
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocyclic peptides that inhibit Wnt signalling via interaction with Wnt3a†
Manuel E.
Otero-Ramirez
 a,
Kyoko
Matoba
b,
Emiko
Mihara
b,
Toby
Passioura
ac,
Junichi
Takagi
*b and
Hiroaki
Suga
*a
a,
Kyoko
Matoba
b,
Emiko
Mihara
b,
Toby
Passioura
ac,
Junichi
Takagi
*b and
Hiroaki
Suga
*a
aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: hsuga@chem.s.u-tokyo.ac.jp
bLaboratory of Protein Synthesis and Expression, Institute for Protein Research, Osaka University, 3-2 Yamadaoka, Suita-shi, Osaka 565-0871, Japan. E-mail: takagi@protein.osaka-u.ac.jp
cSydney Analytical, School of Chemistry and School of Life and Environmental Sciences, The University of Sydney, Sydney 2006, Australia
First published on 24th March 2020
Abstract
Here we report de novo macrocyclic peptide binders to Wnt3a, a member of the Wnt protein family. By means of the Random non-standard Peptides Integrated Discovery (RaPID) system, we have performed in vitro selection against the complex of mouse Wnt3a (mWnt3a) with human afamin (hAFM) to discover macrocyclic peptides that bind mWnt3a with KD values as tight as 110 nM. One of these peptides, WAp-D04 (Wnt–AFM-peptide-D04), was able to inhibit the receptor-mediated signaling process, which was demonstrated in a Wnt3a-dependent reporter cell-line. Based on this initial hit, we applied a block-mutagenesis scanning display to identify a mutant inhibitor, WAp-D04-W10P, with 5-fold greater potency in a reporter assay. This work represents the first instance of molecules capable of inhibiting Wnt signaling through direct interaction with a Wnt protein, a molecular class for which targeting has been challenging due its highly hydrophobic nature.
Introduction
Wnt pathway signalling regulates the expression of genes affecting differentiation and proliferation, and represents one of the best-studied mechanisms underlying development and homeostasis.10,11 In its β-catenin-dependent (so-called “canonical”) pathway, Wnt signalling is initiated by the binding of secreted Wnt proteins to cell-surface Frizzled (Fz) receptors. With the help of co-receptor molecule lipoprotein receptor-related protein (LRP) 5/6, this initiates signalling cascades that elevate the level of free cytoplasmic β-catenin, making it available for translocation to the nucleus, thereby resulting in alteration of gene expression (Fig. 1).16,17 This signalling cascade is complicated by the involvement of numerous Wnt and Fz isoforms (19 Wnts and 10 Fz in mammals), which exhibit differential binding and signalling responses. Both Wnt and Fz proteins are highly conserved among species, and the Wnt pathway plays critical roles not only during development but also in disease states such as oncological processes, particularly colorectal cancer.17,19,20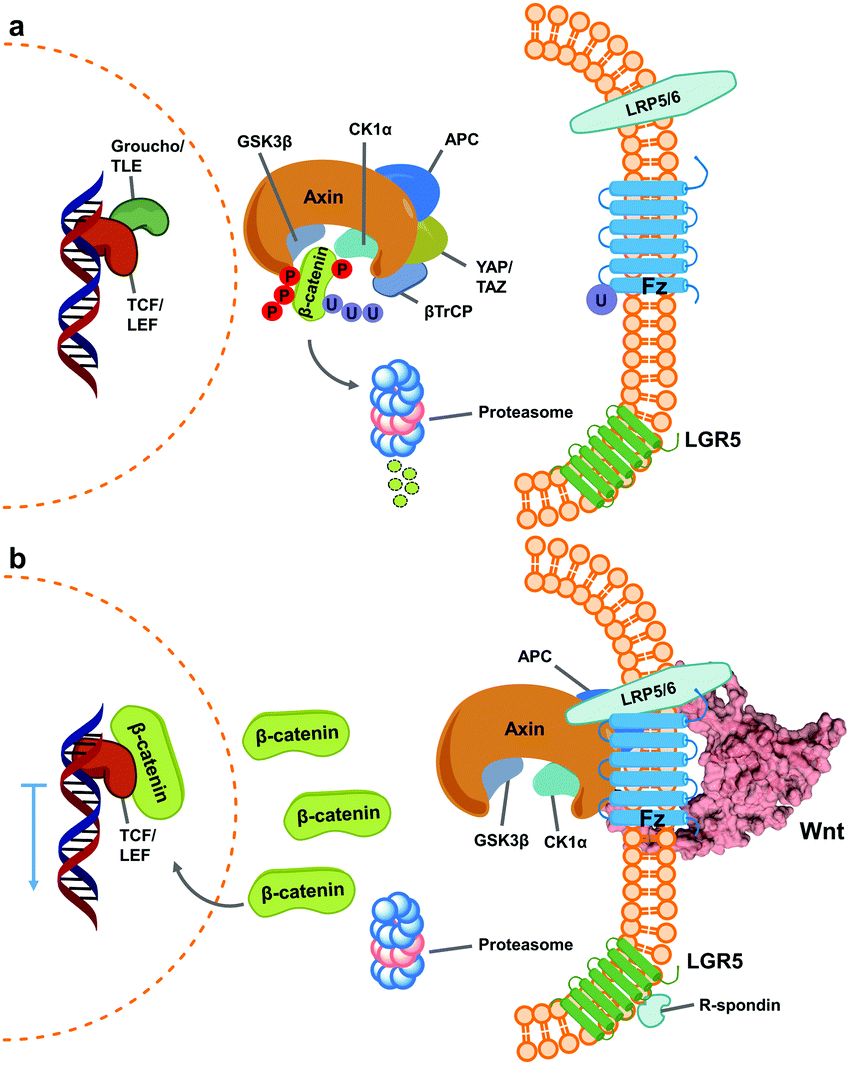 | ||
| Fig. 1 Simplified scheme of canonical Wnt signalling pathway in an inactive and active state. (a) When inactive, cytoplasmic concentration of the protein β-catenin is regulated by the destruction complex, a multi-unit structure that targets it for proteasome degradation.1–3 At this stage, regulated genes’ translation is blocked by Groucho/TLE repressor action on TCF/LEF transcription factors.4,5 (b) Activation of Wnt signalling occurs upon binding of Wnt ligands (shown here as PDB 6AHY) on Fz receptors, followed by oligomerization with LRP 5/6, promoting sequestering of the destruction complex (as one of varied mechanisms for its inactivation) and impeding β-catenin degradation.6–9 Consequently, cytoplasmic β-catenin levels increase and it translocates into the nucleus, binding TCF/LEF and calling other transcription-promoters to allow gene expression.12,13 The mechanism is further regulated by binding of R-spondin agonists on leucine-rich repeat-containing G-protein-coupled receptor (LGR) 5, inhibiting ubiquitin ligase activity on Fz receptors to increase their membrane population.14,15 | ||
A characteristically conserved Ser residue in Wnt proteins, e.g., Ser209 in Wnt3a, is modified by palmitoleic acid by a specific enzyme called Porcupine, and it is responsible for binding to Fz receptors.21–25 A crystal structure of a complex of mouse Fz8's cysteine-rich domain (CRD) with X. laevis Wnt8 has shed light on the functional importance of this lipid, mediating the interaction with the hydrophobic groove on Fz.26 Moreover, a human Wnt3 in complex with mouse Fz8's CRD has also revealed a nearly identical binding mode (Fig. 2a), where their 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex accommodates two acyl chains in-between parallel opposite sides of the Fz–Fz interface, suggesting a possible role in the receptor's homodimerization during signalling.27 Due to this lipidated nature, however, purified Wnt proteins can only be solubilized in the presence of detergents, and are notoriously unstable during storage.23,28 Recently, a method to isolate water-soluble and bioactive Wnt proteins was established by making it complexed with a protein called afamin (AFM) (Fig. 2b).29 AFM is a plasma glycoprotein that belongs to the albumin superfamily and is thought to serve as a transporter of small hydrophobic molecules like vitamin E,30,31 and we have found it is likely the main serum component responsible for Wnt solubilisation in cell culture supernatant.29 It was also found that Wnt3a complexed with AFM was fully soluble in a physiological buffer while maintaining its biological activity, indicating that binding by AFM does not interfere with the binding of Wnt3a to the critical signalling component, e.g., Fz receptors. Thus, the Wnt–AFM complex offers a unique opportunity to present hydrophobic Wnt proteins in a detergent-free environment without shielding its biologically important molecular surfaces.
:2 complex accommodates two acyl chains in-between parallel opposite sides of the Fz–Fz interface, suggesting a possible role in the receptor's homodimerization during signalling.27 Due to this lipidated nature, however, purified Wnt proteins can only be solubilized in the presence of detergents, and are notoriously unstable during storage.23,28 Recently, a method to isolate water-soluble and bioactive Wnt proteins was established by making it complexed with a protein called afamin (AFM) (Fig. 2b).29 AFM is a plasma glycoprotein that belongs to the albumin superfamily and is thought to serve as a transporter of small hydrophobic molecules like vitamin E,30,31 and we have found it is likely the main serum component responsible for Wnt solubilisation in cell culture supernatant.29 It was also found that Wnt3a complexed with AFM was fully soluble in a physiological buffer while maintaining its biological activity, indicating that binding by AFM does not interfere with the binding of Wnt3a to the critical signalling component, e.g., Fz receptors. Thus, the Wnt–AFM complex offers a unique opportunity to present hydrophobic Wnt proteins in a detergent-free environment without shielding its biologically important molecular surfaces.
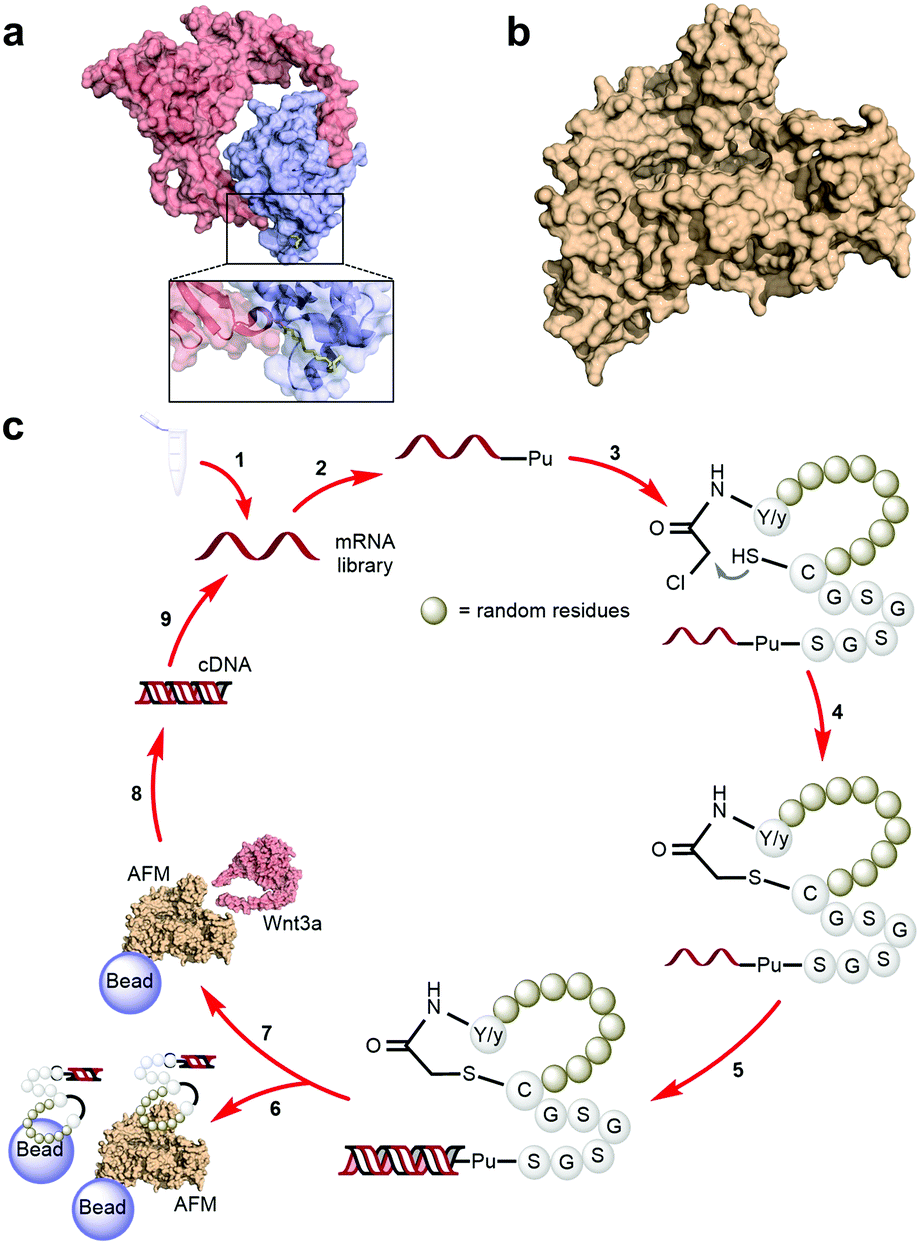 | ||
| Fig. 2 Human Wnt3, human AFM and RaPID selection of macrocyclic peptide ligands against mWnt3a–hAFM. (a) Crystal structure of a Wnt (hWnt3) ligand (red) bound to its natural Fz receptor (mFz8, blue) as identified by Hirai et al. (PDB 6AHY, with an inset highlighting the palmitoleic acid moiety in yellow). (b) Wnt-stabilizing protein partner (hAFM) used in this work (PDB 6FAK). (c) RaPID selection scheme for the identification of macrocyclic peptides capable of binding to mWnt3a: (1) the designed mRNA library is prepared, (2) ligated with Pu using a T4 RNA ligase and (3) translated into peptides using the FIT system to generate peptide-Pu-mRNA fusions, after which (4) macrocyclization would occur by reaction of L-Tyr (Y) or D-Tyr (y) with a downstream Cys at the end of the random region (brown spheres) or within the random region itself. (5) This macrocyclic peptide library is ligated to each cognate RNA through a (Gly-Ser)3 and Pu-linker region, and the RNA template is reverse transcribed into its cDNA. (6) The library is then exposed several times to a combination of unloaded, biotin-saturated and biotinylated hAFM-loaded magnetic beads in order to lower the likelihood of recovering non-specific binders as much as possible. These fractions are discarded and (7) the depleted library is then exposed in a single cycle to the biotinylated mWnt3a–hAFM complex-loaded magnetic beads. (8) cDNA bound to the complex is then recovered and amplified by PCR followed by (9) transcription to generate the enriched mRNA library for the next round. | ||
Great efforts have been carried out recently targeting Wnt signalling for drug discovery. In particular, the pathway's relationship with cancer has been long established, boosting the development of therapeutics including small molecules,32 antibodies,33 stabilizing lipid systems34 or even receptor-dimerizing agonists.35 However, to the best of our knowledge, an isolated Wnt protein itself has not been successfully targeted (or at least such an outcome has not been yet documented).
Macrocyclic peptides are an emerging class of potential pharmacological modulators capable of interacting with a wide range of protein targets.36–38 Among the technologies for screening of such compounds, the RaPID (Random non-standard Peptides Integrated Discovery) system has proven to be highly reliable for uncovering de novo macrocyclic peptides from a pool of random sequences consisting of more than 1012 members. Such peptides have exhibited not only strong affinity and specificity to targets of interest, but also antagonize or even agonize the signalling cascade.39–44 Taking advantage of the RaPID system and the unique AFM-based Wnt-stabilizing technology, here we have challenged discovering macrocyclic peptides that interact with mouse Wnt3a (mWnt3a), leading us to identify modulators of Wnt3a signalling.
Experimental
Protein expression and purification
Biologically active mouse Wnt3a (mWnt3a) in complex with human afamin (hAFM) was prepared according to the method described previously.29 Briefly, N-terminally PA-tagged mWnt3a and N-terminally Target-tagged hAFM were co-transfected into Expi293F cells to establish a stable cell line secreting the Wnt3a–AFM complex, followed by the purification of the complex by using an anti-PA tag NZ-1 antibody column.45 The purified complex dissolved in PBS (20 mM phosphate, 150 mM NaCl, pH 7.0) was biotinylated by EZ-Link NHS-Lc-Biotin (Thermo Fisher Scientific Inc.) to facilitate bead immobilization. Biotin incorporation was estimated to be 1–2 moles of biotin per mole Wnt3a. We also confirmed that the biotinylation did not diminish biological activity of mWnt3a by using a TOP flash reporter assay. Uncomplexed hAFM was expressed in a similar manner and purified by anti-Target tag P20.1 antibody column,46 biotinylated, and immobilized onto the beads for the negative selections. For certain SPR assays, hAFM fused with human IgG1 Fc was used in place of the Target-tagged hAFM described above.mRNA library preparation
The initial randomized library was designed to contain a T7 Polymerase and ribosome binding site, a peptide encoding region constituted by an initiating AUG codon followed by 6 to 15 contiguous NNK codons (in independent libraries), a UGC Cys codon and a (Gly-Ser)3 region, in turn followed by a stop codon and a small sequence region complementary to a DNA splint used for Puromycin ligation. Independent libraries were transcribed and purified through gel electrophoresis, resuspended in water to a 10 μM concentration, and then combined in equal volumes for a total 10 μM concentration. For the WAp-D04-focused library, the inhibitor's structure was varied as shown in Fig. 4a for a total of 50% randomization (corresponding to 5 amino acids) in 5 different groups, each with increasing number of sequential randomized residues. A total of 31 independent libraries were purified separately as stated, and combined again in equal volumes to a total 10 μM concentration.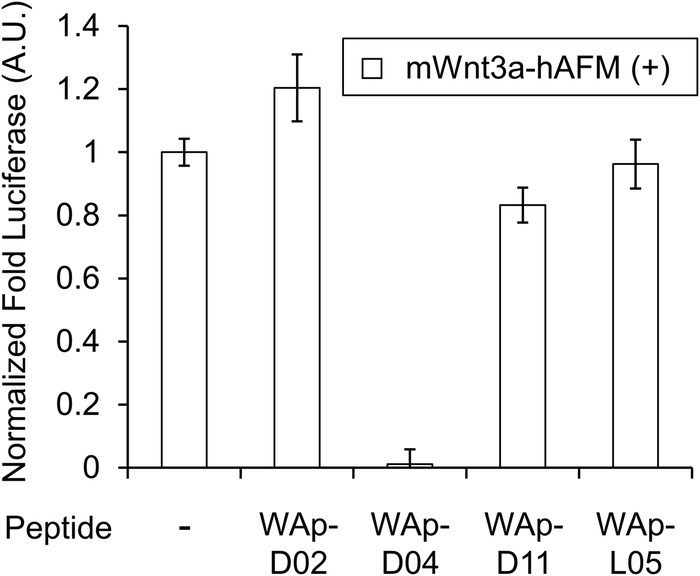 | ||
| Fig. 3 Inhibition of Wnt3a signalling by WAp-D04. Luciferase reporter activity was assessed following cell treatment with mWnt3a–hAFM, in the presence or absence of 1 μM candidate WAp-peptide. Bars show mean values of a set of 4 data points, all relative to a reference signal stimulated by mWnt3a–hAFM in the absence of any peptide, while error bars indicate standard error (n = 4). Data was normalized to a blank signal generated in the presence of hAFM alone and in the absence of any peptide, subtracted from every value. Full data for this experiment is available as ESI.† | ||
RaPID selection
Peptide libraries were prepared from initial 10 μM RNA libraries using the FIT system in a similar manner as previously reported,47,48 aiming for a total 150 μL translation for the first round of selection. Briefly, mRNA libraries were ligated to Puromycin using a T4 RNA Ligase at 25 °C for 30 minutes, then extracted once with 1 volume of phenol-chloroform-isoamyl alcohol (PCI) solution and again with a chloroform-isoamyl alcohol (CI) solution, then precipitated by addition of 1/10 volume of 3 M NaCl and twice its volume of ethanol and finally washed with 70% ethanol, briefly air-dried and resuspended in water. The libraries were then translated using a Met-deficient FIT system in the presence of 50 μM ClAc-D-Tyr-tRNAfMetCAU or ClAc-L-Tyr-tRNAfMetCAU to give the corresponding initiating stereochemistry to the first amino acid in each library. Translation was then carried out at 37 °C for 30 minutes, and after an incubation period of 12 minutes at 25 °C one tenth of its volume of 100 mM EDTA was added to the reaction mix and left incubating again for an additional period of 30 minutes to finalize macrocyclization. mRNA-peptide product libraries were then reverse transcribed to its corresponding cDNA using an MLV (H-) Reverse Transcriptase (Promega) for 1 hour at 42 °C, mixed with acetylated BSA in TBS-T pH 7.5 buffer to a final 1 mg mL−1 concentration, neutralized with the same buffer and desalted using a Sephadex G-25 Fine (GE Healthcare) column previously swelled in the same buffer, to a total of 400 μL. At this point, 0.5 μL of the final library was diluted 1000 times in water to make an input sample reference to calculate recovered cDNA after the selection process. Prepared libraries were then exposed to the target mWnt3a–hAFM complex previously immobilized on M-280 Streptavidin Dynabeads (LifeTechnologies®) for a total of 80 pmol loaded target (200 nM target complex concentration when resuspended in the peptide pool). Peptide pools containing these loaded beads were then incubated at 4 °C for 30 minutes, washed 3 times with cold TBS-T pH 7.5 buffer to discard the unbound peptide fraction and finally resuspended in cold PCR buffer. cDNA cognate to the target-binding peptide fraction was then recovered by heating the samples 95 °C for 5 minutes and separating the supernatant. A small amount of the input and recovered cDNA samples were then analysed using real-time qPCR on a Roche LightCycler® system and the remainder was amplified by PCR. The amplified samples were then purified by one extraction each with PCI and CI solutions, precipitated with ethanol and resuspended in water. The resulting samples were then transcribed to mRNA, forming the initial library for the next round. Following rounds of selection were carried out using the same scheme, though performing translation at a total 5 μL scale and 10 pmol of target protein (still maintaining final 200 nM when exposed to libraries). Additionally, before incubating the library in the target complex, they were first exposed in 10 separate cycles to a mixture containing 25% free M-280 Streptavidin Dynabeads (LifeTechnologies®), 25% of the same beads saturated with biotin and 50% of the same beads loaded only with hAFM in twice the molar amount of the objective target concentration (for a total of 20 pmol). These “negative selection” cycles were performed in order to lower the possibility of recovering non-specific peptides or those with preferential binding to the protein partner instead of the original target mWnt3a. The cycles were carried out for at 4 °C for 30 minutes for the first and last cycles, and otherwise for 15 minutes at the same temperature. Moreover, an additional 20 times the molar amount of free (untagged) hAFM were added to the final samples before exposing them to the target mWnt3a–hAFM complex to further decrease the recovery of non-specific peptides. Samples were then incubated in beads containing 10 pmol of the target and corresponding cDNA was recovered and amplified as before.Peptide synthesis and purification
Selected peptides were synthesized through standard solid-phase peptide synthesis with a Biotage Syro I® parallel synthesis system using a common Fmoc-protected amino acid chemistry on NovaPEG Rink amide resin at a 25 μmol scale. Deprotection of the final N-terminal was carried out as a last step, and the loaded resin was washed 5 times with dimethylformamide (DMF) and dichloromethane (DCM). Resin was then swelled in DCM for 10 minutes, resuspended in 0.2 M diisopropylethylamine (DIPEA) and then washed 6 times with DMF. Chloro-acetylation of the free N-terminal amino acid was carried out by addition of a coupling mixture containing 6.3 equivalents each of chloro-acetic acid (ClAcOH), HOBt and diisopropylcarbodiimide (DIC), reacting for 1 hour at room temperature. Resin was then washed 3 times with DMF, methanol (MeOH) and DCM before drying in vacuo. Deprotection and cleavage of the peptides from the resin was then performed by resuspending the resin in 2 mL of a TFA deprotection cocktail containing 2.5%v/v each of triisopropylsilane (TIS), 3,6-dioxa-1,8-octanedithiol (DODT) and water for 3 hours at room temperature under continuous mixing. Resin was then washed twice with 2 mL TFA, and the peptide solution was then concentrated by centrifugal evaporation, later precipitating the cleaved peptides with cold ether. Collected peptides were then washed with cold ether 5 times and dried in vacuo before dissolving in DMSO. Cyclization of peptides was finally carried out in the presence of trimethylamine (TEA). Peptides were then purified by reverse-phase HPLC in a Shimadzu LC-20AP system using a Merck Cromolith® Column with a gradient of 0.1% TFA/acetonitonitrile containing 0.1% TFA, and collected fractions were then concentrated and freeze-dried. The resulting peptides were dissolved in low-concentration HCl for exchange to an HCl salt and freeze-dried again twice. Final peptides were dissolved in a small volume of DMSO and reserved at −20 °C. Structural identity of each peptide and their evolution through the synthesis scheme was confirmed through MALDI-TOF MS, while the final purity of the samples by UPLC.Surface plasmon resonance
For the evaluation of the interaction between the mWnt3a and the peptides obtained in the initial selection series, purified mWnt3a–hAFM complex or hAFM alone were directly immobilized onto the Series S Sensor Chip CM5 (GE healthcare; 29104988) at a surface density of ∼7800 RU (for complex) or ∼5600 RU (for hAFM), respectively, using an amine coupling kit (GE healthcare; BR100050) according to the manufacturer's protocol. The binding was evaluated by injecting peptide solutions serially diluted using the running buffer (20 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, 0.05% Surfactant P20) maintaining the final concentration of DMSO as 0.1%. The runs were conducted in a single cycle kinetics mode employing the following parameters; flow rate of 30 μl min−1, contact time of 120 s, and dissociation time of 300 s. After each run, the surface was regenerated by injecting 3 M MgCl2 until the response returned to the original baseline level. The binding curves of the measurement cell (mWnt3a–hAFM) were subtracted with that of reference cell (hAFM), and used to derive KD values. Data were obtained using a BIAcoreT200 instrument (GE Healthcare) at 25 °C, and the results were analysed by using BIAcoreT200 evaluation software version 4.1. For the evaluation of the mWnt3a-binding properties of the peptides obtained in the second round of selection from the focused library, hAFM fused with human IgG1 Fc was used in place of the Target-tagged hAFM described above. Thus mWnt3a–hAFM-Fc complex as well as hAFM-Fc were expressed in Expi293F cells and purified by either immobilized NZ-1 antibody resin or rProteinA Sepharose Fast Flow (GE healthcare, 17-1279-03) and immobilized on a series S Sensor Chip CM5 via Human Fc capture kit (GE healthcare; BR100839) at a density of ∼10000 RU. The binding of peptides was evaluated similarly to the method described above, except for the use of steady-state affinity analysis mode in the BIAevaluation 4.1.
Luciferase reporter assay
The effects of each peptide on the biological activity of mWnt3a were evaluated in a paracrine-type reporter cell assay as described previously.29 Briefly, HEK293 cells stably transfected with a firefly luciferase gene under the control of a TCF/LEF response element (BPS Bioscience, #60531) were seeded at 30000 cells per 50 μL per well in 96-well white-clear plates (Corning, #3610), followed by an overnight culture before the assay. 10 μL of PBS containing 100 nM purified mWnt3a–hAFM complex or hAFM alone and 40 μL of peptide solution in 10% serum-containing media were added to the reporter cells, maintaining the final concentration of DMSO at 0.1%. After culturing under 5% CO2 at 37 °C for 6 h, cells were briefly washed with PBS and lysed by adding 20 μL per well of cell lysis buffer (Promega, E153A). Luciferase activities of the lysates were directly measured in the same plate using the Luciferase assay reagent (Promega, E1500) and an SH-9000 microplate reader (Corona Electric, Inc.).
Results
Library design and selection
In order to identify direct inhibitors of Wnt3a signalling, we employed an affinity-based selection by means of the RaPID system. Although Wnt3a alone can readily denature upon isolation, the Wnt ligand can be stabilized through the formation of a complex with AFM, which would give us an opportunity to explore the available interaction surface with Fz receptors. We thus decided to use the bioactive mWnt3a–human AFM (hAFM) complex as a bait protein. We then applied our standard mRNA library consisting of AUG, (NNK)6–15 (N = A, C, G or U and K = U or G), UGC, followed by (GGCAGC)3-UAG to express two independent, thioether-cyclized macrocyclic peptide libraries under a reprogrammed genetic code assisted by flexizymes.48–50 The AUG initiator codon was assigned to L-N-chloroacetyl-Tyr (Y-library) or D-N-chloroacetyl-Tyr (y-library) in order to induce subsequent head-to-sidechain thioether cyclization with a downstream Cys residue, which could appear in the random region and/or the designated UGC codon after the random region.51 A triple repeat of a Gly-Ser peptide linker assigned by (GGCAGC)3 allowed linking to a 3′-puromycin-CC-PEG-5′ attached to the 3′-end of the mRNA library.Upon the ribosome-catalysed specific formation of a peptide linked to its cognate puromycin-mRNA (Pu-mRNA) in the absence of RF-1, the resulting peptide-mRNA fusion was “counter-selected” against immobilized hAFM in order to deplete species that had affinity for hAFM. Following 10 cycles of counter-selection, the peptide library was then panned against a beads-immobilized mWnt3a–hAFM complex in the presence of free soluble hAFM to further reduce the binding of peptides unselective for mWnt3a. We hypothesized that inclusion of these extra steps were necessary since AFM, like serum albumin, has a strong tendency to bind hydrophobic molecules and may give very high background binding to various hydrophobic peptide sequences. The cognate cDNAs for bound peptides were then recovered by PCR amplification and transcribed in vitro to generate an enriched mRNA library for the subsequent round of selection (Fig. 2c). However, despite our extensive precautions to deplete hAFM-binding peptides by the negative (counter) selection procedure, qPCR after 5 sequential rounds of selection indicated similar levels of cDNA recovery from the hAFM-coated counter-selection beads and the mWnt3a/hAFM-coated beads (Fig. S1, ESI†), suggesting that the recovered peptide libraries could still be contaminated by species that bind to hAFM rather than mWnt3a.
To circumvent this problem, we decided to perform a 2-dimensional deep-sequencing analysis comparing the most enriched sequences found to bind hAFM alone or the mWnt3a–hAFM complex. For this analysis, the cDNA libraries in the 5th round targeting either hAFM alone (negative clones) or mWnt3a–hAFM complex (positive clones) were sequenced separately, and the individual sequences of each were compared in terms of identity and frequency (those appearing near the x-axis of “Frequency of positive clones” in Fig. S2 and S3, ESI†). We hoped this 2D analysis would allow us to discriminate macrocyclic peptides that might have specific affinity to mWnt3a over those binding to hAFM.
From the group of enriched peptide sequences that appeared to have higher frequencies of deep-sequencing reads to the mWnt3a–hAFM complex than those to hAFM alone, 14 structurally distinct peptides were chosen for the monoclonal RaPID-display recovery assay (Table S1, ESI†). We found that all of these peptides showed favourable binding to the mWnt3a–hAFM complex over hAFM or mock beads (Fig. S4, ESI†). These were then chemically synthesized by solid-phase peptide synthetic method, purified by HPLC, and their structure and purity were confirmed through MALDI-TOF-MS and UPLC. It is important, however, to note that even with all the cautions taken and analysis performed, our approach does not necessarily ensure all of them could have a biological effect on Wnt signalling, i.e. those capable of modulating the interaction between mWnt3a and cognate Fz, which made necessary further characterization of the individual macrocyclic peptides for not only binding affinity but also bioactivity.
Binding profile and activity of selected peptides
The binding properties of each of the 14 synthesized peptides was assessed using surface plasmon resonance (SPR) against hAFM alone or mWnt3a–hAFM immobilized through amine coupling. However, unambiguous binding affinities could be determined only for 4 of these peptides that yielded reliable SPR profiles (Fig. S5, ESI†). These 4 macrocyclic peptides exhibited specific binding toward mWnt3a with sub-micromolar equilibrium dissociation constants (KD in Table 1).| Peptide name | Sequence | Number of reads | k a × 104 (M−1 s−1) | k d × 10−3 (s−1) | K D (nM) |
|---|---|---|---|---|---|
| WAp-D02 |
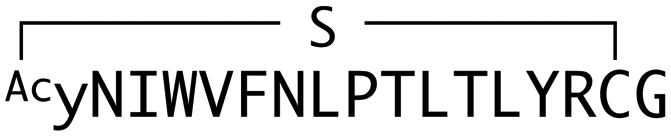
|
3010 | 1.12 | 5.47 | 490 |
| WAp-D04 |
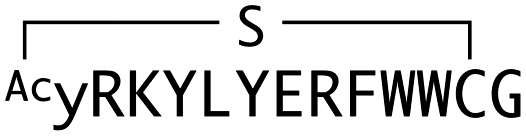
|
440 | 4.05 | 6.92 | 171 |
| WAp-D11 |

|
150 | 13.85 | 44.10 | 318 |
| WAp-L05 |

|
477 | 2.72 | 3.01 | 110 |
We next sought to assess the effect of these peptides on the Wnt3a-mediated signalling in a cell culture model. To validate this, we made use of a TCF/LEF element-dependent Wnt signalling luciferase reporter cells,52 and measured reporter response following exposure to purified mWnt3a–hAFM in the presence of the selected peptides shown in Table 1 (Fig. 3). Notably, this initial bioactivity screening identified a single hit peptide, WAp-D04 (one of the most abundant species in the y-library), which exhibited nearly complete inhibition of Wnt3a signalling at a 1 μM concentration. The activity titration of WAp-D04 in a range of concentrations of 7.8–4000 nM determined an IC50 value of approximately 290 nM (Fig. 5).
WAp-D04 constitutes, to our knowledge, the first instance of a non-naturally occurring molecule that inhibits the Wnt signalling pathway by binding directly to a specific Wnt ligand, mWnt3a. Although the finding of such a unique and bioactive peptide with selective binding to mWnt3a constitutes an advance in developing synthetic modulators of Wnt signalling, we considered its binding and activity properties could be further improved. Notably, our unusual approach targeting mWnt3a in complex with a large stabilizing partner, hAFM, allowed us access to a very rare inhibitory species by our 2D analysis. However, as we could not rule out a possibility that more potent variants of this hit peptide might not have been available in the original library consisting of >1012 members, we were prompted to further investigate on optimizing the initial hit WAp-D04 sequence to one with higher affinity to mWnt3a.
WAp-D04 focused library and re-selection of its active variants
In order to identify more potent analogues of WAp-D04, we next constructed a focused peptide library based on the original WAp-D04 sequence, randomizing 5 amino acid residues of the pharmacophore's total 10 residues. The positions for randomization were chosen as shown in Fig. 4a, where 0 to 5 (X-1 to X-5) consecutive mutations occurred. We prepared individual synthetic DNA template libraries and transcribed them into mRNA libraries. All of the resulting mRNA libraries was then mixed into a focused mRNA library for the expression of macrocyclic peptide libraries. Five rounds of selection were then performed to enrich the active population (Fig. S6, ESI†), and the enriched libraries were finally deep-sequenced to identify improved analogues of WAp-D04 (Fig. 4b).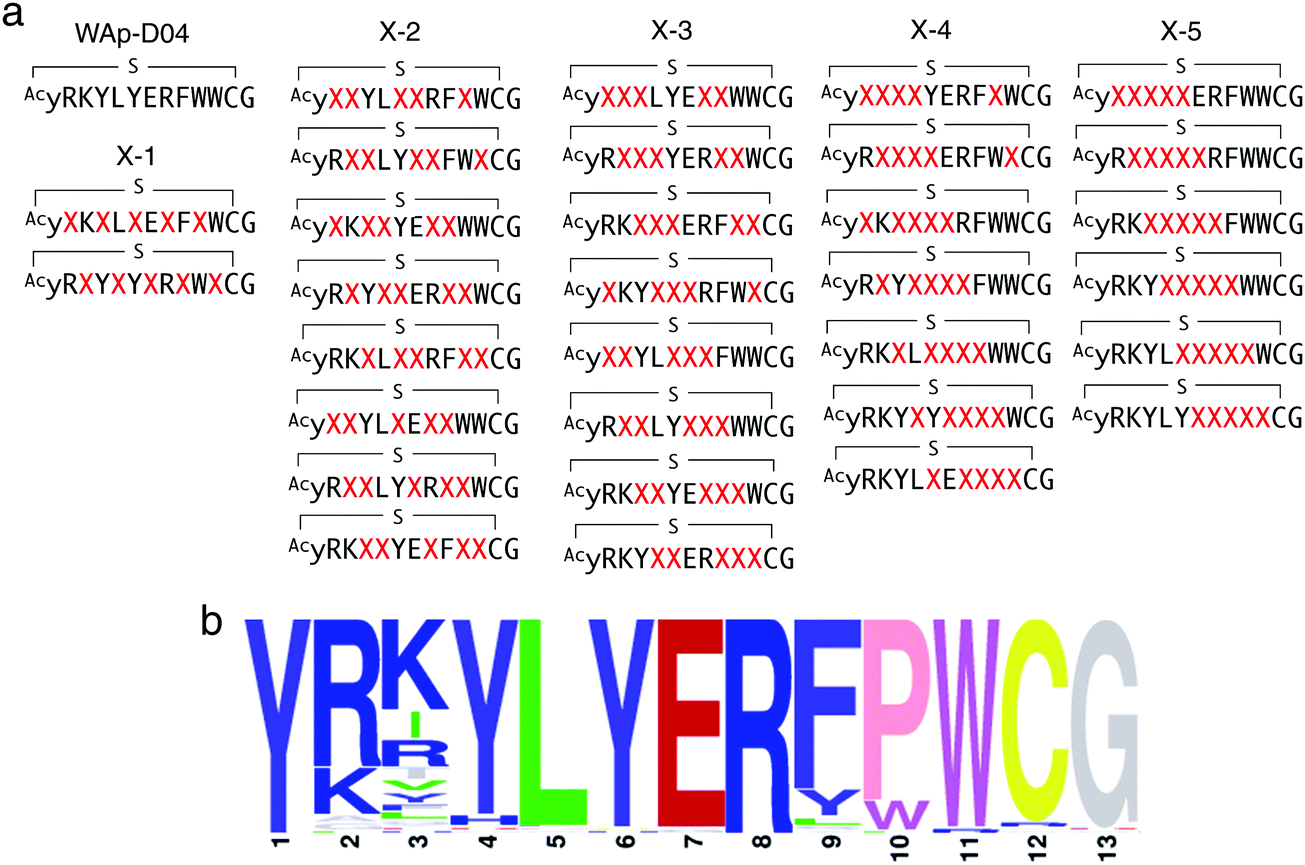 | ||
| Fig. 4 Design and screening of WAp-D04-focused libraries. (a) Design of a focused library based on the WAp-D04 sequence. The library was composed of five groups of structural variations, changing consecutive amino acids according to group number and translocating the variation along the chain. X indicates a random amino acid determined by an NNK codon. (b) Sequence LOGO graphs of the final round of selection, representing the 80 most enriched sequences from the focused macrocyclic peptide library.18 Note that the first Y residue of in the LOGO graph corresponds to D-Tyr (y). | ||
The selection campaign of the focused library yielded six mutant peptides of the parental peptide (WAp-D04). All newly identified peptides contained a highly conserved Trp to Pro substitution at position 10, as represented by the single mutant WAp-D04-W10P, and exhibited higher frequency following isolation with mWnt3a–hAFM as compared to hAFM in 2D analysis (Fig. S7, ESI†). Double mutation occurred either at positions 2 or 3 along with the W10P mutation, with conservative mutations at R2 or K3 to a basic sidechain alternative (R2K or K3R) or hydrophobic sidechain alternative (K3I, K3T, or K3V) (Table 2). Thus, the result of this focused library selection indicated that the original WAp-D04 itself is well optimized, but that the subtle W10P mutation possibly enhanced its inherent binding ability of WAp-D04. Based on these findings, we synthesized all six WAp-D04 mutants and tested their binding and inhibitory activity as in the previous reporter assay.
| Peptide name | Sequence | Number of reads | Steady-state KD (nM) | IC50 (nM) |
|---|---|---|---|---|
| WAp-D04 |

|
125 | 319 | 290 |
| WAp-D04-W10P |
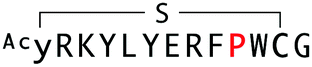
|
8014 | 39.5 | 56 |
| WAp-D04-W10P/R2K |
|
2059 | — | — |
| WAp-D04-W10P/K3I |
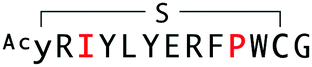
|
1808 | — | — |
| WAp-D04-W10P/K3R |

|
1034 | — | — |
| WAp-D04-W10P/K3T |

|
796 | — | — |
| WAp-D04-W10P/R2K/K3V |

|
395 | — | — |
Inhibitory activity of WAp-D04-W10P mutants
The synthesized WAp-D04-W10P mutants were analysed for Wnt3a signalling inhibitory activity at two concentrations (0.16 and 1.6 μM) in the reporter assay for primary screening (Fig. 5a). All six peptides exhibited stronger inhibition than the parental peptide, particularly at 1.6 μM concentration, at which all peptides shut down Wnt signalling, i.e. they completely inhibited TCF reporter activation caused by exogenously added mWnt3a. Even at 0.16 μM peptide concentration appreciable inhibition occurred, where the single mutant WAp-D04-W10P (Fig. 5b) itself displayed slightly more potent inhibitory activity than the series of double mutants. This data is consistent with the observation that it was the most abundant sequence in the next generation sequencing data (Table 2).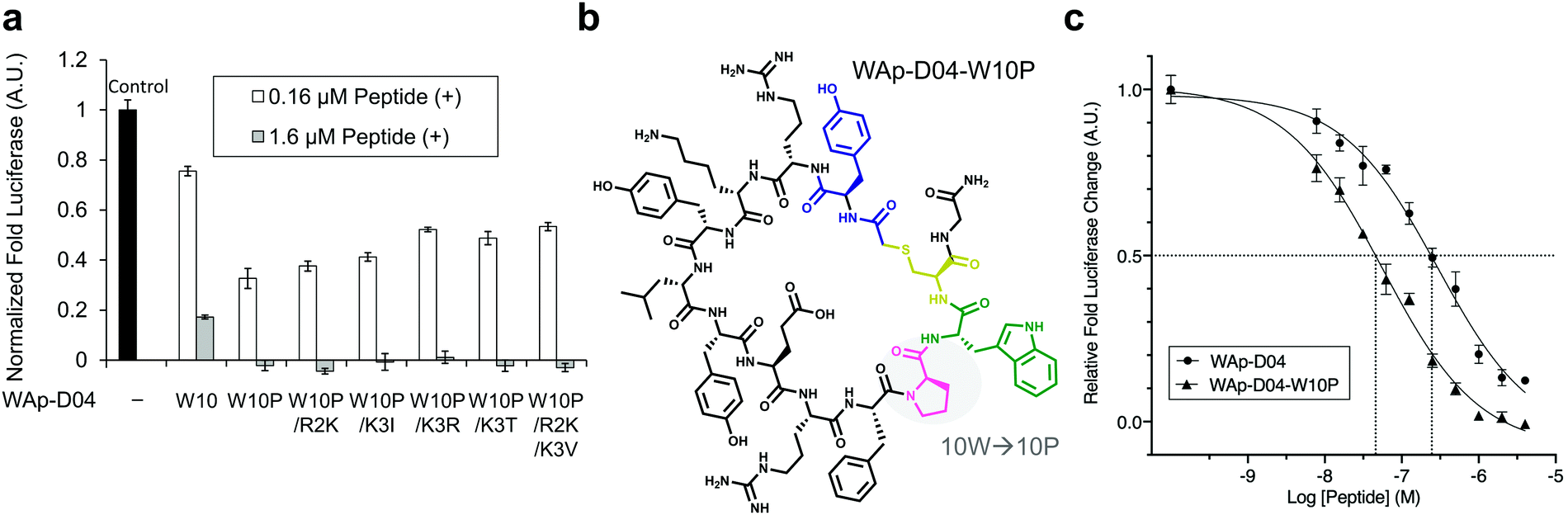 | ||
| Fig. 5 Focused library-selected peptides’ activity and structure and comparison of the strongest peptide inhibitors found in this work. (a) Luciferase reporter assay results for the peptides shown in Table 2. The effect of each peptide on activity was analysed independently at 0.16 and 1.6 μM concentration in the presence of 10 nM Wnt3a–AFM complex. Shown are fold values relative to a basal Wnt signal reference (black bar) using 10 nM mWnt3a–hAFM in the absence of any peptide. All data was normalized to a blank signal generated in the presence of 10 nM hAFM alone and in the absence of any peptide, subtracted from each value. Data are mean ± SD (n = 4). Data for this experiment is available as ESI.† (b) The single W10P mutation (highlighted) was found to dramatically increase the activity of the parental WAp-D04 peptide. (c) Overall inhibition rate properties of WAp-D04 (original) and WAp-D04-W10P. Shown is a sigmoidal fit of luciferase reporter assay results (mean ± SD, n = 4) for the two peptides conducted as in (a). Data are from a representative experiment out of three independent ones. Concentration value of each peptide corresponding to IC50 is marked with dotted lines for comparison. | ||
Further characterization through a titration of WAp-D04-W10P (7.8–4000 nM) demonstrated that inhibition of Wnt3a signalling by this peptide was concentration dependent as with WAp-D04, but approximately 5-fold more potent, i.e. IC50 of 56 nM as compared to 290 nM for WAp-D04 (Fig. 5c). This is a remarkable finding considering that these two compounds differ at only a single amino acid residue. We also modified our SPR condition by employing a more reliable ligand immobilization method (i.e. the Fc-mediated capture of hAFM and mWnt3a–hAFM), which gave us an approximately 90% higher KD value for WAp-D04 than the previous one (319 nM). However, a much smaller KD value of 39.5 nM was obtained with the W10P mutant, i.e. WAp-D04-W10P exhibited an 8-fold enhanced affinity.
Discussion
Wnt signalling is a critical component of embryonic development and post-development homeostasis, and its role in oncogenic processes such as colorectal cancer has led to the development of a number of different Wnt pathway inhibitors.35,53,54 These inhibitors target diverse components of the Wnt signalling mechanism, including Fz receptors, components of the destruction complex and β-catenin.34,35,53–56 However, direct targeting of Wnt ligands themselves has remained challenging due to the relative instability of Wnt proteins and the consequent difficulty in developing agents against them. To the best of our knowledge, the present study describes the first instance of mid-sized molecules acting as direct binders of a Wnt protein.A key advantage of our approach is the use of a recently identified stabilized form of a Wnt ligand, mWnt3a–hAFM, which allowed the direct selection of anti-mWnt3a macrocyclic peptides through the RaPID system. This system is remarkably reliable, and allows us to explore a vast sequence space of macrocyclic peptide libraries, potentially including both canonical and non-canonical amino acids.40,57–59 However, since the selection generally requires “soluble” bait proteins, it is not well applicable to “insoluble” proteins with intrinsic structural fragility that results in self-aggregation.
The unique strategy reported here (i.e. the use of a stabilizing protein partner during the selection process) solved this problem by sequestering such insoluble proteins adequately within a complex, probably shielding their hydrophobic regions and making selection against these targets an achievable task. Even though aggressive counter-selection against the “solubilizing” protein partner hAFM was implemented, the enriched pool of peptides still contained background binders to the partner. This did not occur when we used Fc-tagged fusion proteins in multiple selections where the negative selection cycles against plain Fc effectively suppressed background binders.47,60 Because of this unexpected outcome, we had to look for an alternative approach that could allow us to deduce which peptides bound solely to the target, mWnt3a, in this study. Accordingly, we utilized a 2-dimensional deep-sequencing analysis to distinguishably identify mWnt3a–hAFM binders over hAFM binders, which led us to find specific binders to mWnt3a. Moreover, the functional assay using the Wnt-signalling reporter cell line has allowed the identification of a rare hit peptide (WAp-D04) capable of inhibiting the mWnt3a-induced signal with a sub-μM concentration. Furthermore, the subsequent selection campaign using a “pharmacophore-focused library” enabled us to discover a 5-fold enhanced inhibitor with 56 nM IC50 by the exclusive W10P substitution. Although at this stage we cannot concretely define why the W10P mutation was able to enhance its activity, the fact that the mutation occurred near the cyclizing C12 and Pro is the unique secondary amino acid among other proteinogenic amino acids both point to the possibility that the W10P mutation may have increased the structural rigidity of the parental WAp-D04, giving an overall positive effect on the binding to mWnt3a. Further studies will include additional analysis of this mutation (as well as other silent mutations observed in WAp-D04) and X-ray crystallography, with a view to understanding the molecular mechanism of mWnt3a inhibition and the generation of molecules for application in animal studies.
Conclusions
Here we have discovered de novo macrocyclic peptides that can strongly bind a normally insoluble mWnt3a protein, and thereby potently inhibit Wnt3a signalling. Targeting of Wnt proteins for therapeutic screening has long been considered extremely difficult, mainly due to the loss of structural integrity or activity of such proteins outside their native cell environment and throughout the process of pharmacophore selection schemes. In this work, we addressed this challenge by applying a unique RaPID selection strategy that made use of a protein partner-stabilized Wnt ligand, and performed 2D deep-sequencing analysis to discriminate specific binders of our main target, mWnt3a. Starting from a randomized library of macrocyclic peptides, we identified an initial hit inhibitor that served as the basis for a focused library, in turn allowing the identification of an analogue with improved binding properties and inhibitory activity. The discovery of such molecules constitutes a useful initial tool in the development of a novel class of modulators of the Wnt pathway, which in turn can lead to a deeper understanding of its molecular action. Furthermore, our approach has enabled targeting of a highly hydrophobic and not commonly targeted protein, and although successful for a single Wnt so far, could prove applicable to other Wnt proteins or similar unstable proteins, expanding the scope of display screening techniques such as RaPID and bringing forward a new class of potential pharmacophores for understudied proteins.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by Japan Agency for Medical Research and Development (AMED), Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research) under JP19am0101090 and 19am0101075 to H. S. and J. T., respectively, and by MEXT KAKENHI Grant Number JP17H01420 from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) to J. T.References
- J. L. Stamos and W. I. Weis, Cold Spring Harbor Perspect. Biol., 2013, 5, a007898 CrossRef PubMed.
- D. B. Lybrand, M. Naiman, J. M. Laumann, M. Boardman, S. Petshow, K. Hansen, G. Scott and M. Wehrli, Development, 2019, 146(13), dev164145 CrossRef CAS PubMed.
- A. Mukherjee, N. Dhar, M. Stathos, D. V. Schaffer and R. S. Kane, iScience, 2018, 6, 13–21 CrossRef CAS PubMed.
- J. Sun and W. I. Weis, J. Mol. Biol., 2011, 405, 519–530 CrossRef CAS PubMed.
- D. Hrckulak, M. Kolar, H. Strnad and V. Korinek, Cancers, 2016, 8(7), 70 CrossRef PubMed.
- H. C. Wong, J. Mao, J. T. Nguyen, S. Srinivas, W. Zhang, B. Liu, L. Li, D. Wu and J. Zheng, Nat. Struct. Biol., 2000, 7, 1178–1184 CrossRef CAS PubMed.
- Z. M. Wang, J. Q. Luo, L. Y. Xu, H. H. Zhou and W. Zhang, Pharmacogenomics J., 2018, 18, 351–358 CrossRef CAS PubMed.
- E. C. van Kappel and M. M. Maurice, Br. J. Pharmacol., 2017, 174, 4575–4588 CrossRef CAS PubMed.
- X. Song, S. Wang and L. Li, Protein Cell, 2014, 5, 186–193 CrossRef CAS PubMed.
- R. Nusse, Cell Res., 2005, 15, 28–32 CrossRef CAS PubMed.
- R. P. Sharma and V. L. Chopra, Dev. Biol., 1976, 48, 461–465 CrossRef CAS PubMed.
- K. M. Cadigan and M. L. Waterman, Cold Spring Harbor Perspect. Biol., 2012, 4(11), a007906 Search PubMed.
- W. H. Lien, X. Guo, L. Polak, L. N. Lawton, R. A. Young, D. Zheng and E. Fuchs, Cell Stem Cell, 2011, 9, 219–232 CrossRef CAS PubMed.
- W. de Lau, W. C. Peng, P. Gros and H. Clevers, Genes Dev., 2014, 28, 305–316 CrossRef CAS PubMed.
- H. Ruffner, J. Sprunger, O. Charlat, J. Leighton-Davies, B. Grosshans, A. Salathe, S. Zietzling, V. Beck, M. Therier, A. Isken, Y. Xie, Y. Zhang, H. Hao, X. Shi, D. Liu, Q. Song, I. Clay, G. Hintzen, J. Tchorz, L. C. Bouchez, G. Michaud, P. Finan, V. E. Myer, T. Bouwmeester, J. Porter, M. Hild, F. Bassilana, C. N. Parker and F. Cong, PLoS One, 2012, 7, e40976 CrossRef CAS PubMed.
- R. Nusse and H. Clevers, Cell, 2017, 169, 985–999 CrossRef CAS PubMed.
- T. Zhan, N. Rindtorff and M. Boutros, Oncogene, 2017, 36, 1461–1473 CrossRef CAS PubMed.
- G. E. Crooks, G. Hon, J. M. Chandonia and S. E. Brenner, Genome Res., 2004, 14, 1188–1190 CrossRef CAS PubMed.
- T. Zhan, G. Ambrosi, A. M. Wandmacher, B. Rauscher, J. Betge, N. Rindtorff, R. S. Haussler, I. Hinsenkamp, L. Bamberg, B. Hessling, K. Muller-Decker, G. Erdmann, E. Burgermeister, M. P. Ebert and M. Boutros, Nat. Commun., 2019, 10, 2197 CrossRef PubMed.
- L. Novellasdemunt, P. Antas and V. S. Li, Am. J. Physiol.: Cell Physiol., 2015, 309, C511–C521 CrossRef CAS PubMed.
- H. Jiang, X. Zhang, X. Chen, P. Aramsangtienchai, Z. Tong and H. Lin, Chem. Rev., 2018, 118, 919–988 CrossRef CAS PubMed.
- A. H. Nile and R. N. Hannoush, J. Biol. Chem., 2019, 294, 726–736 CrossRef CAS PubMed.
- G. R. Dhamdhere, M. Y. Fang, J. Jiang, K. Lee, D. Cheng, R. C. Olveda, B. Liu, K. A. Mulligan, J. C. Carlson, R. C. Ransom, W. I. Weis and J. A. Helms, PLoS One, 2014, 9, e83650 CrossRef PubMed.
- J. F. Bazan, C. Y. Janda and K. C. Garcia, Dev. Cell, 2012, 23, 227–232 CrossRef CAS PubMed.
- R. Takada, Y. Satomi, T. Kurata, N. Ueno, S. Norioka, H. Kondoh, T. Takao and S. Takada, Dev. Cell, 2006, 11, 791–801 CrossRef CAS PubMed.
- C. Y. Janda, D. Waghray, A. M. Levin, C. Thomas and K. C. Garcia, Science, 2012, 337, 59–64 CrossRef CAS PubMed.
- H. Hirai, K. Matoba, E. Mihara, T. Arimori and J. Takagi, Nat. Struct. Mol. Biol., 2019, 26, 372–379 CrossRef CAS PubMed.
- K. Willert, J. D. Brown, E. Danenberg, A. W. Duncan, I. L. Weissman, T. Reya, J. R. Yates, 3rd and R. Nusse, Nature, 2003, 423, 448–452 CrossRef CAS PubMed.
- E. Mihara, H. Hirai, H. Yamamoto, K. Tamura-Kawakami, M. Matano, A. Kikuchi, T. Sato and J. Takagi, eLife, 2016, 5, e11621 CrossRef PubMed.
- L. Jerkovic, A. F. Voegele, S. Chwatal, F. Kronenberg, C. M. Radcliffe, M. R. Wormald, E. M. Lobentanz, B. Ezeh, P. Eller, N. Dejori, B. Dieplinger, F. Lottspeich, W. Sattler, M. Uhr, K. Mechtler, R. A. Dwek, P. M. Rudd, G. Baier and H. Dieplinger, J. Proteome Res., 2005, 4, 889–899 CrossRef CAS PubMed.
- H. S. Lichenstein, D. E. Lyons, M. M. Wurfel, D. A. Johnson, M. D. McGinley, J. C. Leidli, D. B. Trollinger, J. P. Mayer, S. D. Wright and M. M. Zukowski, J. Biol. Chem., 1994, 269, 18149–18154 CAS.
- B. Chen, M. E. Dodge, W. Tang, J. Lu, Z. Ma, C. W. Fan, S. Wei, W. Hao, J. Kilgore, N. S. Williams, M. G. Roth, J. F. Amatruda, C. Chen and L. Lum, Nat. Chem. Biol., 2009, 5, 100–107 CrossRef CAS PubMed.
- M. M. Fischer, B. Cancilla, V. P. Yeung, F. Cattaruzza, C. Chartier, C. L. Murriel, J. Cain, R. Tam, C. Y. Cheng, J. W. Evans, G. O'Young, X. Song, J. Lewicki, A. M. Kapoun, A. Gurney, W. C. Yen and T. Hoey, Sci. Adv., 2017, 3, e1700090 CrossRef PubMed.
- N. Tuysuz, L. van Bloois, S. van den Brink, H. Begthel, M. M. Verstegen, L. J. Cruz, L. Hui, L. J. van der Laan, J. de Jonge, R. Vries, E. Braakman, E. Mastrobattista, J. J. Cornelissen, H. Clevers and D. Ten Berge, Nat. Commun., 2017, 8, 14578 CrossRef PubMed.
- C. Y. Janda, L. T. Dang, C. You, J. Chang, W. de Lau, Z. A. Zhong, K. S. Yan, O. Marecic, D. Siepe, X. Li, J. D. Moody, B. O. Williams, H. Clevers, J. Piehler, D. Baker, C. J. Kuo and K. C. Garcia, Nature, 2017, 545, 234–237 CrossRef CAS PubMed.
- T. Passioura and H. Suga, Chem. Commun., 2017, 53, 1931–1940 RSC.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- N. K. Bashiruddin and H. Suga, Curr. Opin. Chem. Biol., 2015, 24, 131–138 CrossRef CAS PubMed.
- Y. Yamagishi, I. Shoji, S. Miyagawa, T. Kawakami, T. Katoh, Y. Goto and H. Suga, Chem. Biol., 2011, 18, 1562–1570 CrossRef CAS PubMed.
- J. Morimoto, Y. Hayashi and H. Suga, Angew. Chem., Int. Ed., 2012, 51, 3423–3427 CrossRef CAS PubMed.
- Y. V. Schlippe, M. C. Hartman, K. Josephson and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 10469–10477 CrossRef PubMed.
- A. Angelini, L. Cendron, S. Chen, J. Touati, G. Winter, G. Zanotti and C. Heinis, ACS Chem. Biol., 2012, 7, 817–821 CrossRef CAS PubMed.
- K. Sakai, T. Passioura, H. Sato, K. Ito, H. Furuhashi, M. Umitsu, J. Takagi, Y. Kato, H. Mukai, S. Warashina, M. Zouda, Y. Watanabe, S. Yano, M. Shibata, H. Suga and K. Matsumoto, Nat. Chem. Biol., 2019, 15, 598–606 CrossRef CAS PubMed.
- T. Passioura, K. Watashi, K. Fukano, S. Shimura, W. Saso, R. Morishita, Y. Ogasawara, Y. Tanaka, M. Mizokami, C. Sureau, H. Suga and T. Wakita, Cell Chem. Biol., 2018, 25, 906–915 CrossRef CAS PubMed , e905.
- Y. Fujii, M. Kaneko, M. Neyazaki, T. Nogi, Y. Kato and J. Takagi, Protein Expression Purif., 2014, 95, 240–247 CrossRef CAS PubMed.
- T. Nogi, T. Sangawa, S. Tabata, M. Nagae, K. Tamura-Kawakami, A. Beppu, M. Hattori, N. Yasui and J. Takagi, Protein Sci., 2008, 17, 2120–2126 CrossRef CAS PubMed.
- K. Ito, K. Sakai, Y. Suzuki, N. Ozawa, T. Hatta, T. Natsume, K. Matsumoto and H. Suga, Nat. Commun., 2015, 6, 6373 CrossRef CAS PubMed.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- H. Xiao, H. Murakami, H. Suga and A. R. Ferre-D'Amare, Nature, 2008, 454, 358–361 CrossRef CAS PubMed.
- H. Murakami, A. Ohta, H. Ashigai and H. Suga, Nat. Methods, 2006, 3, 357–359 CrossRef CAS PubMed.
- Y. Goto, A. Ohta, Y. Sako, Y. Yamagishi, H. Murakami and H. Suga, ACS Chem. Biol., 2008, 3, 120–129 CrossRef CAS PubMed.
- M. Molenaar, M. van de Wetering, M. Oosterwegel, J. Peterson-Maduro, S. Godsave, V. Korinek, J. Roose, O. Destree and H. Clevers, Cell, 1996, 86, 391–399 CrossRef CAS PubMed.
- J. Hu, A. Dong, V. Fernandez-Ruiz, J. Shan, M. Kawa, E. Martinez-Anso, J. Prieto and C. Qian, Cancer Res., 2009, 69, 6951–6959 CrossRef CAS PubMed.
- A. Gurney, F. Axelrod, C. J. Bond, J. Cain, C. Chartier, L. Donigan, M. Fischer, A. Chaudhari, M. Ji, A. M. Kapoun, A. Lam, S. Lazetic, S. Ma, S. Mitra, I. K. Park, K. Pickell, A. Sato, S. Satyal, M. Stroud, H. Tran, W. C. Yen, J. Lewicki and T. Hoey, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11717–11722 CrossRef CAS PubMed.
- N. T. Morrell, P. Leucht, L. Zhao, J. B. Kim, D. ten Berge, K. Ponnusamy, A. L. Carre, H. Dudek, M. Zachlederova, M. McElhaney, S. Brunton, J. Gunzner, M. Callow, P. Polakis, M. Costa, X. M. Zhang, J. A. Helms and R. Nusse, PLoS One, 2008, 3, e2930 CrossRef PubMed.
- R. Takada, Y. Mii, E. Krayukhina, Y. Maruyama, K. Mio, Y. Sasaki, T. Shinkawa, C. G. Pack, Y. Sako, C. Sato, S. Uchiyama and S. Takada, Commun. Biol., 2018, 1, 165 CrossRef PubMed.
- H. Hirose, C. Tsiamantas, T. Katoh and H. Suga, Curr. Opin. Biotechnol., 2019, 58, 28–36 CrossRef CAS PubMed.
- Y. Kato, T. Kuroda, Y. Huang, R. Ohta, Y. Goto and H. Suga, Angew. Chem., Int. Ed., 2020, 59, 684–688, DOI:10.1002/anie.201910894.
- T. Katoh and H. Suga, J. Am. Chem. Soc., 2018, 140, 12159–12167 CrossRef CAS PubMed.
- T. Kawakami, T. Ishizawa, T. Fujino, P. C. Reid, H. Suga and H. Murakami, ACS Chem. Biol., 2013, 8, 1205–1214 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00016g |
| This journal is © The Royal Society of Chemistry 2020 |