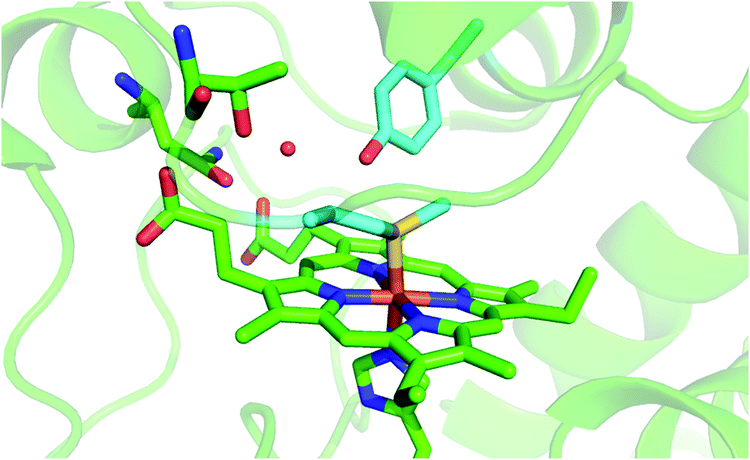 | ||
| Fig. 1 Cartoon representing the heme environment in wild type yeast cytochrome c (PDB 3CYT) and highlighting the Met80 and Tyr67 residues in light blue. | ||
Previous studies showed that removal of the axial Met ligand enhances the pseudoperoxidase and nitrite reductase activity97–99 and facilitates urea-induced unfolding of the SAM-immobilized proteins, preventing coordination of a second axial His ligand at high urea concentrations,77 whereas suppression of Tyr67 induced significant urea-induced changes in protein solvation.77 Moreover, axial heme iron ligation, the protein conformation, the solvation properties, the heme reduction potential and the pseudoperoxidase and nitrite reductase activity of immobilized ycc were found to be affected by the nature of the noncovalent protein–SAM interaction (either electrostatic or hydrophobic).76,77 In this work, we compare the thermodynamics of urea-induced denaturation of ycc in solution versus the immobilized state to test the effect of constrained protein mobility on the susceptibility of ycc to chemical unfolding. We find that such a constraint favors unfolding. This finding recognizes an additional factor that contributes to the functional versatility and tunability of cytochrome c under physiological conditions.
Experimental
Materials
All chemicals were reagent grade. Doubly distilled water was used throughout. 4-Mercapto-pyridine was purchased from Sigma-Aldrich and used without further purification.Protein production and isolation
The M80A and M80A/Y67A mutants of recombinant S. cerevisiae iso-1 cytochrome c were expressed and isolated as described previously.95,97–100 All protein variants are nontrimethylated and carry the C102T mutation to prevent protein dimerization.95,97–100Spectroscopic measurements
Electronic absorption, CD and MCD spectra were recorded with a Jasco J-810 spectropolarimeter. The magnetic field was provided by a GMW magnet system Model 3470 split coil superconductivity magnet with a maximum field of 1 Tesla (T). Both CD and MCD spectra were measured in θ = mdeg. The former were converted to molar ellipticity [θ] using the conversion factor [θ] = θ(deg)·100/(d·c), where c is the protein concentration (mol dm−3) and d is the thickness of the sample (path length, 0.5 cm),21,84,90 while the latter were converted to Δε [M−1 cm−1 T−1] using the conversion factor Δε = θ/(32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 980·c·d·B), where c is the protein concentration, B is the magnetic field (1 T), and d is the thickness of the sample (path length, 0.5 cm).13,101–104 All experiments were carried out at 25 °C with protein solutions freshly prepared before use in 5 mM phosphate buffer pH 7 and the protein concentration was checked spectrophotometrically, using ε405 = 121700 M−1 cm−1 for both the M80A and M80A/Y67A mutants.105,106
980·c·d·B), where c is the protein concentration, B is the magnetic field (1 T), and d is the thickness of the sample (path length, 0.5 cm).13,101–104 All experiments were carried out at 25 °C with protein solutions freshly prepared before use in 5 mM phosphate buffer pH 7 and the protein concentration was checked spectrophotometrically, using ε405 = 121700 M−1 cm−1 for both the M80A and M80A/Y67A mutants.105,106
Electrochemical measurements
Cyclic voltammetry (CV) measurements were carried out with a potentiostat/galvanostat mod. 273A (EG&G PAR, Oak Ridge, USA). Experiments were carried out at different scan rates (0.02–5 V s−1) using a cell for small volume samples (0.5 mL) under argon. A polycrystalline gold wire functionalized with 4-mercapto-pyridine,12,107 a platinum sheet, and a saturated calomel electrode (SCE) were used as the working, counter, and reference electrodes, respectively. The electric contact between the SCE and the working solution was achieved with a Vycor® (from PAR) set. Reduction potentials were calibrated against the ferrocene/ferrocenium couple under all experimental conditions employed in this work to make sure that the effects of liquid junction potentials were negligible. All reduction potentials reported here are given with reference to the standard hydrogen electrode (SHE). Protein solutions were freshly prepared before use in 10 mM phosphate buffer plus 100 mM sodium perchlorate at pH 7.2 and their concentration was carefully checked spectrophotometrically (with a Jasco mod. V-570 spectrophotometer). The urea concentration was varied between 0 and 6 M. The formal potentials E°′ were calculated as the semisum of the anodic and cathodic peak potentials and were found to be almost independent of the scan rate in the range 0.02–2 V s−1. The signals persist for several voltammetric cycles throughout the temperature range investigated. The experiments were performed at least two times and the E°′ values were found to be reproducible within ±0.002 V. The current intensities are proportional to the square root of the scan rate, as expected for a diffusive electrochemical process (not shown).Variable-temperature CV experiments were carried out using a “non-isothermal” cell, in which the reference electrode was kept at a constant temperature (21 ± 0.1 °C), whereas the half-cell containing the working electrode and the Vycor® junction to the reference electrode was under thermostatic control with a water bath.108–113 The temperature was varied from 5 to 35 °C. With this experimental configuration, the standard entropy change for heme Fe(III) to Fe(II) reduction in ycc 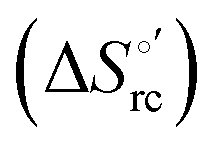 is given by:108–110
is given by:108–110
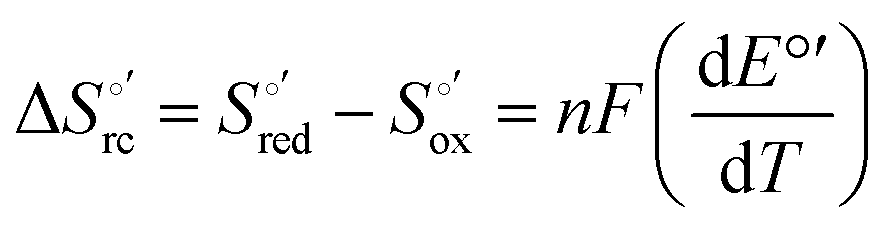 | (1) |
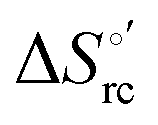 was determined from the slope of the plot of E°′ versus temperature, which turns out to be linear under the assumption that
was determined from the slope of the plot of E°′ versus temperature, which turns out to be linear under the assumption that 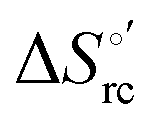 is constant over the limited temperature range investigated. With the same assumption, the enthalpy change
is constant over the limited temperature range investigated. With the same assumption, the enthalpy change 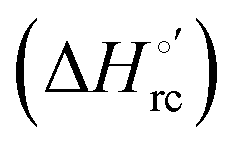 was obtained from the Gibbs–Helmholtz equation, namely as the negative slope of the E°′/T versus 1/T plot.11,111–113 The nonisothermal behavior of the cell was carefully checked by determining the
was obtained from the Gibbs–Helmholtz equation, namely as the negative slope of the E°′/T versus 1/T plot.11,111–113 The nonisothermal behavior of the cell was carefully checked by determining the 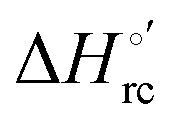 and
and 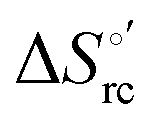 values of the ferricyanide/ferrocyanide couple.110–113
values of the ferricyanide/ferrocyanide couple.110–113
Results and discussion
Absorption and MCD spectra
The electronic absorption and MCD spectra of ferric M80A and M80A/Y67A in the Soret (360–450 nm) and in the visible (450–710 nm) regions at pH 7 in the absence of urea (Fig. 2 and 3 and Table 1) match those reported previously.102 The spectra point to a 6-coordinate His/OH− low spin (LS1) form as the major species.76,77,98,99,102,105,106,114 However, the shoulder at 398 nm observed in the 2nd derivative absorption spectra of both proteins (Fig. 2c and d) indicates that a minor high-spin (HS1) form is also present.13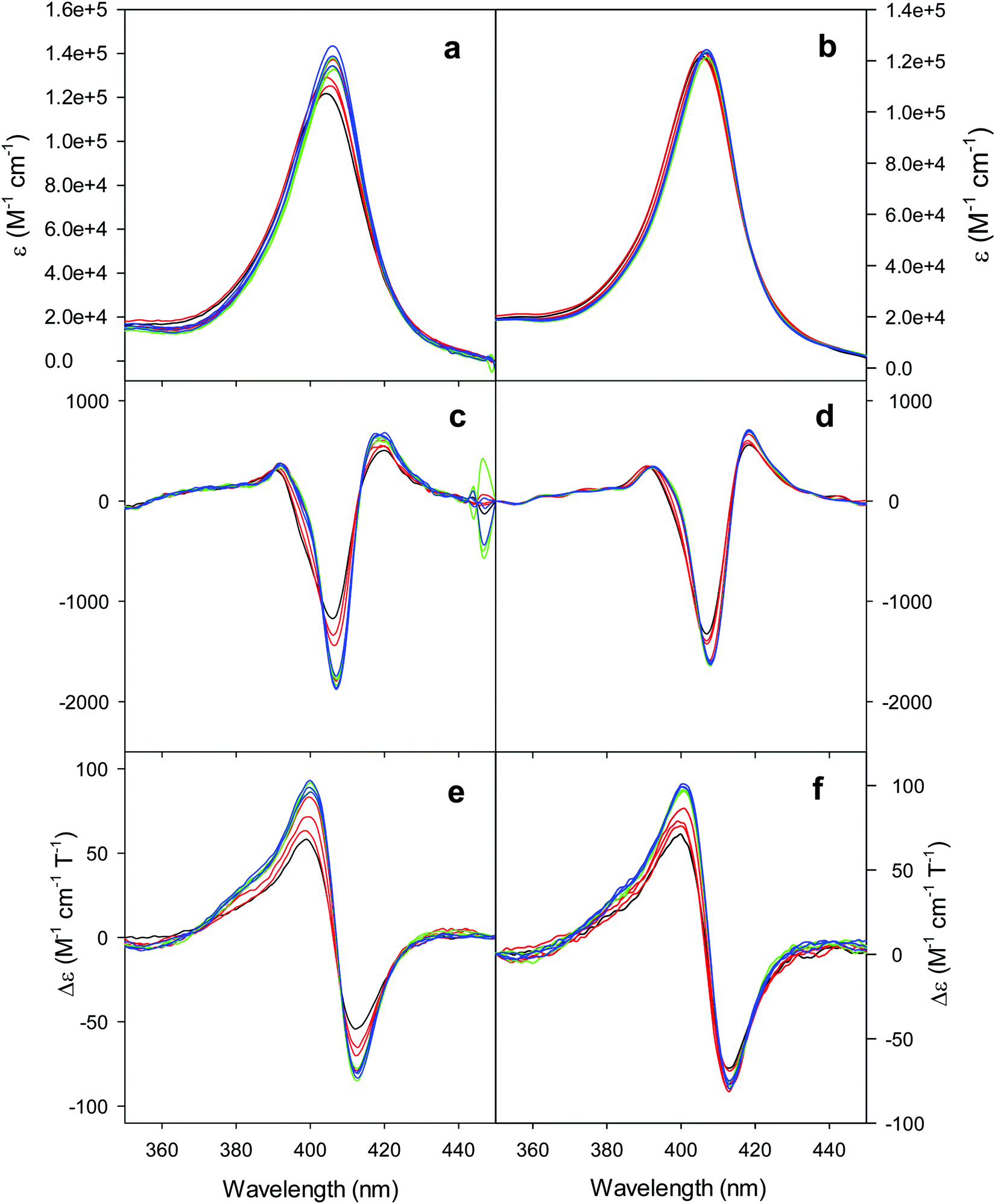 | ||
| Fig. 2 Electronic absorption, 2nd derivative electronic absorption and MCD spectra in the Soret region for the oxidized M80A (a, c and e) and M80AY67A (b, d and f) variants of S. cerevisiae iso-1 cytochrome c in 5 mM phosphate buffer at pH 7 in the presence of increasing urea concentration: 0 M (black); 1, 2, and 3 M (red); 4, 5, and 6 M (green); and 7, 8, and 9 M (blue). | ||
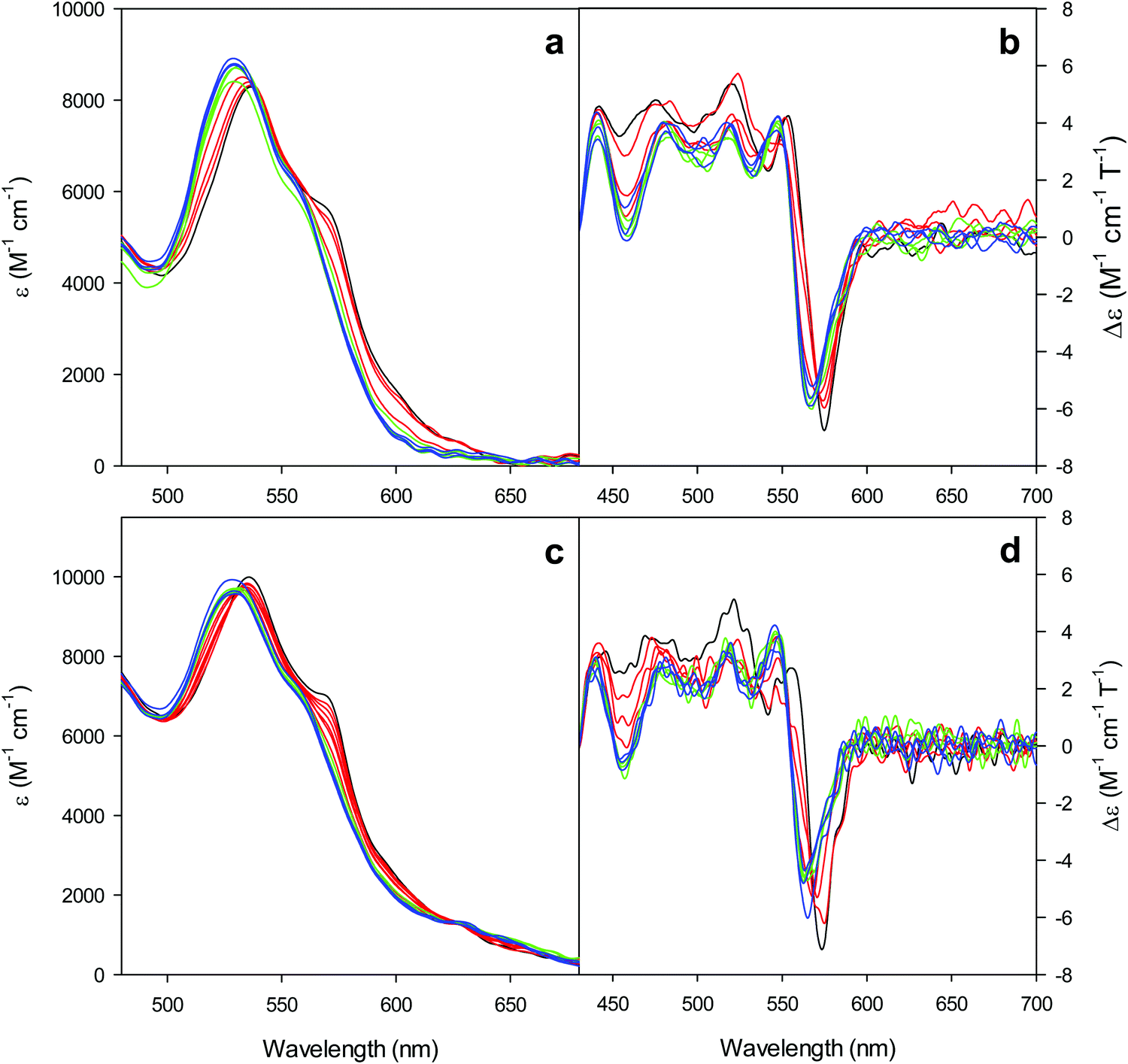 | ||
| Fig. 3 Electronic absorption and MCD spectra in the visible region for the oxidized M80A (a and b) and M80AY67A (c and d) variants of S. cerevisiae iso-1 cytochrome c in 5 mM phosphate buffer at pH 7 in the presence of increasing urea concentration: 0 M (black); 1, 2, and 3 M (red); 4, 5, and 6 M (green); and 7, 8, and 9 M (blue). | ||
| Species | [Urea] | MCD | Absorption | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Soret | Vis | Soret | Vis | |||||||
| Peak | Trough | Zerocross | Peak | Trough | Zerocross | Peak | 2nd derivative | |||
| M80A | 0 | 399 | 413 | 406 | 553 | 575, 542 | 562 | 404 | 406, 398(sh) | 533, 565 |
| 4 | 400 | 413 | 407 | 548 | 568, 532 | 556 | 406 | 407 | 528, 555 | |
| 9 | 400 | 413 | 407 | 547 | 568, 533 | 556 | 406 | 407, 398(sh) | 528, 555 | |
| M80A/Y67A | 0 | 398 | 412 | 405 | 555 | 573 | 566 | 405 | 407, 398(sh) | 532, 562 |
| 5 | 399 | 412 | 406 | 547 | 565 | 555 | 407 | 408 | 526, 555 | |
| 9 | 398 | 412 | 406 | 548 | 563 | 555 | 407 | 408 | 526, 555 | |
For both variants, the Soret band in the absorption spectra and the single trough in the corresponding 2nd derivative spectra slightly redshift upon urea addition (Fig. 2a–d and Table 1), whereas the shoulder in the 2nd derivative spectra at 398 nm disappears above 4 M urea (Fig. 2c and d and Table 1).
The symmetrical S-shaped MCD signal is associated with the Soret band. The Soret band corresponds to a π → π* electronic transition of the porphyrin ring, which would be doubly degenarate under the ideal D4h symmetry of the porphyrin system (acceptor π* orbitals of eg symmetry).115 However, the asymmetry of the protein environment around the heme lifts this degeneracy, producing two different electronic transitions close in energy.116 In the MCD spectra of low spin ferric hemes these two transions gain intensity through the C-term mechanism but have opposite signs and hence generate the characteristic S-shaped MCD signal of the Soret band.117,118 The position and the overall shape of this MCD feature are not influenced by urea addition (Fig. 2e and f and Table 1), yet the peak-to-trough distance increases up to 4 M and 5 M urea for M80A and M80A/Y67A, respectively (Fig. 2e and f).
Furthermore, the α and β bands of both variants shift to shorter wavelengths (Fig. 3a and c and Table 1) and their intensity increases up to 5 M urea (Fig. 3a and c); the trough in the MCD spectra at 575 (M80A) and 573 (M80A/Y67A) nm is progressively replaced by a new trough at 568 and 565 nm, respectively, whereas the peak at 553 nm is replaced by a new peak at 547 nm (Fig. 3b and d and Table 1). No further spectral changes are observed above 4 M urea for M80A, while M80A/Y67A displays an intensity decrease above 7 M urea (Fig. 3). These spectral changes show that the major low-spin His/OH−-ligated form (LS1) and the minor high-spin form (HS1) observed in the absence of urea both transform into a second LS conformer (LS2) above 4 M urea (M80A) and 5 M urea (M80A/Y67A), whose spectroscopic features are consistent with the replacement of the axial hydroxide ligand by a His ligand (either His26 or His33),1,19,27,33,34,39,43,44,62,66,119 as previously observed for wt ycc and its K72A/K73A/K79A mutant.14,21 Hence, the behavior of freely diffusing M80A and M80A/Y67A markedly differs from that of the same species electrostatically immobilized on a MUA/MU SAM, which showed no change in the His/OH− axial heme iron coordination up to 7 M urea.77 The absence of further spectral changes at higher urea concentrations indicates that the His/His axial coordination of the LS2 conformer of both mutants is remarkably stable.
The urea concentration that realizes half of the total absorbance changes at 570 and 520 nm in Fig. 4 (which would correspond to a 50% population of LS1 and LS2) is about 2.2–2.3 M for both variants. These values are significantly lower than those reported for wt ycc (3.2 M and 3.5 M, at 25 °C and 5 °C, respectively14,35) and its K72A/K73A/K79A and K72A/K73H/K79A mutants (3.2 M and 3.1 M) at 5 °C,14,75 indicating that removal of the Fe—Met80 bond favors the unfolding effect of urea. Hence, reduction of the structural constraints that connect the heme center to the polypeptide matrix due to removal of the Fe–Met80 bond lowers the protein resistance to chemical unfolding as found previously for the same immobilized on a MUA–MU SAM. This view is supported by the thermodynamics of unfolding (vide infra). The additional alteration of the network of H-bonding in the heme crevice due to the suppression of Tyr67 apparently exerts a negligible effect on the resistance of the freely diffusing protein to chemical unfolding.
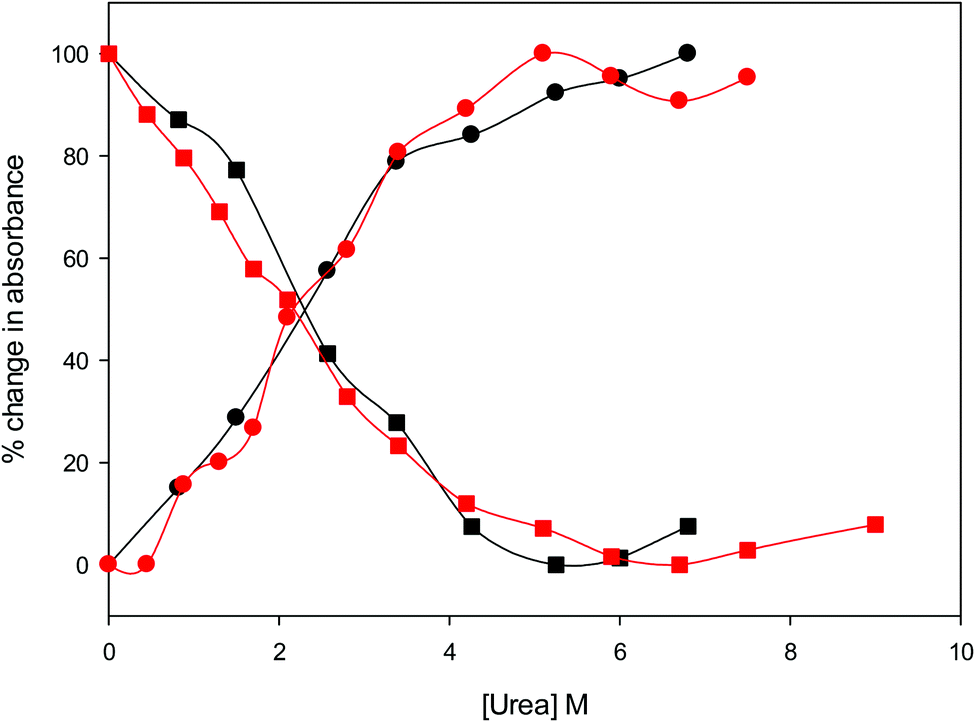 | ||
| Fig. 4 Relative change in absorbance at 520 nm (circles) and 570 nm (squares) for the oxidized M80A (black) and M80AY67A (red) variants of S. cerevisiae iso-1 cytochrome c in 5 mM phosphate buffer at pH 7 in the presence of increasing urea concentration. | ||
Near-UV CD spectra
The near-UV (250–330 nm) CD spectrum of proteins consists of optically active heme transitions120 and vibronic transition bands of the aromatic side chains.121 This technique is particularly sensitive to structural changes in the environment of aromatic side chains in cytochromes c.21,84,90,122–124 The CD spectrum of wt ferric ycc contains a number of small positive bands around 256 nm, arising from tyrosine side chains,21 a broad positive band around 264 nm, attributed to porphyrin transitions,122 and two sharp negative bands at 282 and 289 nm, assigned to transitions involving the Trp-59 side chain.21,84,122 The near-UV CD spectra of M80A and M80A/Y67A (Fig. 5) are almost superimposable on that of wt ycc, indicating that neither deletion of the axial Met80 ligand nor changes in the H-bond network surrounding the heme induced by Tyr67 replacement significantly modify the protein folding.21,84,90 Urea addition simplifies the CD spectrum of both mutants (Fig. 5), as the two sharp negative bands at 282 and 289 nm progressively decrease in intensity and disappear above 4 M urea, while the two maxima at 255 and 259 nm are replaced by a more intense maximum around 258 nm. These changes suggest that disruption of the tight packing of core residues occurs in both mutants upon increasing the urea concentration up to 5 M. No significant changes are observed at higher urea concentrations.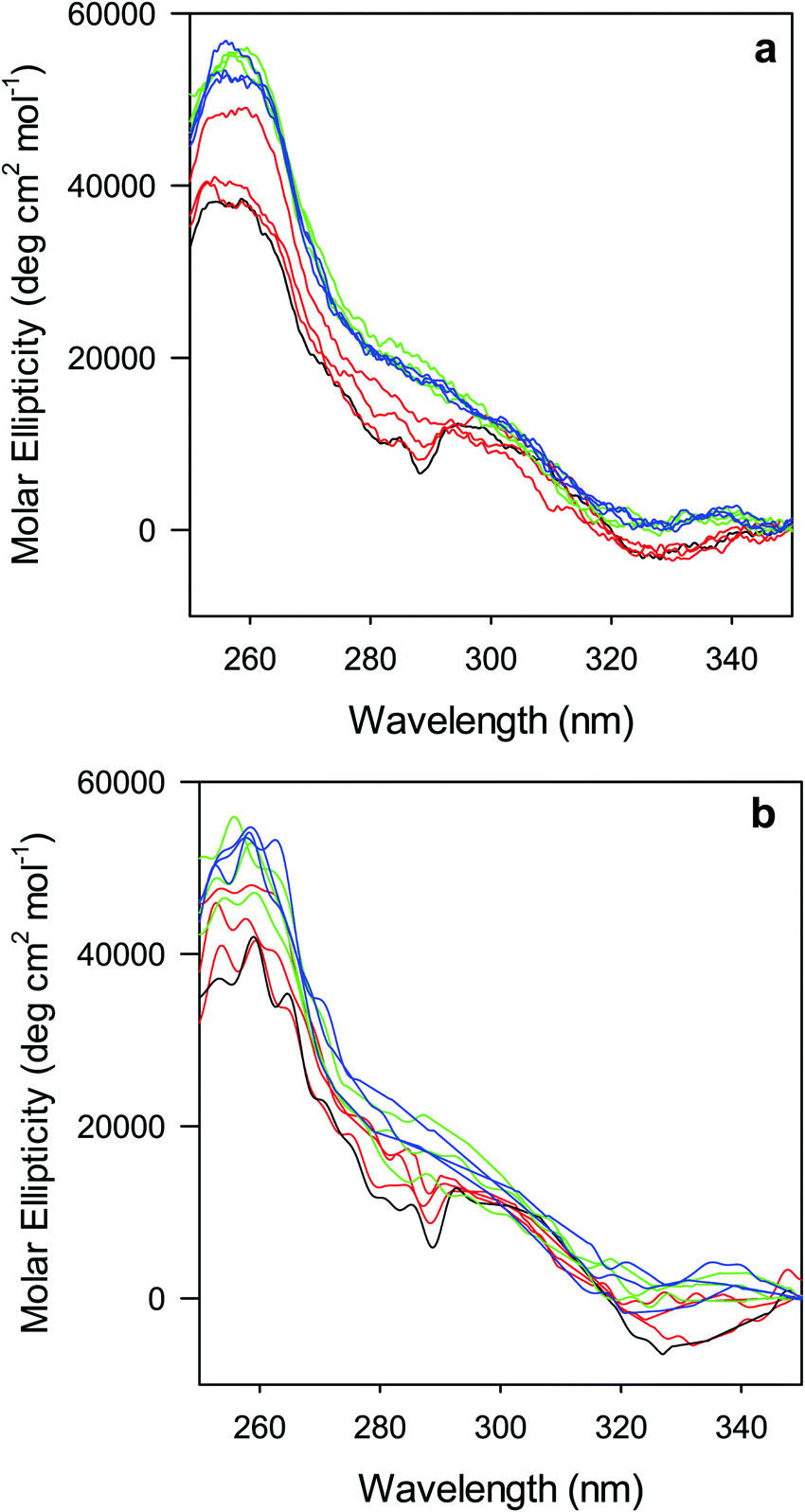 | ||
| Fig. 5 Near UV CD spectra for the oxidized M80A (a) and M80AY67A (b) variants of S. cerevisiae iso-1 cytochrome c in 5 mM phosphate buffer at pH 7 in the presence of increasing urea concentration: 0 M (black); 1, 2, and 3 M (red); 4, 5, and 6 M (green); and 7, 8, and 9 M (blue). | ||
Voltammetric response
The cyclic voltammograms for freely diffusing M80A and M80A/Y67A at pH 7.2 show quasi-reversible signals arising from the one-electron reduction/oxidation of the heme iron (Fig. 6). The E°′ values of −0.170 and −0.196 V obtained at pH 7.2 and 293 K for M80A and M80A/Y67A, respectively (Table 2), compare well with previous data for the same species immobilized on a polycrystalline gold electrode coated with a 1:1 mixed SAM of MUA and MU,77,98,99 if we take into account the electrostatic stabilization of the ferric form by the negatively charged SAM.98,99,125 Both the freely diffusing and immobilized variants feature a His/OH− axial heme iron coordination set.76,77,97–99 The voltammetric response and the temperature dependence of E°′ were studied at varying urea concentration up to 6 M in the working solution. A typical CV is shown in Fig. 6. Urea addition does not perturb the quasi-reversible electrochemical response but induces an anodic shift of E°′ up to 0.024 V and 0.046 V for M80A and M80A/Y67A, respectively, at 6 M urea (Fig. 7 and Table 2). As no further E°′ and spectroscopic changes occur at larger urea concentration, 6 M urea is taken as the condition that yields the fully unfolded ycc form. This urea-induced anodic shift is larger than that previously found for the same species immobilized on MUA–MU (0.011 V for M80A and 0.022 V for M80A/Y67A), which SERRS data showed to retain the His/OH− axial heme iron coordination.77 This fits with the urea-induced replacement of the axial hydroxide ion with an as yet unknown histidine residue to yield a bis-His axial ligand set. Indeed, the E°′ values of both variants in 6 M urea are similar to those determined for urea unfolded wt ycc and its K72A/K73A/K79A mutant,14 which were shown to possess His/His axial coordination.
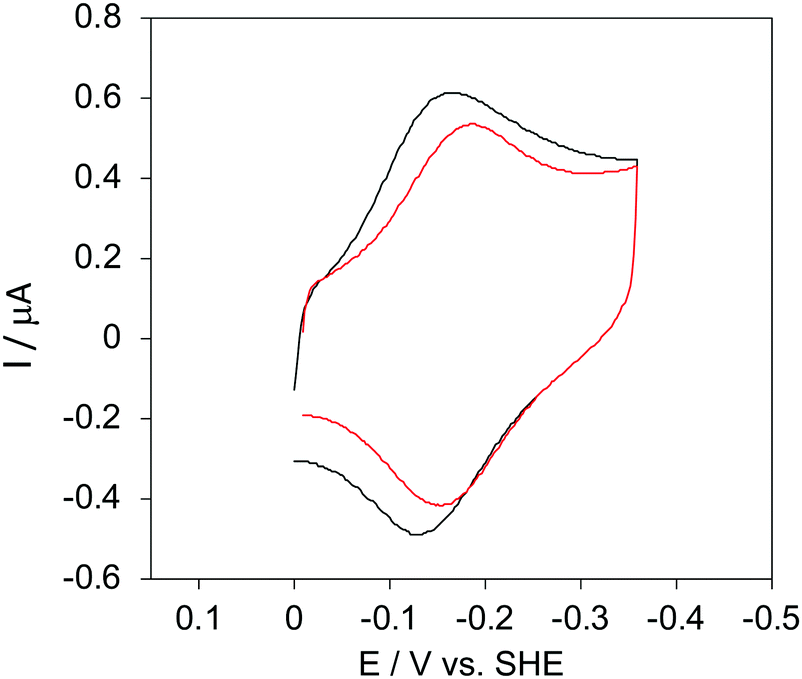 | ||
| Fig. 6 Cyclic voltammograms for the M80A variant of yeast iso-1-cytochrome c in the presence of varying urea concentrations at pH 7.2 under diffusive conditions. 0 M urea (red) and 6 M urea (black). Working electrode: polycrystalline gold wire functionalized with 4-mercapto-pyridine. Working solution: 10 mM phosphate buffer plus 100 mM sodium perchlorate, pH 7.2. T = 293 K. Sweep rate: 0.5 V s−1. Similar CVs were obtained for the M80A/Y67A variant. | ||
| [Urea] (M) | M80A | M80AY67A | ||||
|---|---|---|---|---|---|---|
| E°′b (V) |
|
|
E°′b (V) |
|
|
|
a Working electrode: polycrystalline gold electrode coated with a SAM of 4-mercapto-pyridine; working solution: 10 mM phosphate buffer plus 100 mM sodium perchlorate at pH 7.2. E°′ is measured at T = 20 °C.
b The average errors on E°′, 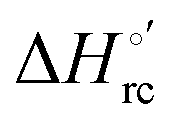 and and 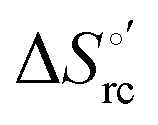 are ±0.002 V, ±0.3 kJ mol−1 and ±2 J mol−1 K−1, respectively. are ±0.002 V, ±0.3 kJ mol−1 and ±2 J mol−1 K−1, respectively.
|
||||||
| 0 | −0.170 | 41.8 | 85 | −0.196 | 37.6 | 63 |
| 1 | −0.165 | 37.5 | 72 | −0.187 | 36.4 | 61 |
| 2 | −0.157 | 34.3 | 64 | −0.181 | 34.7 | 59 |
| 3 | −0.155 | 32.3 | 59 | −0.170 | 33.1 | 57 |
| 4 | −0.152 | 29.8 | 51 | −0.162 | 31.8 | 55 |
| 5 | −0.147 | 28.0 | 46 | −0.156 | 30.8 | 53 |
| 6 | −0.146 | 26.9 | 43 | −0.150 | 30.0 | 52 |
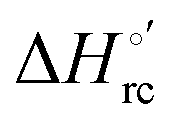
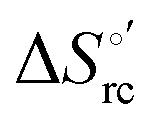
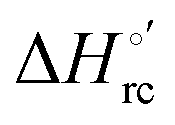
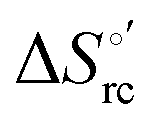
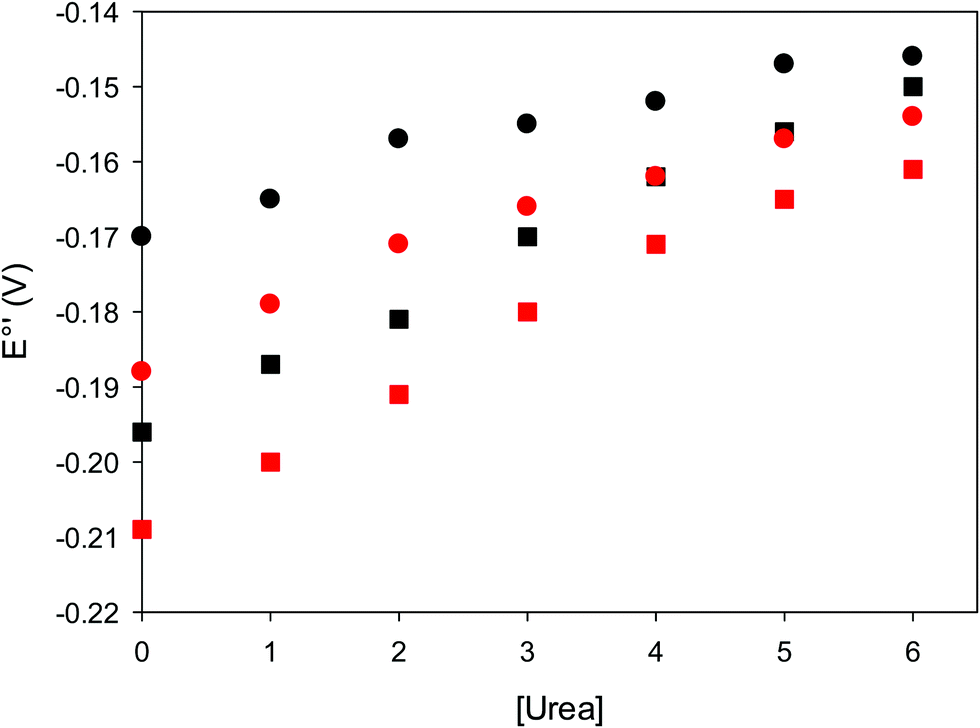 | ||
| Fig. 7 Urea-induced changes in E°′ for the M80A (circles) and M80AY67A (squares) variants of S. cerevisiae iso-1 cytochrome c at 5 °C (red) and 25 °C (black). Working electrode: polycrystalline gold wire functionalized with 4-mercapto-pyridine. 10 mM phosphate buffer plus 100 mM sodium perchlorate, pH 7.2. | ||
Thermodynamics of heme iron reduction
Valuable information on the mechanism of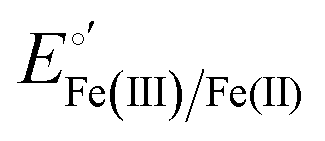 modulation in heme proteins has been obtained from the enthalpy
modulation in heme proteins has been obtained from the enthalpy 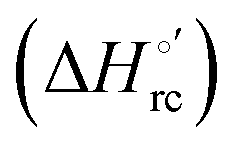 and entropy
and entropy 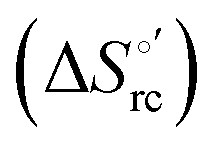 changes accompanying the Fe3+ → Fe2+ reduction, determined by analyzing the temperature dependence of E°′.14,74–77,103,109–111,126–147 These thermodynamic data contain contributions from protein-based “intrinsic” factors
changes accompanying the Fe3+ → Fe2+ reduction, determined by analyzing the temperature dependence of E°′.14,74–77,103,109–111,126–147 These thermodynamic data contain contributions from protein-based “intrinsic” factors 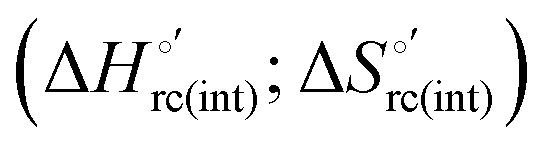 and solvent reorganization effects within the hydration sphere of the molecule
and solvent reorganization effects within the hydration sphere of the molecule 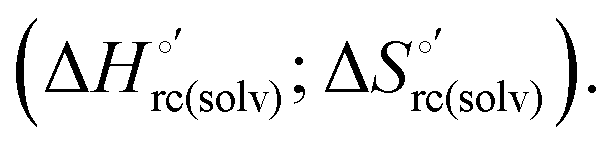 33,74,75,103,126,128–133,135,138,139,145,146,148,149 The intrinsic enthalpic contribution depends on the donor properties of the axial heme ligands, the polarity and the electrostatics at the redox center, whereas the intrinsic entropic term is mainly controlled by oxidation-state dependent differences in the conformational degrees of freedom of the polypeptide chain.74–77,98,99,103,126,128–133,135,137–139,144–149 For both variants, the E°′ values increase linearly with increasing temperature with and without urea (Fig. 8a and b) and both display positive
33,74,75,103,126,128–133,135,138,139,145,146,148,149 The intrinsic enthalpic contribution depends on the donor properties of the axial heme ligands, the polarity and the electrostatics at the redox center, whereas the intrinsic entropic term is mainly controlled by oxidation-state dependent differences in the conformational degrees of freedom of the polypeptide chain.74–77,98,99,103,126,128–133,135,137–139,144–149 For both variants, the E°′ values increase linearly with increasing temperature with and without urea (Fig. 8a and b) and both display positive 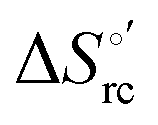 and
and 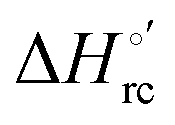 values (Table 2). Hence, the enthalpic contribution disfavors Fe(III) reduction and is the main determinant of the negative E°′ values, while Fe(III) reduction is favored entropically. In the absence of urea, the enthalpic stabilization of the oxidized heme in both mutants is largely the result of the strong electron donor character of the hydroxide ion and the mutation-induced increase in the exposure of the heme center to solvent resulting in greater solvation of the heme–protein interface.76,77,98,99,147 The positive reduction entropy of both variants is consistent with the electrostatically driven increase in the disorder of the water molecules in the heme cavity due to the reduction of the net charge of the heme group (from 1+ to zero) upon Fe(III) reduction. A further contribution conceivably arises from the reduction-induced release of the axial hydroxide ligand.76,77,98,99,147 Upon increasing the urea concentration, the E°′ values increase while the reduction enthalpy and entropy values decrease. The following changes occur from 0 to 6 M urea for M80A and M80AY67A, respectively (Table 2): ΔE°′ = 0.024 and 0.046 V,
values (Table 2). Hence, the enthalpic contribution disfavors Fe(III) reduction and is the main determinant of the negative E°′ values, while Fe(III) reduction is favored entropically. In the absence of urea, the enthalpic stabilization of the oxidized heme in both mutants is largely the result of the strong electron donor character of the hydroxide ion and the mutation-induced increase in the exposure of the heme center to solvent resulting in greater solvation of the heme–protein interface.76,77,98,99,147 The positive reduction entropy of both variants is consistent with the electrostatically driven increase in the disorder of the water molecules in the heme cavity due to the reduction of the net charge of the heme group (from 1+ to zero) upon Fe(III) reduction. A further contribution conceivably arises from the reduction-induced release of the axial hydroxide ligand.76,77,98,99,147 Upon increasing the urea concentration, the E°′ values increase while the reduction enthalpy and entropy values decrease. The following changes occur from 0 to 6 M urea for M80A and M80AY67A, respectively (Table 2): ΔE°′ = 0.024 and 0.046 V, 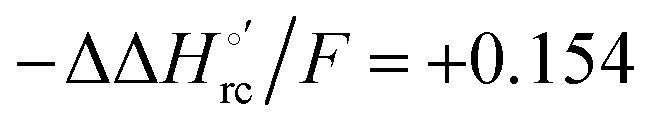 and +0.079 V,
and +0.079 V, 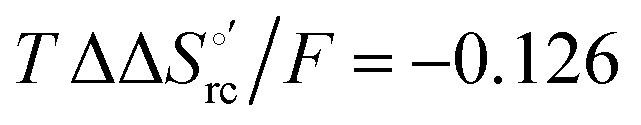 and −0.033 V (at 293 K). Therefore, the change in the entropic term opposes but does not offset the enthalpic stabilization of the reduced form due to urea-induced unfolding and axial OH− for His ligand swapping. Such a compensatory effect has been thoroughly described for several events in biomolecules and has been the subject of several theories and controversies.18,20,74,76,77,133,146,150–164 For redox processes involving metal centers in proteins it can be considered the hallmark of the reduction-induced reorganization of the hydrogen bonding network within the hydration sphere of the molecule.20,74,76,77,133,146,150–152,162,165–167 The linear
and −0.033 V (at 293 K). Therefore, the change in the entropic term opposes but does not offset the enthalpic stabilization of the reduced form due to urea-induced unfolding and axial OH− for His ligand swapping. Such a compensatory effect has been thoroughly described for several events in biomolecules and has been the subject of several theories and controversies.18,20,74,76,77,133,146,150–164 For redox processes involving metal centers in proteins it can be considered the hallmark of the reduction-induced reorganization of the hydrogen bonding network within the hydration sphere of the molecule.20,74,76,77,133,146,150–152,162,165–167 The linear 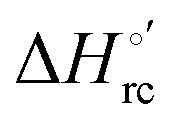 versus
versus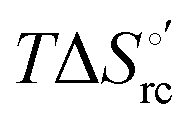 plots at 293 K for both mutants at different urea concentrations are reported in Fig. 9. The least-square fittings yield slopes of 1.23 and 2.40 and regression coefficients (r2) of 0.999 and 0.995 for the M80A and M80A/Y67A mutants, respectively. As the changes in
plots at 293 K for both mutants at different urea concentrations are reported in Fig. 9. The least-square fittings yield slopes of 1.23 and 2.40 and regression coefficients (r2) of 0.999 and 0.995 for the M80A and M80A/Y67A mutants, respectively. As the changes in 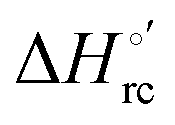 and
and 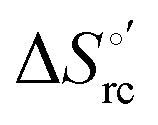 arising from the reduction-induced reorganization of the H-bond network at the protein–solvent interface (
arising from the reduction-induced reorganization of the H-bond network at the protein–solvent interface (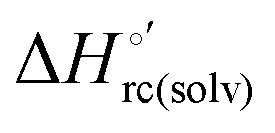 and
and 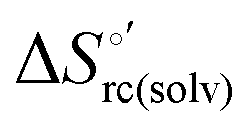 ) are fully compensative,20,34,74,75,103,104,130–133,140,141,146–150 the absence of perfect compensation (i.e. slopes greater than one) indicates that, beside solvent reorganization effects, protein-based “intrinsic” factors significantly contribute to the urea-induced changes in the reduction thermodynamics. In particular, axial ligand swapping should lower the reduction enthalpy due to the slightly lower electron donor character of the His ligand compared to the hydroxide anion and to the resulting decreased exposure of the metal center to solvent. The less positive
) are fully compensative,20,34,74,75,103,104,130–133,140,141,146–150 the absence of perfect compensation (i.e. slopes greater than one) indicates that, beside solvent reorganization effects, protein-based “intrinsic” factors significantly contribute to the urea-induced changes in the reduction thermodynamics. In particular, axial ligand swapping should lower the reduction enthalpy due to the slightly lower electron donor character of the His ligand compared to the hydroxide anion and to the resulting decreased exposure of the metal center to solvent. The less positive 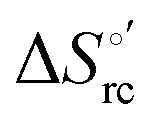 values of the His/His conformer fit with decreased solvent accessibility of the metal center, leading to a lower increase in the disorder of the water molecules in the heme cavity upon Fe(III) reduction.
values of the His/His conformer fit with decreased solvent accessibility of the metal center, leading to a lower increase in the disorder of the water molecules in the heme cavity upon Fe(III) reduction.
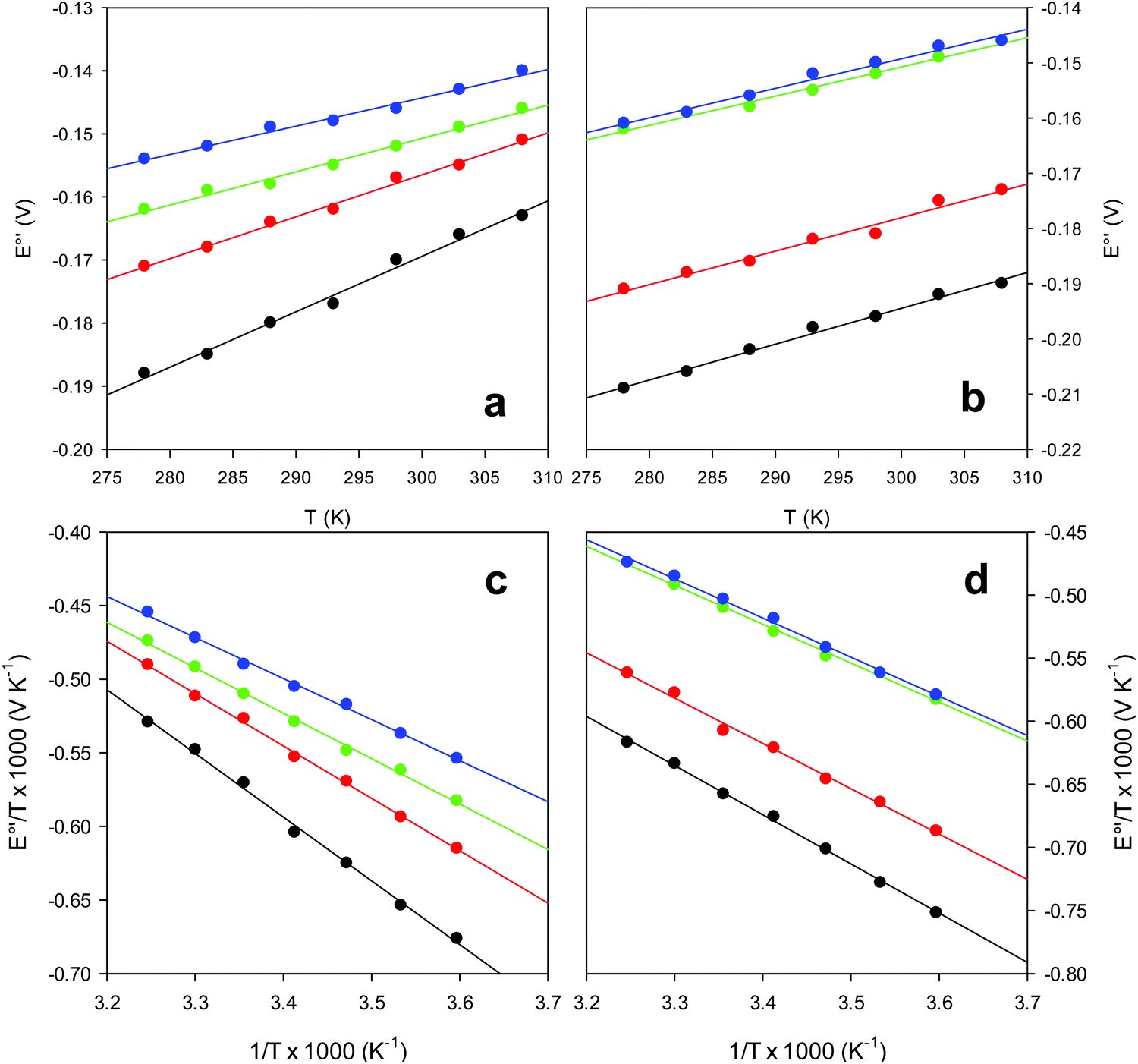 | ||
| Fig. 8 Plot of E°′ versus T and E°′/T versus 1/T for the M80A (a and c) and the M80A/Y67A (b and d) variants of yeast iso-1-cytochrome c in the presence of urea 0 M (black), 2 M (red), 4 M (green) and 6 M (blue), in 10 mM phosphate buffer plus 100 mM sodium perchlorate, pH 7.2. Solid lines are least-squares fits to the data points. | ||
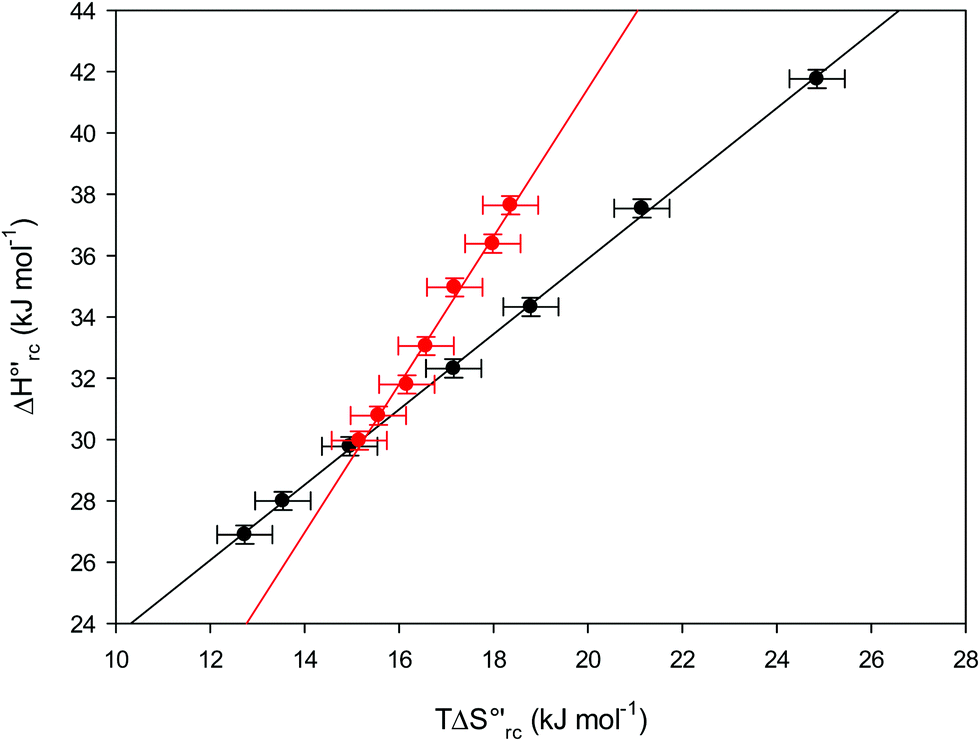 | ||
Fig. 9 Enthalpy–entropy compensation plots for the reduction thermodynamics of the M80A (black) and the M80A/Y67A (red) variants of yeast iso-1-cytochrome c, in the presence of varying urea concentrations: 0 M, 1 M, 2 M, 3 M, 4 M, 5 M and 6 M in 10 mM phosphate buffer plus 100 mM sodium perchlorate, pH 7.2. Solid lines are least-squares fits to the data points. T = 293 K. Error bars were calculated from the average errors on ΔH°′ and 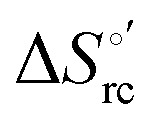 reported in Table 2. reported in Table 2. | ||
The slopes of the compensation plots indicate that protein-based “intrinsic” factors exert a greater influence on the urea-induced changes in reduction enthalpy 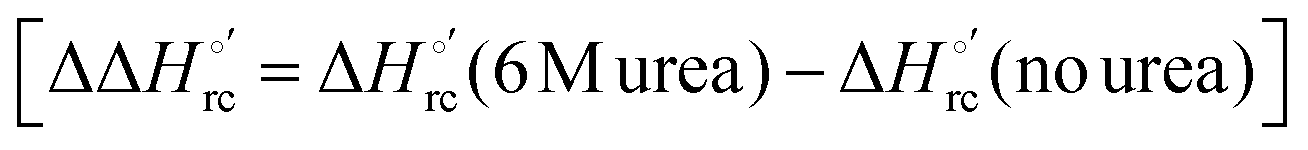 than the corresponding changes in reduction entropy
than the corresponding changes in reduction entropy 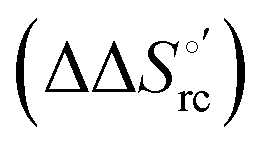 and that their effect is larger for the M80A/Y67A variant. This is also confirmed by the high compensation temperatures Tc featured by the M80A (359 K) and M80A/Y67A (695 K) mutants, which were calculated from the crossing point of the linear plots in Fig. 8. As the two mutants share the same axial heme coordination in both the folded (His/OH−) and unfolded states (His/His), this behavior confirms that the alteration of the H-bonding network in the distal heme site due to the Tyr67 to Ala substitution generates specific mutation-induced changes in the electrostatics and polarity of the metal site of the folded protein, which apparently tend to disappear upon urea unfolding (Table 2).
and that their effect is larger for the M80A/Y67A variant. This is also confirmed by the high compensation temperatures Tc featured by the M80A (359 K) and M80A/Y67A (695 K) mutants, which were calculated from the crossing point of the linear plots in Fig. 8. As the two mutants share the same axial heme coordination in both the folded (His/OH−) and unfolded states (His/His), this behavior confirms that the alteration of the H-bonding network in the distal heme site due to the Tyr67 to Ala substitution generates specific mutation-induced changes in the electrostatics and polarity of the metal site of the folded protein, which apparently tend to disappear upon urea unfolding (Table 2).
Thermodynamics of urea-induced unfolding
The equation: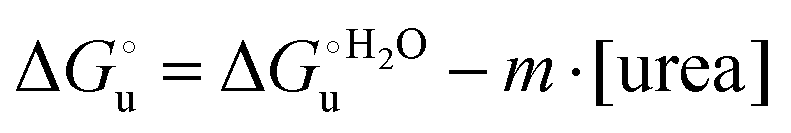 | (2) |
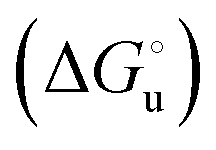 and at denaturant infinite dilution (
and at denaturant infinite dilution (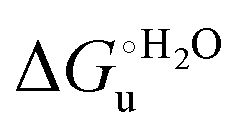 ) for a two-state denaturation event, where m is a parameter proportional to the increase in the solvent-exposed surface area of the denatured state compared to the folded protein and provides an estimation of residual structure in the denatured state.16,19,35,75,77,168,169
) for a two-state denaturation event, where m is a parameter proportional to the increase in the solvent-exposed surface area of the denatured state compared to the folded protein and provides an estimation of residual structure in the denatured state.16,19,35,75,77,168,169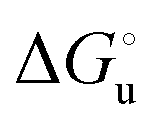 can be calculated from the equilibrium constant, Ku, of the denaturation process at a given urea concentration:
can be calculated from the equilibrium constant, Ku, of the denaturation process at a given urea concentration: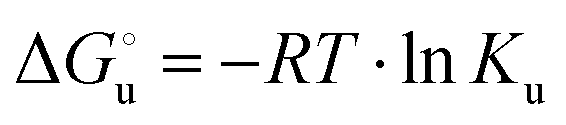 | (3) |
| Ku = aunfolded protein/afolded protein ≈ [unfolded protein]/[folded protein] | (4) |
The concentration of folded and unfolded protein at each urea concentration was determined according to the following equations
| [unfolded protein] = α[total protein concentration] | (5) |
| [folded protein] = (1 − α)[total protein concentration] | (6) |
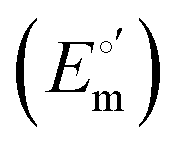 and from the limit E°′ values for the folded
and from the limit E°′ values for the folded 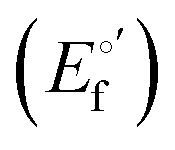 and unfolded forms
and unfolded forms 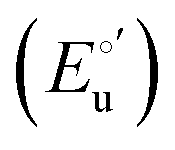 measured at 0 and 6 M urea, respectively, and (ii) the intensity of the UV-vis and MCD spectra at selected wavelengths (520/570 nm and 575–573 nm, respectively) at each urea concentration (Im) and for the folded (Imf) and unfolded forms (Imu) measured at 0 and 6 M urea, respectively, according to the following equations
measured at 0 and 6 M urea, respectively, and (ii) the intensity of the UV-vis and MCD spectra at selected wavelengths (520/570 nm and 575–573 nm, respectively) at each urea concentration (Im) and for the folded (Imf) and unfolded forms (Imu) measured at 0 and 6 M urea, respectively, according to the following equations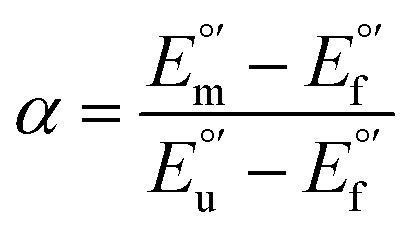 | (7) |
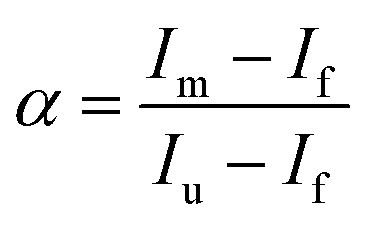 | (8) |
The plots of 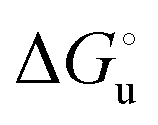 vs. urea concentration for both proteins are shown in Fig. 10. The
vs. urea concentration for both proteins are shown in Fig. 10. The 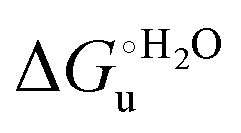 and m values obtained from the intercept and the slope of the least-squares linear fit of the data points to eqn (2), respectively, are listed in Table 3, along with the data obtained previously for wt ycc and its K72A/K73A/K79A and K72A/K73H/K79A mutants.14,75 The variable-temperature voltammetric measurements carried out at different urea concentrations allow the
and m values obtained from the intercept and the slope of the least-squares linear fit of the data points to eqn (2), respectively, are listed in Table 3, along with the data obtained previously for wt ycc and its K72A/K73A/K79A and K72A/K73H/K79A mutants.14,75 The variable-temperature voltammetric measurements carried out at different urea concentrations allow the 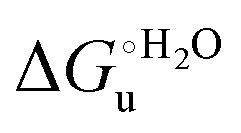 and m values from 5 to 35 °C to be determined. Both terms slightly decrease with increasing temperature (Table 3). The
and m values from 5 to 35 °C to be determined. Both terms slightly decrease with increasing temperature (Table 3). The 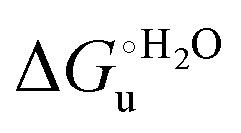 values for both variants are significantly lower than those for the species showing intact Met/His coordination, as previously observed for the same species immobilized on a MUA/MU SAM at the same T (5 °C).77 This indicates that the axial Fe–(S)Met bond plays a significant role in determining the stability of cytochrome c against chemical denaturation, as its removal invariably “relaxes” the 3D structure of the protein and facilitates unfolding. The additional Y67A mutation disfavors to some extent protein unfolding, since the
values for both variants are significantly lower than those for the species showing intact Met/His coordination, as previously observed for the same species immobilized on a MUA/MU SAM at the same T (5 °C).77 This indicates that the axial Fe–(S)Met bond plays a significant role in determining the stability of cytochrome c against chemical denaturation, as its removal invariably “relaxes” the 3D structure of the protein and facilitates unfolding. The additional Y67A mutation disfavors to some extent protein unfolding, since the 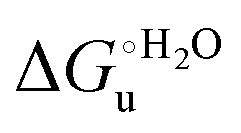 values for the M80A/Y67A variant are slightly larger (0.7–0.8 kJ mol−1) than for M80A. Hence, the alteration of the H-bonding network that stabilizes the heme crevice due to the Y67A mutation85–96 results in a larger inherent thermodynamic stability of the protein. This finding is rather surprising, since Tyr67 mutation would be expected to facilitate unfolding, but this conflict is only apparent because the difference in free energy of unfolding between the single and double mutant turns out to be an entropic effect, as discussed below.
values for the M80A/Y67A variant are slightly larger (0.7–0.8 kJ mol−1) than for M80A. Hence, the alteration of the H-bonding network that stabilizes the heme crevice due to the Y67A mutation85–96 results in a larger inherent thermodynamic stability of the protein. This finding is rather surprising, since Tyr67 mutation would be expected to facilitate unfolding, but this conflict is only apparent because the difference in free energy of unfolding between the single and double mutant turns out to be an entropic effect, as discussed below.
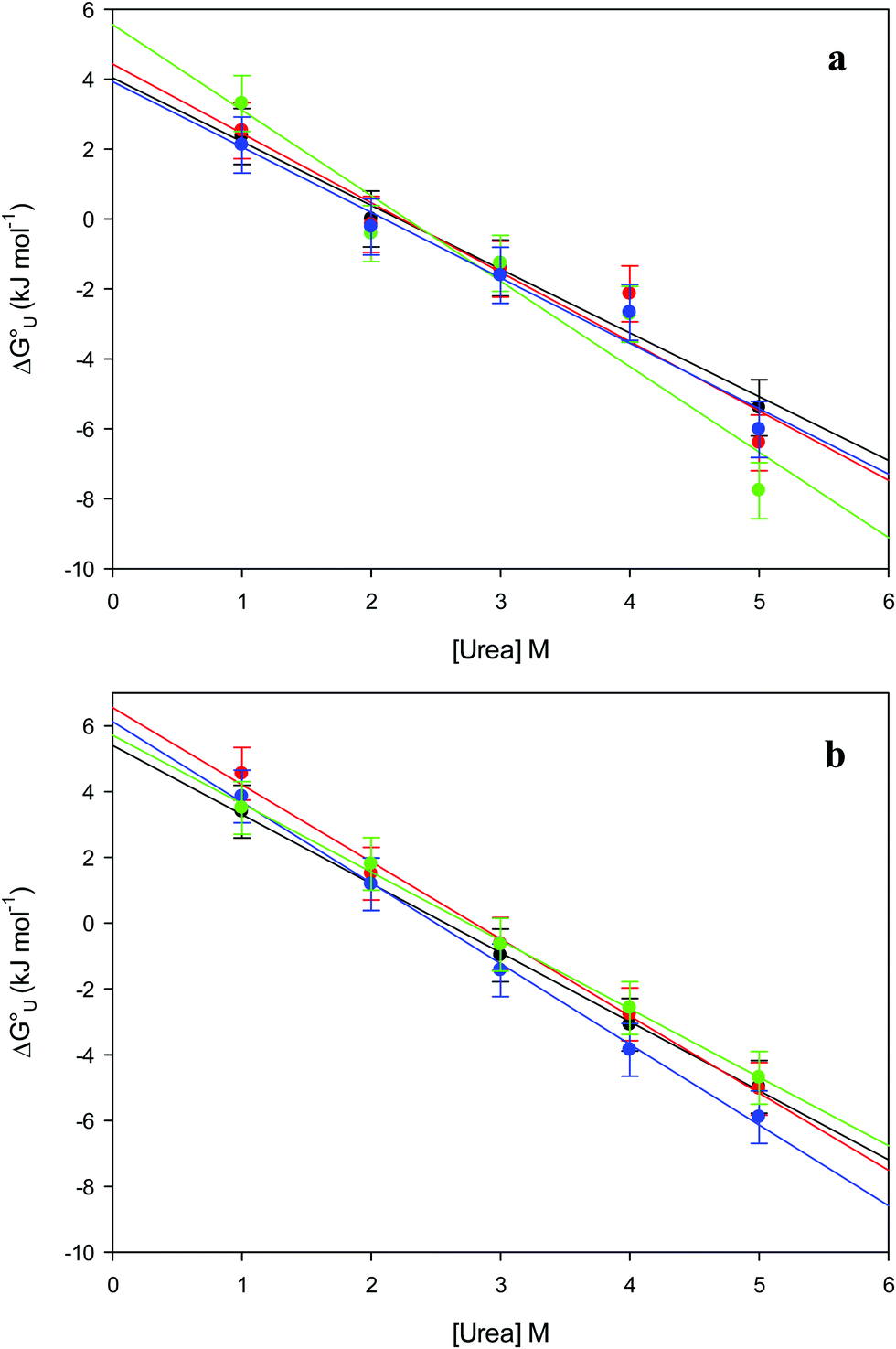 | ||
Fig. 10 Plots of 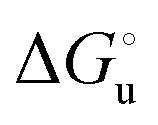 versus molar urea concentration for the M80A (a) and the M80A/Y67A (b) variants of yeast iso-1-cytochrome c in 10 mM phosphate buffer plus 100 mM sodium perchlorate, pH 7.2. T = 278 K (black), 288 K (red), 298 K (green) and 308 K (blue). According to eqn (2), versus molar urea concentration for the M80A (a) and the M80A/Y67A (b) variants of yeast iso-1-cytochrome c in 10 mM phosphate buffer plus 100 mM sodium perchlorate, pH 7.2. T = 278 K (black), 288 K (red), 298 K (green) and 308 K (blue). According to eqn (2), 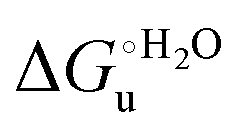 and m are obtained from the intercept and the slope of the least-squares linear fit of the data points, respectively. and m are obtained from the intercept and the slope of the least-squares linear fit of the data points, respectively. | ||
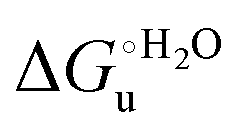 and m values at different temperatures for the urea-induced unfolding of the M80A and M80AY67A variants of yeast iso-1-cytochrome c
and m values at different temperatures for the urea-induced unfolding of the M80A and M80AY67A variants of yeast iso-1-cytochrome c
| Protein | T (°C) |
|
m (kJ mol−1 M−1) |
|---|---|---|---|
a The errors on 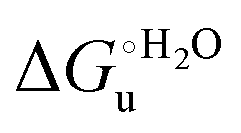 and m are ±0.80 kJ mol−1 and ±10% (relative error), respectively.
b From voltammetric experiments carried out in 10 mM phosphate buffer plus 100 mM sodium perchlorate at pH 7.2.
c Average values calculated from the changes in the UV-vis and MCD spectra at selected wavelengths (520/570 nm and 575–573 nm, respectively) observed in the presence of an increasing concentration of urea in 5 mM phosphate buffer pH 7.
d From ref. 77.
e From ref. 75.
f Calculated form data in ref. 4.
g From ref. 35. and m are ±0.80 kJ mol−1 and ±10% (relative error), respectively.
b From voltammetric experiments carried out in 10 mM phosphate buffer plus 100 mM sodium perchlorate at pH 7.2.
c Average values calculated from the changes in the UV-vis and MCD spectra at selected wavelengths (520/570 nm and 575–573 nm, respectively) observed in the presence of an increasing concentration of urea in 5 mM phosphate buffer pH 7.
d From ref. 77.
e From ref. 75.
f Calculated form data in ref. 4.
g From ref. 35.
|
|||
| M80Asol | 5 | 5.98b | 2.64b |
| 10 | 5.65b | 2.59b | |
| 15 | 5.44b | 2.38b | |
| 20 | 5.33b | 2.47b | |
| 25 | 5.20b | 2.15b | |
| 30 | 4.42b | 1.93b | |
| 35 | 4.60b | 2.07b | |
| 25 | 6.16c | 2.51c | |
| M80Aads | 5 | 4.9d | 2.16d |
| 25 | 0.95d | 1.52d | |
| M80AY67Asol | 5 | 6.72b | 2.64b |
| 10 | 6.31b | 2.39b | |
| 15 | 6.27b | 2.03b | |
| 20 | 6.13b | 1.81b | |
| 25 | 6.05b | 2.10b | |
| 30 | 5.92b | 1.87b | |
| 35 | 5.80b | 2.13b | |
| 25 | 6.87c | 2.98c | |
| M80AY67Aads | 5 | 6.6d | 2.52d |
| 25 | 3.75d | 1.71d | |
| K72A/K73H/K79Asol | 5 | 12.7e | 4.2e |
| K72A/K73A/K79Asol | 5 | 8.1f | 2.3f |
| wtsol | 5 | 7.6f | 1.9f |
| 25 | 20.8g | 6.90g | |
| K73H | 25 | 15.2g | 5.1g |
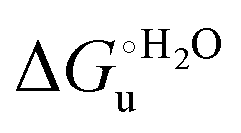
The M80A and M80AY67A variants feature quite similar m values (Table 3) throughout the temperature range investigated. We note that at 5 °C the m values are comparable to those for the Met/His-ligated species (wt and variants to surface lysines), but lower than that of the K72A/K73H/K79A variant. Therefore, it appears that at low temperature neither removal of the axial Fe–(S)Met bond, also coupled to alteration of the H-bonding network stabilizing the heme crevice (M80A/Y67A), nor deletion of surface charges (K72A/K73A/K79A) significantly influences the structural impact of urea on the overall ycc folding, which instead is affected by insertion of a new potential axial ligand (K72A/K73H/K79A). On the contrary, at 25 °C the m values for M80A and M80AY67A are much lower compared to the wt (Table 3), indicative of a smaller increase in the solvent-exposed surface area. However, this effect is not the result of less pronounced protein unfolding as the near-UV CD spectra (Fig. 5) indicate that the variants and wt both undergo disruption of the tight packing of the core residues.84,90,122 A possible role in this effect is played by the different urea-induced axial ligand swapping occurring in the variants compared to the wt.
The enthalpy and entropy changes associated with the unfolding process calculated using the van’t Hoff equation from the temperature dependence of 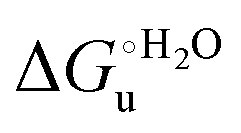 are positive for both species (Fig. 11 and Table 4). Hence, the urea-induced denaturation realizes an entropic gain but is disfavored on enthalpic grounds. The enthalpy change is the main determinant of the positive
are positive for both species (Fig. 11 and Table 4). Hence, the urea-induced denaturation realizes an entropic gain but is disfavored on enthalpic grounds. The enthalpy change is the main determinant of the positive 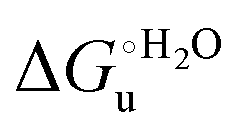 values (Fig. 12 and Table 4).
values (Fig. 12 and Table 4).
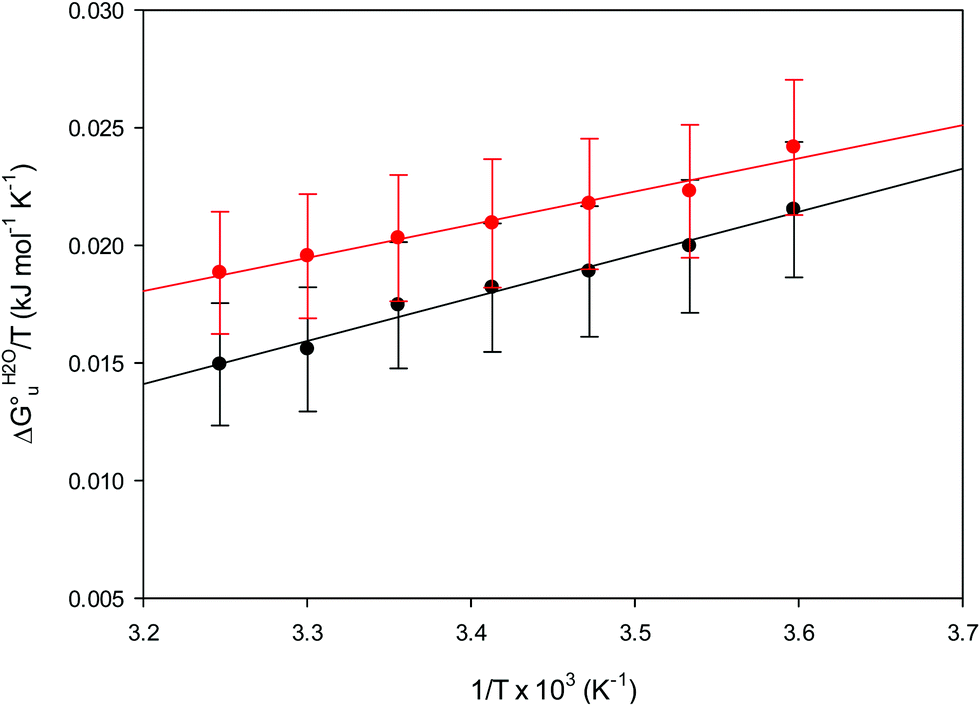 | ||
| Fig. 11 vant’Hoff plots for the urea-induced unfolding of the M80A (black) and the M80A/Y67A (red) variants of yeast iso-1-cytochrome c in 10 mM phosphate buffer plus 10 mM sodium perchlorate, pH 7.2. Solid lines are least-squares fits to the data points. | ||
| Protein | T |
|
|
|
|
m (kJ mol−1 M−1) |
|---|---|---|---|---|---|---|
a The average errors on 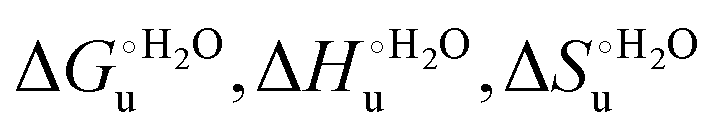 and m are ±0.8 kJ mol−1, ±8% (relative error), ±10% (relative error) and ±10% (relative error), respectively.
b At 5 °C.
c Present work, freely diffusing species in 10 mM phosphate buffer plus 100 mM sodium perchlorate, pH 7.2.
d Protein adsorbed on a polycrystalline gold electrode coated with a SAM of MUA/MU, from ref. 77.
e Values calculated from data in ref. 74. and m are ±0.8 kJ mol−1, ±8% (relative error), ±10% (relative error) and ±10% (relative error), respectively.
b At 5 °C.
c Present work, freely diffusing species in 10 mM phosphate buffer plus 100 mM sodium perchlorate, pH 7.2.
d Protein adsorbed on a polycrystalline gold electrode coated with a SAM of MUA/MU, from ref. 77.
e Values calculated from data in ref. 74.
|
||||||
| M80A in solutionc | 5 | 6.0 | 18.3 | 44.4 | 12 | 2.64 |
| M80AY67A in solutionc | 5 | 6.7 | 14.1 | 27.1 | 8 | 2.64 |
| M80A immobilizedd | 5 | 4.9 | 58.5 | 191.5 | 53 | 2.19 |
| M80AY67A immobilizedd | 5 | 6.6 | 46.6 | 142.4 | 40 | 2.61 |
| wt immobilizedd,e | 5 | 13.4 | 91.8 | 286.6 | 77 | 3.24 |
| K72A/K73A/K79A immobilizedd,e | 5 | 11.3 | 69.9 | 209.3 | 58 | 2.95 |
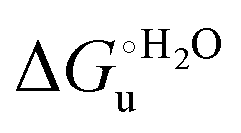
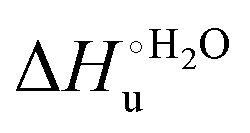
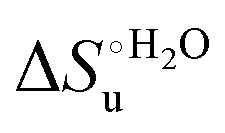
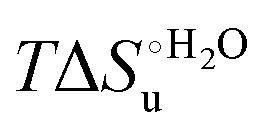
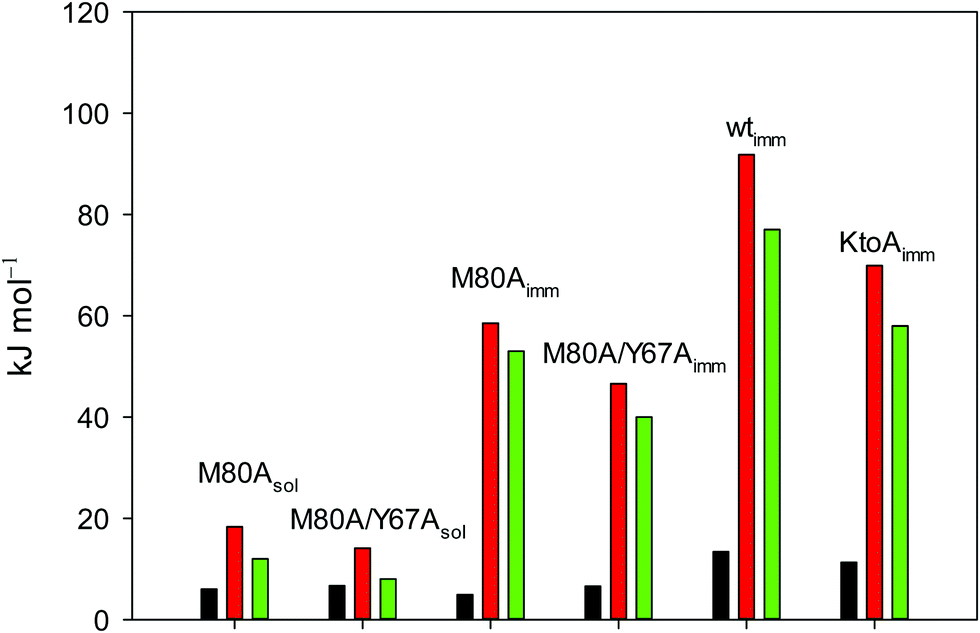 | ||
Fig. 12 Histograms depicting the thermodynamic parameters of urea-induced unfolding for yeast iso-1-cytochrome c and its variants in solution and in the immobilized state reported in Table 4. Black, red and green bars correspond to 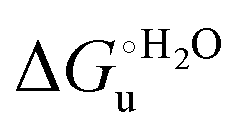 , , 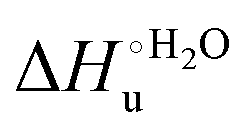 and T (5 °C)· and T (5 °C)·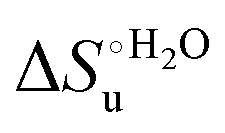 values, respectively. values, respectively. | ||
As suggested previously,77 the enthalpic cost of urea unfolding results from the balance of bond breaking and formation, involving heme axial ligand swapping and intramolecular and intermolecular protein–solvent van der Waals interactions and H-bonding, associated with urea intrusion into the tertiary structure followed by hydration and loss of the native structure.170 The positive unfolding entropy possibly results from the urea-induced weakening of the structural constraints and intramolecular bonding interactions in the secondary and tertiary structure of the polypeptide chain, which results in increased molecular accessible states and degrees of freedom. Likewise, elimination of constrained water assemblies on the protein surface due to hydrophobic hydration upon displacement by the larger urea molecules possibly contributes to the entropy gain.77,171 The opposite contributions of the enthalpic and entropic terms to 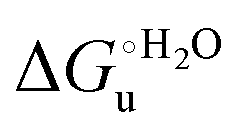 (H–S compensation) (Table 4 and Fig. 12) confirm that changes in the hydrogen bonding network in the hydration sphere of the protein upon unfolding strongly affect the thermodynamics of the denaturation process. The increase of
(H–S compensation) (Table 4 and Fig. 12) confirm that changes in the hydrogen bonding network in the hydration sphere of the protein upon unfolding strongly affect the thermodynamics of the denaturation process. The increase of 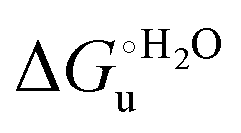 for M80A/Y67A compared to M80A is entropy-driven (Table 4 and Scheme 1). The similarity of the urea-induced changes in the spectroscopic properties of the two mutants suggests that such an effect has more to do with the increased hydrophobicity of the heme cavity following the substitution of a hydrophilic Tyr with a hydrophobic Ala residue than to mutation-specific differences in the structural changes upon urea-induced unfolding.
for M80A/Y67A compared to M80A is entropy-driven (Table 4 and Scheme 1). The similarity of the urea-induced changes in the spectroscopic properties of the two mutants suggests that such an effect has more to do with the increased hydrophobicity of the heme cavity following the substitution of a hydrophilic Tyr with a hydrophobic Ala residue than to mutation-specific differences in the structural changes upon urea-induced unfolding.
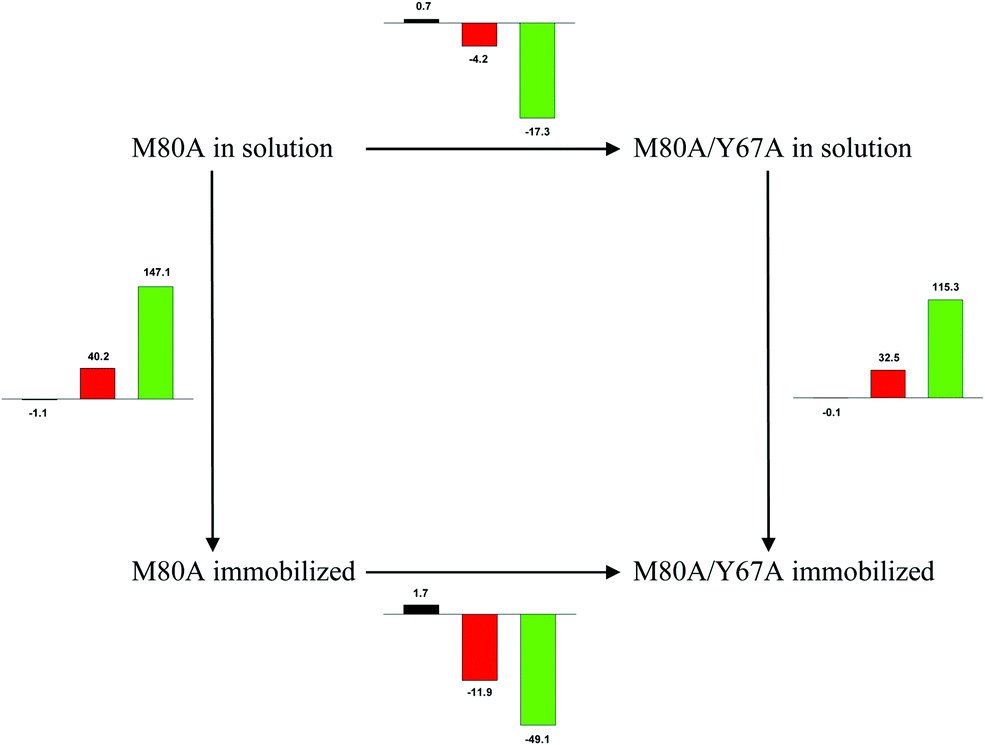 | ||
Scheme 1 Cycle summarizing (i) the effects of the Y67A mutation on the unfolding thermodynamics of the M80A variant of yeast iso-1-cytochrome c in freely diffusing conditions (upper line) and immobilized on a MUA/MU SAM (bottom line) and (ii) the effect of immobilization on a MUA/MU SAM on the unfolding thermodynamics of the M80A and M80A/Y67A variants of yeast iso-1-cytochrome c (left and right column, respectively). 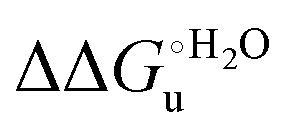 (kJ mol−1, black column), (kJ mol−1, black column), 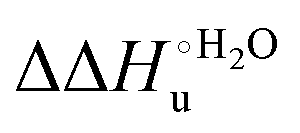 (kJ mol−1, red column) and (kJ mol−1, red column) and 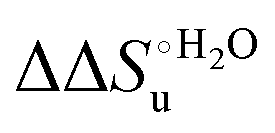 (J K−1 mol−1, green column) were calculated form the data reported in Table 4. (J K−1 mol−1, green column) were calculated form the data reported in Table 4. | ||
Solution versus immobilized state
A comparison of the present data with those obtained previously for the same mutants adsorbed electrostatically on a MUA/MU SAM allows examination of how the thermodynamics of urea-induced unfolding are affected by protein adsorption. The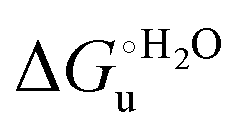 values for the solution variants are larger with respect to those for the same electrode-immobilized species, especially at 25 °C. Therefore, immobilization favors the chemically-induced protein unfolding (Table 4 and Scheme 1). Both proteins feature much less positive
values for the solution variants are larger with respect to those for the same electrode-immobilized species, especially at 25 °C. Therefore, immobilization favors the chemically-induced protein unfolding (Table 4 and Scheme 1). Both proteins feature much less positive 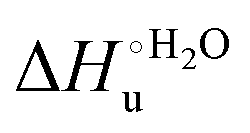 and
and  values in freely diffusing conditions than in the immobilized state (Table 4 and Scheme 1). The changes in the unfolding enthalpy and entropy are much larger than the changes in the unfolding free energy (Scheme 1). Once again, this H–S compensation indicates that differences in solvent reorganization effects accompanying denaturation of the protein under freely diffusing conditions or subjected to motional restriction are the main factor responsible for the differences in
values in freely diffusing conditions than in the immobilized state (Table 4 and Scheme 1). The changes in the unfolding enthalpy and entropy are much larger than the changes in the unfolding free energy (Scheme 1). Once again, this H–S compensation indicates that differences in solvent reorganization effects accompanying denaturation of the protein under freely diffusing conditions or subjected to motional restriction are the main factor responsible for the differences in 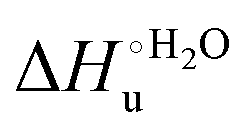 and
and 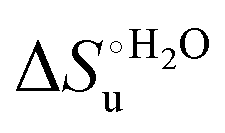 . This balance however changes with temperature. In particular, at 5 °C the enthalpy/entropy compensation is almost perfect, resulting in very limited (M80A) or negligible (M80AY67A) free energy differences, but decreases with increasing temperature, leading to sizeable changes in the
. This balance however changes with temperature. In particular, at 5 °C the enthalpy/entropy compensation is almost perfect, resulting in very limited (M80A) or negligible (M80AY67A) free energy differences, but decreases with increasing temperature, leading to sizeable changes in the 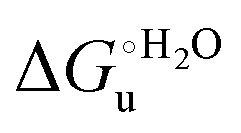 values for immobilized and freely diffusing proteins at room temperature (Table 3). Analogously, Δm [=m(ads) − m(sol)] becomes more negative with increasing temperature. At 25 °C, electrostatic adsorption on MUA/MU induces a progressive lowering of the inherent thermodynamic stability of the three-dimensional structure of both proteins and reduces the increase in the solvent-exposed surface area of the denatured state compared to the folded protein. It is tempting to speculate that a non-negligible contribution to the higher
values for immobilized and freely diffusing proteins at room temperature (Table 3). Analogously, Δm [=m(ads) − m(sol)] becomes more negative with increasing temperature. At 25 °C, electrostatic adsorption on MUA/MU induces a progressive lowering of the inherent thermodynamic stability of the three-dimensional structure of both proteins and reduces the increase in the solvent-exposed surface area of the denatured state compared to the folded protein. It is tempting to speculate that a non-negligible contribution to the higher 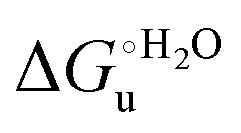 values of the freely diffusing species at room temperature results from the urea-induced replacement of the axial OH− by an endogenous histidine, which does not occur upon protein immobilization.
values of the freely diffusing species at room temperature results from the urea-induced replacement of the axial OH− by an endogenous histidine, which does not occur upon protein immobilization.
The negative Δm [=m(ads) − m(sol)] values for both the M80A and M80AY67A variants indicate that the urea-induced increase in the solvent-exposed surface area is less pronounced for the immobilized species. Although this effect does not match intuitively with the larger thermodynamic tendency of the latter species to unfold (lower 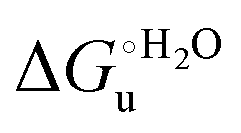 values), it fits well with the hypothesis that both mutants undergo non-negligible unfolding upon adsorption on a MUA/MU SAM, which lowers the effects of structure opening to solvent due to eventual urea unfolding, thereby preventing the coordination of a second His ligand at high urea concentrations.77
values), it fits well with the hypothesis that both mutants undergo non-negligible unfolding upon adsorption on a MUA/MU SAM, which lowers the effects of structure opening to solvent due to eventual urea unfolding, thereby preventing the coordination of a second His ligand at high urea concentrations.77
Conclusions
Partial protein unfolding due to a chemical stimulus, in general represented by the binding of an effector molecule or adsorption onto a supramolecular assembly (e.g. cell or organelle membrane), is a strategy widely employed by biological systems to transmit a chemical message or trigger a biochemical event. Cytochrome c participates in this kind of mechanisms. In fact, upon binding to cardiolipin, a component of the inner mitochondrial membrane (IMM), cytc is subjected to a structural change that results in oxidative reactions leading to its release from the mitochondrion to the cytosol whereby it activates caspases that initiate the cell apoptotic cascade. Here we have presented evidence that at room (or physiological) temperature the susceptibility to unfolding of ycc – represented by the free energy of unfolding at denaturant infinite dilution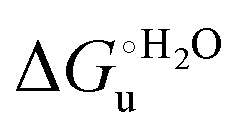 – is affected by the motional regime of the protein, namely it differs depending on whether the protein is freely diffusing or subjected to a motional restriction due to the interaction with a molecular construct. In particular, electrostatic adsorption onto a negatively charged molecular surface (a MUA/MU SAM, which would mimic the negatively charged phospholipidic inner mitochondrial membrane) renders ycc more susceptible to chemical unfolding compared to the solution state. This finding has physiological relevance related to the cytochrome c interaction with cardiolipin at the IMM. Such motional state-dependent behavior results in different molecular events involving the change in axial ligands of the heme iron and different unfolding-induced changes in the hydrogen bonding network within the hydration sphere of the molecule. As we have also found previously that the unfolding behavior of immobilized ycc changes with the nature of the adsorbing construct,76,77 it is apparent how, from a more general perspective, the motional properties of the protein and the nature of the adsorbing surface constitute additional means that Nature can exploit to modulate protein unfolding and the eventual molecular response.5,13,31
– is affected by the motional regime of the protein, namely it differs depending on whether the protein is freely diffusing or subjected to a motional restriction due to the interaction with a molecular construct. In particular, electrostatic adsorption onto a negatively charged molecular surface (a MUA/MU SAM, which would mimic the negatively charged phospholipidic inner mitochondrial membrane) renders ycc more susceptible to chemical unfolding compared to the solution state. This finding has physiological relevance related to the cytochrome c interaction with cardiolipin at the IMM. Such motional state-dependent behavior results in different molecular events involving the change in axial ligands of the heme iron and different unfolding-induced changes in the hydrogen bonding network within the hydration sphere of the molecule. As we have also found previously that the unfolding behavior of immobilized ycc changes with the nature of the adsorbing construct,76,77 it is apparent how, from a more general perspective, the motional properties of the protein and the nature of the adsorbing surface constitute additional means that Nature can exploit to modulate protein unfolding and the eventual molecular response.5,13,31
Funding information
This work was supported by the University of Modena and Reggio Emilia FAR 2019 funding program.Conflicts of interest
There are no conflicts of interest to declare.References
- D. Alvarez-Paggi, L. Hannibal, M. A. Castro, S. Oviedo-Rouco, V. Demicheli, V. Tórtora, F. Tomasina, R. Radi and D. H. Murgida, Chem. Rev., 2017, 117, 13382–13460 CrossRef CAS.
- S. Zaidi, M. I. Hassan, A. Islam and F. Ahmad, Cell. Mol. Life Sci., 2014, 71, 229–255 CrossRef CAS.
- I. Bertini, G. Cavallaro and A. Rosato, Chem. Rev., 2006, 106, 90–115 CrossRef CAS.
- M. Hüttemann, P. Pecina, M. Rainbolt, T. H. Sanderson, V. E. Kagan, L. Samavati, J. W. Doan and I. Lee, Mitochondrion, 2011, 11, 369–381 CrossRef.
- R. Schweitzer-Stenner, Biophys. Rev., 2018, 10, 1151–1185 CrossRef CAS.
- G. W. Moore and G. R. Pettigrew, Cytochromes c. Evolutionary, Structural, and Physicochemical Aspects, Springer-Verlag, Berlin, Germany, 1990 Search PubMed.
- Cytochrome c – A Multidisciplinary Approach, ed. A. G. Scott and R. A. Mauk, University Science Books, Sausalito, CA, 1996 Search PubMed.
- G. Battistuzzi, M. Borsari, L. Loschi, A. Martinelli and M. Sola, Biochemistry, 1999, 38, 7900–7907 CrossRef CAS.
- G. Battistuzzi, M. Borsari, G. Rossi and M. Sola, Inorg. Chim. Acta, 1998, 272, 168–175 CrossRef CAS.
- G. Battistuzzi, M. Borsari, A. Ranieri and M. Sola, Arch. Biochem. Biophys., 2002, 404, 227–233 CrossRef CAS.
- G. Battistuzzi, M. Borsari and M. Sola, Eur. J. Inorg. Chem., 2001, 2989 CrossRef CAS.
- G. Battistuzzi, M. Borsari, F. De Rienzo, G. Di Rocco, A. Ranieri and M. Sola, Biochemistry, 2007, 46, 1694–1702 CrossRef CAS.
- A. Paradisi, M. Bellei, L. Paltrinieri, C. A. Bortolotti, G. Di Rocco, A. Ranieri, M. Borsari, M. Sola and G. Battistuzzi, JBIC, J. Biol. Inorg. Chem., 2020, 25, 467–487 CrossRef CAS.
- S. Monari, A. Ranieri, G. Di Rocco, G. van der Zwan, S. Peressini, C. Tavagnacco, D. Millo and M. Borsari, J. Appl. Electrochem., 2009, 39, 2181–2190 CrossRef CAS.
- N. A. Belikova, Y. A. Vladimirov, A. N. Osipov, A. A. Kapralov, V. A. Tyurin, M. V. Potapovich, L. V. Basova, J. Peterson, I. V. Kurnikov and V. E. Kagan, Biochemistry, 2006, 45, 4998–5009 CrossRef CAS.
- S.-R. Yeh, S. Han and D. L. Rousseau, Acc. Chem. Res., 1998, 31, 727–736 CrossRef CAS.
- S. W. Englander, T. R. Sosnick, L. C. Mayne, M. Shtilerman, P. X. Qi and Y. Bai, Acc. Chem. Res., 1998, 31, 737–744 CrossRef CAS.
- L. Hoang, S. Bédard, M. M. G. Krishna, Y. Lin and S. W. Englander, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12173–12178 CrossRef CAS.
- S.-R. Yeh and D. L. Rousseau, Nat. Struct. Biol., 1998, 5, 222–228 CrossRef CAS.
- M. Fedurco, J. Augustynski, C. Indiani, G. Smulevich, M. Antalík, M. Bánó, E. Sedlák, M. C. Glascock and J. H. Dawson, J. Am. Chem. Soc., 2005, 127, 7638–7646 CrossRef CAS.
- M. Fedurco, J. Augustynski, C. Indiani, G. Smulevich, M. Antalík, M. Bánó, E. Sedlák, M. C. Glascock and J. H. Dawson, Biochim. Biophys. Acta, Proteins Proteomics, 2004, 1703, 31–41 CrossRef CAS.
- S. Baddam and B. E. Bowler, Biochemistry, 2005, 44, 14956–14968 CrossRef CAS.
- R. Schweitzer-Stenner, New J. Sci., 2014, 2014, 1–28 CrossRef.
- N. J. O’Reilly and E. Magner, Langmuir, 2005, 21, 1009–1014 CrossRef.
- S. Crilly and E. Magner, Chem. Commun., 2009, 535–537 RSC.
- G. Battistuzzi, M. Borsari and M. Sola, Trends Inorg. Chem., 1996, 4, 1–8 Search PubMed.
- G. Battistuzzi, M. Borsari, D. Dallari, I. Lancellotti and M. Sola, Eur. J. Biochem., 1996, 241, 208–214 CrossRef CAS.
- A. Díaz-Quintana, G. Pérez-Mejías, A. Guerra-Castellano, M. A. De la Rosa and I. Díaz-Moreno, Oxid. Med. Cell. Longev., 2020, 2020, 1–20 CrossRef.
- S. Oellerich, H. Wackerbarth and P. Hildebrandt, J. Phys. Chem. B, 2002, 106, 6566–6580 CrossRef CAS.
- P. Ascenzi, M. Coletta, M. T. Wilson, L. Fiorucci, M. Marino, F. Polticelli, F. Sinibaldi and R. Santucci, IUBMB Life, 2015, 67, 98–109 CrossRef CAS.
- J. Muenzner and E. V. Pletneva, Chem. Phys. Lipids, 2014, 179, 57–63 CrossRef CAS.
- A. Ranieri, D. Millo, G. Di Rocco, G. Battistuzzi, C. A. Bortolotti, M. Borsari and M. Sola, JBIC, J. Biol. Inorg. Chem., 2015, 20, 531–540, DOI:10.1007/s00775-015-1238-6.
- A. Ranieri, G. Di Rocco, D. Millo, G. Battistuzzi, C. A. Bortolotti, L. Lancellotti, M. Borsari and M. Sola, Electrochim. Acta, 2015, 176, 1019–1028 CrossRef CAS.
- A. Ranieri, G. Battistuzzi, M. Borsari, C. A. Bortolotti, G. Di Rocco, S. Monari and M. Sola, Electrochem. Commun., 2012, 14, 29–31 CrossRef CAS.
- S. Godbole, A. Dong, K. Garbin and B. E. Bowler, Biochemistry, 1997, 36, 119–126 CrossRef CAS.
- S. Baddam and B. E. Bowler, Biochemistry, 2006, 45, 4611–4619 CrossRef CAS.
- M. G. Duncan, M. D. Williams and B. E. Bowler, Protein Sci., 2009, 18, 1155–1164, DOI:10.1002/pro.120.
- M. M. Elmer-Dixon and B. E. Bowler, Biochemistry, 2018, 57, 5683–5695 CrossRef CAS.
- G. Battistuzzi, M. Borsari, A. Ranieri and M. Sola, J. Biol. Inorg. Chem., 2004, 9, 781–787 CrossRef CAS.
- V. E. Kagan, H. A. Bayir, N. A. Belikova, O. Kapralov, Y. Y. Tyurina, V. A. Tyurin, J. Jiang, D. A. Stoyanovsky, P. Wipf, P. M. Kochanek, J. S. Greenberger, B. Pitt, A. A. Shvedova and G. Borisenko, Free Radicals Biol. Med., 2009, 46, 1439–1453 CrossRef CAS.
- M. M. Cherney and B. E. Bowler, Coord. Chem. Rev., 2011, 255, 664–677 CrossRef CAS.
- B. Milorey, R. Schweitzer-Stenner, R. Kurbaj and D. Malyshka, ACS Omega, 2019, 4, 1386–1400 CrossRef CAS.
- L. Hannibal, F. Tomasina, D. A. Capdevila, V. Demicheli, V. Tórtora, D. Alvarez-Paggi, R. Jemmerson, D. H. Murgida and R. Radi, Biochemistry, 2016, 55, 407–428 CrossRef CAS.
- D. A. Capdevila, S. Oviedo Rouco, F. Tomasina, V. Tortora, V. Demicheli, R. Radi and D. H. Murgida, Biochemistry, 2015, 54, 7491–7504 CrossRef CAS.
- L. A. Pandiscia and R. Schweitzer-Stenner, J. Phys. Chem. B, 2015, 119, 12846–12859 CrossRef CAS.
- L. Pandiscia and R. Schweitzer-Stenner, Biophys. J., 2015, 106, 517a CrossRef.
- M. Li, A. Mandal, V. A. Tyurin, M. DeLucia, J. Ahn, V. E. Kagan and P. C. A. van der Wel, Structure, 2019, 1–11 Search PubMed.
- D. Mohammadyani, N. Yanamala, A. K. Samhan-Arias, A. A. Kapralov, G. Stepanov, N. Nuar, J. Planas-Iglesias, N. Sanghera, V. E. Kagan and J. Klein-Seetharaman, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1057–1068 CrossRef CAS.
- B. Milorey, D. Malyshka and R. Schweitzer-Stenner, J. Phys. Chem. Lett., 2017, 8, 1993–1998 CrossRef CAS.
- J. M. Bradley, G. Silkstone, M. T. Wilson, M. R. Cheesman and J. N. Butt, J. Am. Chem. Soc., 2011, 133, 19676–19679 CrossRef CAS.
- B. S. Rajagopal, G. G. Silkstone, P. Nicholls, M. T. Wilson and J. A. R. Worrall, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 780–791 CrossRef CAS.
- J. Hanske, J. R. Toffey, A. M. Morenz, A. J. Bonilla, K. H. Schiavoni and E. V. Pletneva, Proc. Natl. Acad. Sci. U. S. A., 2011, 109, 125–130 CrossRef.
- Y. Hong, J. Muenzner, S. K. Grimm and E. V. Pletneva, J. Am. Chem. Soc., 2012, 134, 18713–18723 CrossRef CAS.
- C. Kawai, J. C. Ferreira, M. S. Baptista and I. L. Nantes, J. Phys. Chem. B, 2014, 118, 11863–11872 CrossRef CAS.
- J. Muenzner, J. R. Toffey, Y. Hong and E. V. Pletneva, J. Phys. Chem. B, 2013, 117, 12878–12886 CrossRef CAS.
- E. K. J. Tuominen, C. J. A. Wallace and P. K. J. Kinnunen, J. Biol. Chem., 2002, 277, 8822–8826 CrossRef CAS.
- E. Kalanxhi and C. J. A. Wallace, Biochem. J., 2007, 407, 179–187 CrossRef CAS.
- F. Sinibaldi, L. Fiorucci, A. Patriarca, R. Lauceri, T. Ferri, M. Coletta and R. Santucci, Biochemistry, 2008, 47, 6928–6935 CrossRef CAS.
- F. Sinibaldi, B. D. Howes, M. C. Piro, F. Polticelli, C. Bombelli, T. Ferri, M. Coletta, G. Smulevich and R. Santucci, J. Biol. Inorg. Chem., 2010, 15, 689–700 CrossRef CAS.
- F. Sinibaldi, E. Droghetti, F. Polticelli, M. C. Piro, D. Di Pierro, T. Ferri, G. Smulevich and R. Santucci, J. Inorg. Biochem., 2011, 105, 1365–1372 CrossRef CAS.
- M. C. Piro, R. Santucci, E. Droghetti, F. Sinibaldi, L. Fiorucci, M. Coletta, D. Di Pierro, B. D. Howes, F. Polticelli and G. Smulevich, Biochemistry, 2013, 52, 4578–4588 CrossRef.
- L. Milazzo, L. Tognaccini, B. D. Howes, F. Sinibaldi, M. C. Piro, M. Fittipaldi, M. C. Baratto, R. Pogni, R. Santucci and G. Smulevich, Biochemistry, 2017, 56, 1887–1898 CrossRef CAS.
- G. Silkstone, S. M. Kapetanaki, I. Husu, M. H. Vos and M. T. Wilson, Biochemistry, 2012, 51, 6760–6766 CrossRef CAS.
- I. Husu, S. Kapetanaki, G. Silkstone, U. Liebl, M. T. Wilson and M. Vos, Biophys. J., 2010, 98, 631a CrossRef.
- S. M. Kapetanaki, G. Silkstone, I. Husu, U. Liebl, M. T. Wilson and M. H. Vos, Biochemistry, 2009, 48, 1613–1619 CrossRef CAS.
- M. M. Elmer-Dixon and B. E. Bowler, Biochemistry, 2017, 56, 4830–4839 CrossRef CAS.
- L. A. Pandiscia and R. Schweitzer-Stenner, Chem. Commun., 2014, 50, 3674–3676 RSC.
- F. Sinibaldi, L. Milazzo, B. D. Howes, M. C. Piro, L. Fiorucci, F. Polticelli, P. Ascenzi, M. Coletta, G. Smulevich and R. Santucci, JBIC, J. Biol. Inorg. Chem., 2017, 22, 19–29 CrossRef CAS.
- P. Ascenzi, D. Sbardella, F. Sinibaldi, R. Santucci and M. Coletta, JBIC, J. Biol. Inorg. Chem., 2016, 21, 421–432 CrossRef CAS.
- L. Zeng, L. Wu, L. Liu and X. Jiang, Anal. Chem., 2016, 88, 11727–11733 CrossRef CAS.
- S. S. Paul, P. Sil, S. Haldar, S. Mitra and K. Chattopadhyay, J. Biol. Chem., 2015, 290, 14476–14490 CrossRef CAS.
- L. C. Godoy, C. Muñoz-Pinedo, L. Castro, S. Cardaci, C. M. Schonhoff, M. King, V. Tórtora, M. Marín, Q. Miao, J. F. Jiang, A. Kapralov, R. Jemmerson, G. G. Silkstone, J. N. Patel, J. E. Evans, M. T. Wilson, D. R. Green, V. E. Kagan, R. Radi and J. B. Mannick, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2653–2658 CrossRef CAS.
- M. Abe, R. Niibayashi, S. Koubori, I. Moriyama and H. Miyoshi, Biochemistry, 2011, 50, 8383–8391 CrossRef CAS.
- S. Monari, D. Millo, A. Ranieri, G. Di Rocco, G. van der Zwan, C. Gooijer, S. Peressini, C. Tavagnacco, P. Hildebrandt and M. Borsari, JBIC, J. Biol. Inorg. Chem., 2010, 15, 1233–1242 CrossRef CAS.
- A. Ranieri, C. A. Bortolotti, G. Battistuzzi, M. Borsari, L. Paltrinieri, G. Di Rocco and M. Sola, Metallomics, 2014, 6, 874–884 RSC.
- L. Lancellotti, M. Borsari, A. Bonifacio, C. A. Bortolotti, G. Di Rocco, S. Casalini, A. Ranieri, G. Battistuzzi and M. Sola, Bioelectrochemistry, 2020, 136, 107628, DOI:10.1016/j.bioelechem.2020.107628.
- L. Lancellotti, M. Borsari, M. Bellei, A. Bonifacio, C. A. Bortolotti, G. Di Rocco, A. Ranieri, M. Sola and G. Battistuzzi, submitted.
- M. M. G. Krishna, Y. Lin, J. N. Rumbley and S. W. Englander, J. Mol. Biol., 2003, 331, 29–36 CrossRef CAS.
- L. Hoang, H. Maity, M. M. G. Krishna, Y. Lin and S. W. Englander, J. Mol. Biol., 2003, 331, 37–43 CrossRef CAS.
- E. Droghetti, S. Sumithran, M. Sono, M. Antalík, M. Fedurco, J. H. Dawson and G. Smulevich, Arch. Biochem. Biophys., 2009, 489, 68–75 CrossRef CAS.
- S. R. Yeh and D. L. Rousseau, J. Biol. Chem., 1999, 274, 17853–17859 CrossRef CAS.
- R. Pietri, A. Lewis, R. G. León, G. Casabona, L. Kiger, S.-R. Yeh, S. Fernandez-Alberti, M. C. Marden, C. L. Cadilla and J. López-Garriga, Biochemistry, 2009, 48, 4881–4894 CrossRef CAS.
- J. S. Milne, L. Mayne, H. Roder, A. J. Wand and S. W. Englander, Protein Sci., 1998, 7, 739–745 CrossRef CAS.
- T. J. T. Pinheiro, G. A. Elöve, A. Watts and H. Roder, Biochemistry, 1997, 36, 13122–13132 CrossRef CAS.
- T. Ying, Z. H. Wang, Y. W. Lin, J. Xie, X. Tan and Z. X. Huang, Chem. Commun., 2009, 4512–4514 RSC.
- C. M. Lett, A. M. Berghuis, H. E. Frey, J. R. Lepock and J. G. Guillemette, J. Biol. Chem., 1996, 271, 29088–29093 CrossRef CAS.
- M. Cervelli, P. Mariottini, G. Smulevich, M. Coletta and L. Fiorucci, J. Inorg. Biochem., 2017, 169, 86–96 CrossRef.
- J. Gu, D. W. Shin and E. V. Pletneva, Biochemistry, 2017, 56, 2950–2966 CrossRef CAS.
- C. M. Lett, M. D. Rosu-Myles, H. E. Frey and J. G. Guillemette, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1999, 1432, 40–48 CrossRef CAS.
- A. M. Berghuis, J. G. Guillemette, M. Smith and G. D. Brayer, J. Mol. Biol., 1994, 235, 1326–1341 CrossRef CAS.
- S. R. Singh, S. Prakash, V. Vasu and C. Karunakaran, J. Mol. Graphics. Modell., 2009, 28, 270–277 CrossRef CAS.
- A. Schejter, T. L. Luntz, T. I. Koshy and E. Margoliash, Biochemistry, 1992, 31, 8336–8343 CrossRef CAS.
- T. L. Luntz, A. Schejter, E. A. E. Garber and E. Margoliash, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 3524–3528 CrossRef CAS.
- B. A. Feinberg, L. Petro, G. Hock, W. Qin and E. Margoliash, J. Pharm. Biomed. Anal., 1999, 19, 115–125, DOI:10.1016/S0731-7085(98)00291-X.
- G. Battistuzzi, C. A. Bortolotti, M. Bellei, G. Di Rocco, J. Salewski, P. Hildebrandt and M. Sola, Biochemistry, 2012, 51, 5967–5978 CrossRef CAS.
- L. Tognaccini, C. Ciaccio, V. D’Oria, M. Cervelli, B. D. Howes, M. Coletta, P. Mariottini, G. Smulevich and L. Fiorucci, J. Inorg. Biochem., 2016, 155, 56–66 CrossRef CAS.
- S. Casalini, G. Battistuzzi, M. Borsari, A. Ranieri and M. Sola, J. Am. Chem. Soc., 2008, 130, 15099–15104 CrossRef.
- S. Casalini, G. Battistuzzi, M. Borsari, C. A. Bortolotti, A. Ranieri and M. Sola, J. Phys. Chem. B, 2008, 112, 1555–1563 CrossRef CAS.
- S. Casalini, G. Battistuzzi, M. Borsari, C. A. Bortolotti, G. Di Rocco, A. Ranieri and M. Sola, J. Phys. Chem. B, 2010, 114, 1698–1706 CrossRef CAS.
- C. A. Bortolotti, G. Battistuzzi, M. Borsari, P. Facci, A. Ranieri and M. Sola, J. Am. Chem. Soc., 2006, 128, 5444–5451 CrossRef CAS.
- F. Paulat and N. Lehnert, Inorg. Chem., 2008, 47, 4963–4976 CrossRef CAS.
- G. Battistuzzi, C. A. Bortolotti, M. Bellei, G. Di Rocco, J. Salewski, P. Hildebrandt and M. Sola, Biochemistry, 2012, 51, 5967–5978 CrossRef CAS.
- M. Bellei, C. A. Bortolotti, G. Di Rocco, M. Borsari, L. Lancellotti, A. Ranieri, M. Sola and G. Battistuzzi, J. Inorg. Biochem., 2018, 178, 70–86 CrossRef CAS.
- G. Di Rocco, G. Battistuzzi, C. A. Bortolotti, M. Borsari, E. Ferrari, S. Monari and M. Sola, JBIC, J. Biol. Inorg. Chem., 2011, 16, 461–471 CrossRef CAS.
- K. L. Bren and H. B. Gray, J. Am. Chem. Soc., 1993, 115, 10382–10383 CrossRef CAS.
- Y. Lu, D. R. Casimiro, K. L. Bren, J. H. Richards and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11456–11459 CrossRef CAS.
- G. Battistuzzi, M. Borsari, D. Dallari, S. Ferretti and M. Sola, Eur. J. Biochem., 1995, 233, 335–339 CrossRef CAS.
- E. L. Yee and M. J. Weaver, Inorg. Chem., 1980, 19, 1077–1079 CrossRef CAS.
- E. L. Yee, R. J. Cave, K. L. Guyer, P. D. Tyma and M. J. Weaver, J. Am. Chem. Soc., 1979, 101, 1131–1137, DOI:10.1021/ja00499a013.
- V. T. Taniguchi, N. Sailasuta-Scott, F. C. Anson and H. B. Gray, Pure Appl. Chem., 1980, 52, 2275–2281 CAS.
- G. Battistuzzi, M. Borsari, M. Sola and F. Francia, Biochemistry, 1997, 36, 16247–16258 CrossRef CAS.
- G. Battistuzzi, M. Borsari, L. Loschi and M. Sola, JBIC, J. Biol. Inorg. Chem., 1997, 2, 350–359 CrossRef CAS.
- G. Battistuzzi, M. Borsari, L. Loschi, F. Righi and M. Sola, J. Am. Chem. Soc., 1999, 121, 501–506 CrossRef CAS.
- L. Banci, I. Bertini, K. L. Bren, H. B. Gray and P. Turano, Chem. Biol., 1995, 2, 377–383 CrossRef CAS.
- M. R. Cheesman, C. Greenwood and A. J. Thomson, Adv. Inorg. Chem., 1991, 36, 201–255 CrossRef CAS.
- R. Schweitzer-Stenner, J. Phys. Chem. B, 2008, 112, 10358–10366 CrossRef CAS.
- L. Vickery, T. Nozawa and K. Sauer, J. Am. Chem. Soc., 1976, 98, 351–357 CrossRef CAS.
- L. Vickery, K. Sauer and T. Nozawa, J. Am. Chem. Soc., 1976, 98, 343–350 CrossRef CAS.
- N. Tomášková, R. Varhač, V. Lysáková, A. Musatov and E. Sedlák, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 1073–1083 CrossRef.
- D. W. Urry, J. Biol. Chem., 1967, 242, 4441–4448 CAS.
- E. H. Strickland and S. Beychok, CRC Crit. Rev. Biochem., 1974, 2, 113–175 CrossRef CAS.
- A. M. Davies, J. G. Guillemette, M. Smith, C. Greenwood, A. G. P. Thurgood, A. G. Mauk and G. R. Moore, Biochemistry, 1993, 32, 5431–5435 CrossRef CAS.
- H. R. Schroeder, F. A. McOdimba, J. G. Guillemette and J. A. Kornblatt, Biochem. Cell Biol., 1997, 75, 191–197 CrossRef CAS.
- N. Sanghera and T. J. T. Pinheiro, Protein Sci., 2009, 9, 1194–1202 CrossRef.
- M. J. Tarlov and E. F. Bowden, J. Am. Chem. Soc., 1991, 113, 1847–1849 CrossRef CAS.
- G. Battistuzzi, M. Bellei, M. Borsari, G. Di Rocco, A. Ranieri and M. Sola, JBIC, J. Biol. Inorg. Chem., 2005, 10, 643–651 CrossRef CAS.
- M. Bellei, C. Jakopitsch, G. Battistuzzi, M. Sola and C. Obinger, Biochemistry, 2006, 45, 4768–4774 CrossRef CAS.
- G. Battistuzzi, M. Bellei, M. Zederbauer, P. G. Furtmüller, M. Sola and C. Obinger, Biochemistry, 2006, 45, 12750–12755 CrossRef CAS.
- G. Battistuzzi, M. Bellei, L. Casella, C. A. Bortolotti, R. Roncone, E. Monzani and M. Sola, JBIC, J. Biol. Inorg. Chem., 2007, 12, 951–958 CrossRef CAS.
- G. Battistuzzi, M. Bellei, J. Vlasits, S. Banerjee, P. G. Furtmüller, M. Sola and C. Obinger, Arch. Biochem. Biophys., 2010, 494, 72–77 CrossRef CAS.
- J. Vlasits, M. Bellei, C. Jakopitsch, F. De Rienzo, P. G. Furtmüller, M. Zamocky, M. Sola, G. Battistuzzi and C. Obinger, J. Inorg. Biochem., 2010, 104, 648–656 CrossRef CAS.
- G. Battistuzzi, J. Stampler, M. Bellei, J. Vlasits, M. Soudi, P. G. Furtmüller and C. Obinger, Biochemistry, 2011, 50, 7987–7994 CrossRef CAS.
- S. Hofbauer, K. Gysel, M. Bellei, A. Hagmüller, I. Schaffner, G. Mlynek, J. Kostan, K. F. Pirker, H. Daims, P. G. Furtmüller, G. Battistuzzi, K. Djinović-Carugo and C. Obinger, Biochemistry, 2014, 53, 77–89 CrossRef CAS.
- M. Paumann-Page, R.-S. S. Katz, M. Bellei, I. Schwartz, E. Edenhofer, B. Sevcnikar, M. Soudi, S. Hofbauer, G. Battistuzzi, P. G. Furtmüller and C. Obinger, J. Biol. Chem., 2017, 292, 4583–4592 CrossRef CAS.
- G. Di Rocco, A. Ranieri, C. A. Bortolotti, G. Battistuzzi, A. Bonifacio, V. Sergo, M. Borsari and M. Sola, Phys. Chem. Chem. Phys., 2013, 15, 13499 RSC.
- X. Liu, Y. Huang, W. Zhang, G. Fan, C. Fan and G. Li, Langmuir, 2005, 21, 375–378 CrossRef CAS.
- S. Monari, G. Battistuzzi, M. Borsari, G. Di Rocco, L. Martini, A. Ranieri and M. Sola, J. Phys. Chem. B, 2009, 113, 13645–13653 CrossRef CAS.
- G. Battistuzzi, M. Bellei, C. A. Bortolotti and M. Sola, Arch. Biochem. Biophys., 2010, 500, 21–36 CrossRef CAS.
- S. Monari, G. Battistuzzi, M. Borsari, D. Millo, C. Gooijer, G. Van Der Zwan, A. Ranieri and M. Sola, J. Appl. Electrochem., 2008, 38, 885–891 CrossRef CAS.
- G. Battistuzzi, M. Borsari, J. A. Cowan, C. Eicken, L. Loschi and M. Sola, Biochemistry, 1999, 38, 5553–5562 CrossRef CAS.
- B. Dangi, S. Sarma, C. Yan, D. L. Banville and R. D. Guiles, Biochemistry, 1998, 2960, 8289–8302, DOI:10.1021/bi9801964.
- G. Di Rocco, F. Bernini, M. Borsari, I. Martinelli, C. A. Bortolotti, G. Battistuzzi, A. Ranieri, M. Caselli, M. Sola and G. Ponterini, Zeitschrift fur Phys. Chemie, 2016, 230, 1329–1349 CAS.
- G. Battistuzzi, M. Borsari, A. Ranieri and M. Sola, J. Am. Chem. Soc., 2002, 124, 26–27 CrossRef CAS.
- G. Battistuzzi, M. Borsari, C. A. Bortolotti, G. Di Rocco, A. Ranieri and M. Sola, J. Phys. Chem. B, 2007, 111, 10281–10287 CrossRef CAS.
- S. Hofbauer, M. Bellei, A. Sündermann, K. F. Pirker, A. Hagmüller, G. Mlynek, J. Kostan, H. Daims, P. G. Furtmüller, K. Djinović-Carugo, C. Oostenbrink, G. Battistuzzi and C. Obinger, Biochemistry, 2012, 51, 9501–9512 CrossRef CAS.
- V. Pfanzagl, M. Bellei, S. Hofbauer, C. V. F. P. Laurent, P. G. Furtmüller, C. Oostenbrink, G. Battistuzzi and C. Obinger, J. Inorg. Biochem., 2019, 199, 110761 CrossRef CAS.
- G. Battistuzzi, M. Borsari, J. A. Cowan, A. Ranieri and M. Sola, J. Am. Chem. Soc., 2002, 124, 5315–5324 CrossRef CAS.
- G. Battistuzzi, M. Bellei, F. De Rienzo and M. Sola, JBIC, J. Biol. Inorg. Chem., 2006, 11, 586–592 CrossRef CAS.
- G. Battistuzzi, M. Borsari, G. Di Rocco, A. Ranieri and M. Sola, JBIC, J. Biol. Inorg. Chem., 2004, 9, 23–26 CrossRef CAS.
- E. Grunwald, J. Am. Chem. Soc., 1986, 108, 5726–5731 CrossRef CAS.
- L. Liu and Q.-X. Guo, Chem. Rev., 2001, 101, 673–696 CrossRef CAS.
- L. Liu, C. Yang and Q.-X. Guo, Biophys. Chem., 2000, 84, 239–251 CrossRef CAS.
- G. Battistuzzi, M. Bellei, C. Dennison, G. Di Rocco, K. Sato, M. Sola and S. Yanagisawa, JBIC, J. Biol. Inorg. Chem., 2007, 12, 895–900 CrossRef CAS.
- E. B. Starikov, Chem. Phys. Lett., 2013, 564, 88–92 CrossRef CAS.
- E. B. Starikov and B. Norden, J. Phys. Chem. B, 2007, 14431–14435, DOI:10.1021/jp075784i.
- E. B. Starikov and B. Nordén, Chem. Phys. Lett., 2012, 538, 118–120 CrossRef CAS.
- L. Movileanu and E. A. Schiff, Monatshefte für Chemie – Chem. Mon., 2013, 144, 59–65 CrossRef CAS.
- G. Battistuzzi, M. Borsari, G. W. Canters, E. De Waal, A. Leonardi, A. Ranieri and M. Sola, Biochemistry, 2002, 41, 14293–14298 CrossRef CAS.
- M. Bellei, G. Battistuzzi, S. pao Wu, S. S. Mansy, J. A. Cowan and M. Sola, J. Inorg. Biochem., 2010, 104, 691–696 CrossRef CAS.
- J. B. Soffer and R. Schweitzer-Stenner, J. Biol. Inorg. Chem., 2014, 19, 1181–1194 CrossRef CAS.
- J. M. Fox, M. Zhao, M. J. Fink, K. Kang and G. M. Whitesides, Annu. Rev. Biophys., 2018, 47, 223–250 CrossRef CAS.
- G. Battistuzzi, M. Bellei, M. Borsari, G. W. Canters, E. De Waal, L. J. C. Jeuken, A. Ranieri and M. Sola, Biochemistry, 2003, 42, 9214–9220 CrossRef CAS.
- S. Monari, A. Ranieri, C. A. Bortolotti, S. Peressini, C. Tavagnacco and M. Borsari, Electrochim. Acta, 2011, 56, 6925–6931 CrossRef CAS.
- E. Grunwald and C. Steel, J. Am. Chem. Soc., 1995, 117, 5687–5692 CrossRef CAS.
- D. N. LeBard and D. V. Matyushov, J. Chem. Phys., 2008, 128, 155106 CrossRef.
- D. V. Matyushov, J. Chem. Phys., 2013, 139, 25102 CrossRef.
- G. Battistuzzi, M. Borsari, G. Di Rocco, A. Ranieri and M. Sola, JBIC, J. Biol. Inorg. Chem., 2004, 9, 23–26 CrossRef CAS.
- D. Shortle, in Protein Stability, ed. C. B. Anfinsen, F. M. Richards, J. T. Edsall and D. S. B. T.-A., P. C. Eisenberg, Academic Press, 1995, vol. 46, pp. 217–247 Search PubMed.
- K. Dill, Annu. Rev. Biochem., 1991, 60, 795–825 CrossRef CAS.
- L. Hua, R. Zhou, D. Thirumalai and B. J. Berne, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16928–16933 CrossRef CAS.
- P. J. Rossky, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16825–16826 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |