
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic tissue models reveal the role of hyaluronan in melanoma proliferation and invasion†
Jiranuwat
Sapudom‡
 abc,
Khiet-Tam
Nguyen‡
c,
Steve
Martin
a,
Tom
Wippold
c,
Stephanie
Möller
d,
Mathias
Schnabelrauch
d,
Ulf
Anderegg
c and
Tilo
Pompe
*a
abc,
Khiet-Tam
Nguyen‡
c,
Steve
Martin
a,
Tom
Wippold
c,
Stephanie
Möller
d,
Mathias
Schnabelrauch
d,
Ulf
Anderegg
c and
Tilo
Pompe
*a
aInstitute of Biochemistry, Faculty of Life Sciences, Universität Leipzig, Leipzig 04103, Germany. E-mail: tilo.pompe@uni-leipzig.de; Fax: +49 341 97 36931/9; Tel: +49 341 97 36931/9
bLaboratory for Immuno Bioengineering Research and Applications, Division of Engineering, New York University Abu Dhabi, Abu Dhabi, United Arab Emirates
cDepartment of Dermatology, Venerology and Allergology, Medical Faculty, Universität Leipzig, Leipzig 04103, Germany
dINNOVENT e. V., Biomaterials Department, Prüssingstraße 27B, 07745 Jena, Germany
First published on 8th January 2020
Abstract
Interactions of hyaluronan (HA) and tumor and stromal cells are highly discussed as one of the major contributors in tumor progression and metastasis. The balance of HA in the tissue is highly regulated by two key enzyme classes; hyaluronan synthases (HAS) and hyaluronidases (HYAL). Current reports hint that the HA amount in the tissue is correlated with poor prognosis in melanoma, the most life-threatening skin tumor. In this work, we generated in vivo mouse models with low and high expression of Has2 and used the models for studying melanoma proliferation of the B78D14 melanoma cell line. We found that a strong reduction of HA amount in the skin was correlated to decreased tissue stiffness and a reduction in tumor weight. Since tumor cells have a direct contact to the HA in the tumor and at the stroma interface, we reconstituted different biomimetic in vitro models using fibroblasts derived from a mouse model to recapitulate melanoma cell behavior at the tumor boundary, namely, (i) decellularized fibroblast matrix (FbECM), (ii) fibroblast embedded into 3D collagen matrices (FbColl), and (iii) well-defined HA-functionalized 3D collagen matrices (HAColl). We found no considerable effect of high and low amounts of fibroblast-derived HA in the matrices on melanoma proliferation and invasion. However, HYAL1-treated FbECM and FbColl, as well as HAColl functionalized with low molecular weight HA (34 kDa) promoted proliferation and invasion of melanoma cells in a concentration dependent manner. Our results emphasize the molecular weight specific effects of HA in regulation of melanoma behavior and provide an alternative explanation for the in vivo observation of HA dependent tumor growth.
1. Introduction
Tumor progression relies on a close interaction of tumor cells, different cell types in the tumor, and the surrounding stromal matrix. The stroma of solid tumors represents the benign vicinity of tumor cells, as also in malignant melanoma. However, compared to normal tissue, tumor stroma is different and originates from cells which are altered by paracrine interactions with tumor cells. It consists of the stromal matrix with (micro-)vessels and embedded stromal cells (immune cells, macrophages, fibroblasts, myofibroblasts) being controlled by paracrine mediators as well as deposited matrix components. Bidirectional interactions of tumor cells and the stroma decisively influence growth and metastatic behavior of many solid tumors. Cells or matrix components of the tumor stroma contribute to important aspects of tumor progression including proliferation, migration, and induction of angiogenesis.1–4Hyaluronan (HA) is a major glycosaminoglycan of the extracellular matrix (ECM) fulfilling essential structural and functional roles during development,5 inflammation,3,6 repair7,8 and tumor progression.4,9–11 HA accumulation in tumors is often associated with poor prognosis. It is synthesized by three HA synthases (HAS1–3), where HAS2 is most strongly expressed in many tissues.12,13 Many effects of HA in tumor biology rely on the interaction with its main cellular receptors CD44 (and tumor specific splice variants) and HA-mediated motility receptor (RHAMM).14,15
HA degradation is mediated either by free radical-related depolymerization occurring in the presence of reactive oxygen species (ROS)16 and Maillard products17 or enzymatically by hyaluronidases.4,18 Several enzymes are described (HYAL1–3, PH20, CEMIP, TMEM2),19–23 and the impact of tumor-derived or stroma-associated hyaluronidases can be both: pro- and anti-tumorigenic.24 HYAL1 on one hand hydrolyses pro-tumorigenic HA, but as a result small HA-oligosaccharides may activate cells in the tumor and the stroma, thus supporting tumor progression and angiogenesis25 by induction of matrix metalloproteinases,26,27 as well as lymph node metastasis.28 HA-oligosaccharides may act via toll like receptor signaling27,29,30 or by interfering with the interaction of high molecular weight HA (HMW-HA) and its receptors CD44 and RHAMM.31,32 Moreover, the relevance of small HA-oligosaccharide products was suggested when Schmaus et al.33 quantified these in tumor interstitial fluids and found a correlation with invasion of tumor cells into lymphatic vessels and the formation of lymph node metastases in colorectal cancer patients. However, recent data suggest that HYAL1 may support tumor metastasis independently of the generation of small HA-oligosaccharide products thus leaving alternative ways of action conceivable.34 The prevalence of other HA degrading enzymes, like CEMIP and TMEM2, obviously also correlates with progression or metastasis in tumor patients,22,35 whereas the small HA-oligosaccharide molecules where not quantified in these studies. Thus, today it remains to prove that really the HA-degradation products are the tumor promoting molecules or whether other mechanisms involving these proteins lead to cancer progression.
Due to its high water-binding capacity, HA deposition strongly contributes to specific physico-chemical properties of the surrounding stroma sensed by the tumor cells. Besides paracrine signals, the behavior of tumor cells is strongly affected by changes of mechanics and network microstructure of the fibrillar collagen matrix, as well as its compositions.36 We have previously demonstrated that changing physical properties of a three-dimensional (3D) collagen biomimetic matrix can influence tumor cells’ behavior, while the chemical composition of the model matrix remained unchanged.37,38 Such findings resulted in the awareness that more complex 3D culture models are essential in order to receive data better resembling the in vivo situation than the commonly used 2D cultures on tissue culture plastics.36,39,40
In order to investigate the impact of stromal HA on the physico-chemical properties of the stroma and on tumor cells facing this defined matrix, we combined model systems of graded complexity and in vitro experiments using stromal mouse fibroblasts with strongly decreased HA-synthesis due to genetic knockdown of the main HA synthase 2 (Has2). The impact of stromal HA on melanoma cells was characterized in respect to invasive behavior, proliferation and gene expression of the tumor cells and compared to in vivo findings in a Has2 knockdown mouse model on melanoma tumor growth.
2. Experimental
2.1. Generation of inducible Has2 knockout mice
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Leipzig University and approved by the Animal Ethics Committee of Saxony/Germany (TVV 57/14, T26/16). Has2-Exon2-floxed, C57BL/6N-embryonic-cell-line JM8A3.N1 was purchased from KOMP Repository, University of California Davis and Children's Hospital Oakland, Research Institute. Chimeric C57BL/6 mice were generated by Konstantinos Anastassiadis (TU Dresden, BIOTEC, Genetic Engineering of Stem Cells Group) and crossed with Flp-deleter C57BL/6 mice to generate offspring with deleted neomycin resistance and deleted lacZ reporter gene but carrying heterozygous floxed Has2-Exon2. Exon 2 was chosen, as it contains the start of translation and a 209 amino acid sequence which is the major part of the HAS2 proteins, see also Ensembl (ensembl.org). This region contains domains with the core enzymatic activity (nucleotide-diphospho-sugar transferases). Backcrossed homozygous mice carrying the Has2-Exon2-floxed alleles develop and reproduce without any obvious phenotype (ESI Fig. S1†). Has2-Exon2-floxed mice were backcrossed with ubiquitous Cre-deleter C57BL/6 mice (human ubiquitin C (UBC) promoter sequence upstream of a Cre-ERT2 fusion gene; Stock no.: 008085; Jackson Laboratory, USA) to Has2flox/flox-Cre-ERT2+/− strain, ready to induce Has2-Exon2 deletion upon tamoxifen application (mice and fibroblasts were named ‘Has2-KD’ after 4-hydroxy-tamoxifen treatment). Because characterization of the intended Has2-knockout mice and fibroblasts thereof showed remaining Has2 expression, they were regarded as a Has2-knockdown and hence named ‘Has2-KD’ throughout this work. Control mice were only Has2flox/flox without inducible Cre expression and mice and fibroblasts were named ‘Ctrl’ after 4-hydroxy-tamoxifen induction.2.2. Isolation of primary fibroblast from mouse skin tissue
Primary fibroblasts were isolated from full thickness skin from the back of the mice, from both Has2flox/flox-Cre-ERT2+/− and Has2flox/flox. Shaved back skin was disinfected with 70% isopropanol before approximately 4 cm2 of the skin was excised and cut into small pieces. To isolate fibroblasts from the tissue, 26 U of Liberase™ DL Research Grade (Sigma-Aldrich, Steinheim, Germany) were added to 3 mL serum-free DMEM (Biochrom, Berlin, Germany) and incubated over 2 h at 37 °C while stirring. Afterwards, the cell suspension was filtered through a 70 μm cell strainer (Becton Dickinson, Heidelberg, Germany) and the filtrate centrifuged at 300g for 4 minutes at room temperature. Collected cells were plated on cell culture Petri dishes and cultivated in DMEM (Biochrom) supplemented with 10% fetal calf serum (Biochrom) and 1% ZellShield (Biochrom) at 37 °C, 5% CO2 and 95% humidity for 24 h. Then, cells were rinsed with PBS to remove any dead and unattached cells. After another 24 h of cultivation, cells were used for in vitro experiments up to the 4th passage.2.3. Induction and characterization of Has2-KD fibroblasts
Isolated fibroblasts from Has2flox/flox-Cre-ERT2+/− and Has2flox/flox were treated with 2 μM 4-hydroxy-tamoxifen (4-OHT, Sigma-Aldrich) in DMEM (Biochrom) supplemented with 10% fetal calf serum (Biochrom) and 1% ZellShield (Biochrom) for 48 h at 37 °C, 5% CO2 and 95% humidity to induce Cre-recombinase translocation into the nucleus in Has2flox/flox-Cre-ERT2+/− fibroblasts for Has2-Exon2 deletion (Has2-KD fibroblasts). To verify the deletion, DNA and RNA were analyzed after 48 h culture without 4-OHT. Presence of Has2-Exon2 was investigated by amplification of Has2-Exon2 gene locus and agarose-gel-electrophoresis. Has2 mRNA expression of was quantified using RT-qPCR and. Sequences of primers using for the PCR are listed in the ESI Table 1.†2.4. Tumor injection in mouse models
Has2 flox/flox-Cre-ERT2+/− and Has2flox/flox mice at the age of 8–12 weeks were intraperitoneally injected with 1 mg 4-OHT dissolved in 100 μL 10% absolute ethanol (vwr, Dresden, Germany) in sunflower seed oil from Helianthus annuus (Sigma-Aldrich) per day for 8 days. At the end of 4-OHT treatment, the mice recovered for 5 days before 1.0 × 106 B78D14 cells (resuspended in 50 μL PBS (Biochrom)) were injected intradermally into the lower back. B78D14 tumors grew for 16 days, until tumor diameter reached 10 mm. Healthy skin from the back and tumor samples were taken for further analysis.2.5. Reconstitution of fibroblast-derived matrix (FbECM)
Fibroblast-derived matrix (FbECM) were reconstituted according to an established protocol.41 Briefly, cover slips (13 mm in diameter, VWR) were coated with cross-linked gelatin before seeding 1.6 × 105Has2-KD or Ctrl fibroblasts on top at 100% confluency. After 24 h, cell culture medium was replaced with DMEM media supplemented with 50 μg mL−1 ascorbic acid for 9 days. Cell culture media was changed every 3 day of culture to enhance ECM production. Afterwards, fibroblasts were lysed using ammonium hydroxide (20 mM) and Triton X-100 (0.5% v/v) in PBS. Extracellular DNA was digested with 100 U mL−1 DNase (Roche, Mannheim, Germany) in DNase buffer (50 mM Tris, 10 mM MnCl2, 1 mM CaCl2, at pH 7.5) for 15 min at RT. After carefully washing with PBS, intact FbECMs were kept in PBS containing penicillin/streptomycin (100 U mL−1, 100 μg mL−1 respectively, Biochrom) prior to use for cell experiments. As HA-negative controls, FbECMs were incubated with HYAL1 (2 U mL−1, Hyaluronidase-1 from bovine testis, Sigma-Aldrich) in 20 mM sodium acetate buffer for 2 h at 37 °C after DNAse treatment.2.6. Reconstitution of fibroblast-embedded 3D collagen matrices (FbColl)
Fibroblast-embedded 3D collagen matrices were reconstituted on 13 mm poly(styrene-alt-maleic anhydride) (PSMA; MW 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000 g mol−1, Sigma-Aldrich) coated coverslip by embedding 1 × 104Has2-KD and Ctrl fibroblasts into 3D collagen matrices. To reconstitution of collagen matrices, rat-tail type I collagen (Corning, NY, USA) was mixed with 250 mM phosphate buffer at pH 7.5, as previously reported.37,38 The collagen fibrillation was introduced by incubation at 37 °C and 95% humidity. After 5 days of cultivation, cells were lysed with double-distilled water for 1 h at room temperature. As HA-negative controls, FbColl were incubated with HYAL1 (Hyaluronidase from bovine testis, Sigma-Aldrich) for 2 h at 37 °C after DNAse treatment.
000–30000 g mol−1, Sigma-Aldrich) coated coverslip by embedding 1 × 104Has2-KD and Ctrl fibroblasts into 3D collagen matrices. To reconstitution of collagen matrices, rat-tail type I collagen (Corning, NY, USA) was mixed with 250 mM phosphate buffer at pH 7.5, as previously reported.37,38 The collagen fibrillation was introduced by incubation at 37 °C and 95% humidity. After 5 days of cultivation, cells were lysed with double-distilled water for 1 h at room temperature. As HA-negative controls, FbColl were incubated with HYAL1 (Hyaluronidase from bovine testis, Sigma-Aldrich) for 2 h at 37 °C after DNAse treatment.
2.7. Reconstitution of defined HA-immobilized collagen matrices (HAColl)
Firstly, 3D collagen matrices were reconstituted on 13 mm poly(styrene-alt-maleic anhydride) (PSMA; MW 20000–30000 g mol−1, Sigma-Aldrich) coated coverslip. Rat-tail type I collagen (Corning, USA) was mixed with 250 mM phosphate buffer at pH 7.5 to archive collagen concentration of 2 mg ml−1, as previously reported.37,38 Matrices were fibrillated by incubation at 37 °C and 95% humidity. HA with a molecular weight of 34 kDa (low molecular weight HA, LMW-HA) and 1170 kDa (HMW-HA) were prepared in 100 mM MES buffer at pH 5. To immobilize defined HA amounts of 1.3 μg per matrix (0.02 μg HA per μg Coll) and 3.3 μg per matrix (0.41 μg HA per μg Coll), HA solutions in MES were prepared at 20 and 50 μg ml−1 (LMW-HA) and 10 and 30 μg ml−1 (HMW-HA), respectively. Reconstituted collagen matrices were incubated with 500 μl of HA solution for 2 h at room temperature. Afterwards, HA solution was discarded and 500 μl of 4 mg mL−1 of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC; Sigma-Aldrich) dissolved in 100 mM in 2-(N-morpholino)ethanesulfonic acid (MES; Sigma-Aldrich) at pH 5 was directly added without washing. Matrices were incubated with EDC crosslinking solution for 2 h at room temperature and followed by washing 3 times each 5 min with PBS. Prior to cell experiments, matrices were equilibrated in cell culture medium for 1 h.
2.8. Topological characterization of reconstituted matrices
To visualize and analyze topological parameters, cell-free FbColl and HAColl matrices were stained with 50 μM 5-(and 6)-carboxytetramethylrhodamine succinimidyl ester (5(6)-TAMRA-SE) (Invitrogen, Carlsbad, USA) at room temperature for 1 h and rinsed 3 times with PBS (Biochrom). Matrices were imaged using a confocal laser scanning microscope (cLSM, LSM700, Zeiss, Jena, Germany) using 40×/NA 1.3 oil immersion objective (Zeiss). Acquired images were 1024 × 1024 pixels in resolution (xyz-voxel size: 0.13 × 0.13 × 0.5 μm) and a vertical stack size of 200 images (equivalent to 100 μm). Pore size and fibril diameter were determined as previously described in ref. 38 and 42 using a home-built image analysis tool using an erosion algorithm and autocorrelation analysis, respectively.2.9. Mechanical characterization of full skin tissue and reconstituted matrices
Mechanical properties of full skin sample cross sections and reconstituted cell-free matrices were quantified by colloidal probe force spectroscopy using a scanning force microscope (NanoWizard 3, JPK Instruments, Berlin, Germany), as previously reported.37,38 Briefly, a 50 μm glass microbead (Polyscience Europe GmbH, Eppelheim, Germany) was attached to a tipless HQ-CSC38 cantilever (NanoAndMore, Wetzlar, Germany) with a spring constant of approximately 0.1 N. At least 50 force–distance curves were mapped at an area of 100 × 100 μm at 3 positions of each sample from 3 independent tumor samples with indentation rate of 5 μm s−1 in PBS buffer (Biochrom) at room temperature. An effective Young's modulus was determined as elastic modulus by fitting the retract part of force distance curves (typical indentation depth 5 μm) using the Hertz model.2.10. Quantification and visualization of HA
For quantitative analysis, full-thickness skin samples, FbECM, and FbColl were lysed with 20 U mL−1 protease from Streptomyces griseus (Sigma-Aldrich) and 5 mM deferoxamine mesylate salt (Sigma-Aldrich) solved in HA-prep buffer (150 mM Tris, 150 mM NaCl, 10 mM CaCl2, at pH 8.3). After shaking, overnight incubation at 55 °C, protease was inactivated by heating at 95 °C for 10 min. HA amount was quantified using commercial HA-ELISA kit (TECOmedical Group, Switzerland).For visualization of HA, full-thickness skin samples, FbECM, and FbColl were stained with biotinylated hyaluronan binding protein (HABP, Sigma-Aldrich) followed by streptavidin-conjugated Alexa Fluor 488 (Invitrogen). HABP-stained matrices were imaged using cLSM (LSM700, Zeiss) using 40×/NA 1.3 oil immersion objective (Zeiss). Staining specificity was controlled both by pre-treatment with hyaluronidase or by omitting HAbP incubation during staining procedure.
For HA optical quantification of HAColl, ATTO565- labelled HA was used to functionalize reconstituted matrix, as previously described.43 Briefly, HMW-HA or LMW-HA derived from ozonolysis of high molecular weight HA (from Streptococcus, Aqua Biochem, Dessau, Germany) was conjugated with Atto565-NH2 (ATTO-TEC, Siegen, Germany) at carboxylic groups along the HA chain. HAColl were digested with papain and fluorescence signal were obtained at excitation wavelength of 565 nm and emission wavelength of 590 nm using fluorescence spectrometer (TECAN Infinite 200 PRO, TECAN, Grödig, Austria). The fluorescence value was related to HA amount using a standard curve. Immobilized HA were visualized using cLSM (LSM700 (Zeiss)) using 40×/NA 1.3 oil immersion objective.
2.11. Melanoma cell proliferation and invasion in animal models, FbECM, FbColl, HAColl matrices
B78D14 murine malignant melanoma (MM) cell line was kindly provided by P. Holst (University Kopenhagen, Denmark) (Becker et al. 1996).67 B78D14 cell line is a derivative of the model cell line B16F10 characterized by slower growth behavior in vivo and in vitro. Cells were grown in DMEM supplemented with FCS (10%) (Biochrom). For experiments, cells were seeded on top of reconstituted matrices and cultivated in DMEM (Biochrom) supplemented with 10% fetal calf serum (Biochrom) and 1% ZellShield (Biochrom) at 37 °C, 5% CO2 and 95% humidity for 3 days. Cell proliferation was determined either by Ki67 mRNA expression (primer sequence is listed in ESI Table 1†) or by using commercially available EdU cell proliferation kits (Baseclick GmbH, Neuried, Germany).Ki67 antigen was detected in cryostat sections of experimental tumors that were fixed with ice-cold acetone (Carl-Roth, Germany). Anti-Ki67 antibody (Bethyl Laboratories Inc., USA, IHC-00375) was added overnight (1:150). Bound antibody was detected with Alexa546 labelled goat-anti-rabbit secondary antibody (ThermoFisherSctientific). Nuclei were stained with 4′,6-diamidino-2-phenyl-indoldihydrochlorid (DAPI, Merck) and sections were mounted with Prolong Gold (ThermoFisherScientific). Microscopy was performed with Keyence BZ-9000E and corresponding software BZ-II Viewer, BZ-II Analyzer (Keyence, Neu Isenburg, Germany).
For cell invasion analysis, GFP-expressing B78D14 cells (transfected with pGFPmax (amaxa, Cologne, Germany) and FACS-sorted for strong expressing cells) or DAPI-stained cells were imaged by an epifluorescence AxioObserver Z1 microscope with scanning stage (Zeiss, Jena, Germany) using a 10× objective (Zeiss, Germany) in bright field and fluorescent mode (DAPI and Alexa Fluor 488). Cell invasion parameters, namely percentage of invaded cells and maximal invasion distance, were quantified using home-built MATLAB script (MATLAB 2019a, MathWorks, USA), as previously described in ref. 37 and 44. At least 600 cells from 3 positions per matrix conditions were analyzed.
2.12. Data analysis and statistics
Experiments were performed at least in triplicate, if not otherwise stated. Levels of statistical significance were determined by one-way ANOVA followed by Tukey's post hoc analysis using GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA, USA). Significance level was set at 0.05.3. Results and discussion
In this work, we aimed to decipher the impact of HA in melanoma progression by means of both in vivo mouse model and in vitro models. Ctrl and Has2-KD mice with injected B78D14 mouse melanoma cells and analyzed concerning tumor growth. These results were compared to three different biomimetic in vitro ECM models, namely (i) decellularized fibroblast matrix (FbECM), (ii) fibroblast embedded into 3D collagen matrices (FbColl), and (iii) well-defined HA-functionalized 3D collagen matrices (HAColl). Used fibroblasts were derived from the mouse models to recapitulate Has2-KD in ECM production and modification. B78D14 mouse melanoma cells were used to demonstrate the impact of HA in terms of cell proliferation and invasive behavior.3.1. Animal model: in vivo mouse model with designated Has2-knockout
The Has2-knockout mouse was generated and characterized regarding HA production and mechanical properties of full-thickness skin. A significant, but incomplete knockout was achievable in these mice (hence, termed Has2-knockdown (Has2-KD) in the following sections). B78D14 mouse melanoma cells were injected into the lower back of Ctrl and Has2-KD mice and were analyzed regarding proliferation by means of tumor weight and Ki67 staining.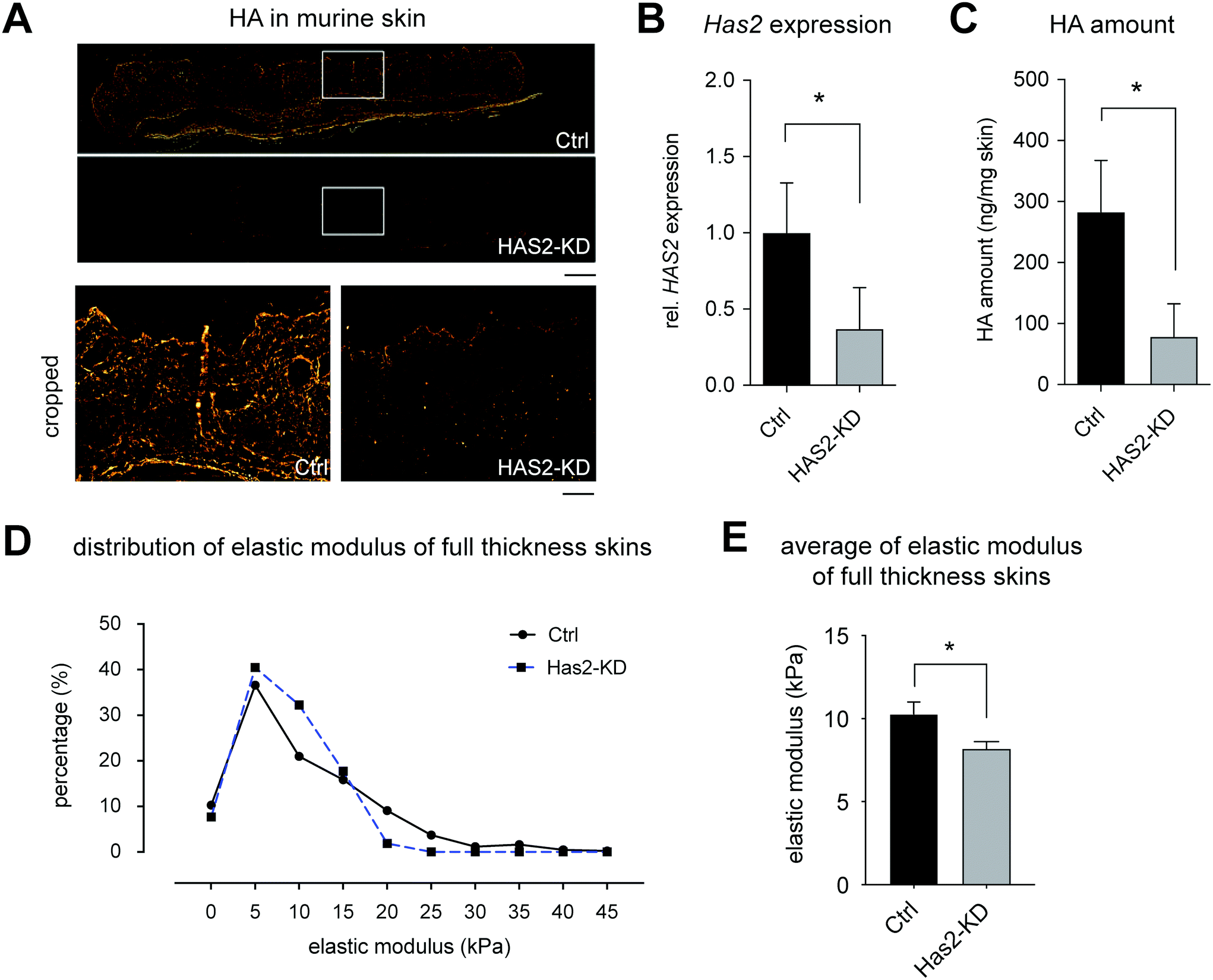 | ||
| Fig. 1 Has2-knockdown led to depletion of hyaluronan and reduction of tissue stiffness. (A) Representative image of HA distribution in murine skin. HA was stained using HABP (scale bar = 1 mm and 100 μm for upper and cropped images, respectively). (B) Relative expression of Has2 gene in murine skin using RT-qPCR. (C) Quantification of HA amount in murine skin using HA-ELISA. (D) Distribution of elastic modulus of representative single murine skin samples determined at 50 different positions. (E) Elastic modulus of full thickness murine skin. The experiment was performed from 3 murine skins from Ctrl and Has2-KD mice, with a mean elastic modulus evaluated for each skin sample from force mapping as shown in (D) (data are represented as mean ± SD; * – significance level of p < 0.05). | ||
These results led to the conclusion of a significant but incomplete knockout (Has2-KD), where at least a fraction of fibroblasts was not affected by the 4-OHT induction. Although, HAS isoenzymes may still contribute to a small amount of HA production, there was no compensatory upregulation of either Has1 or Has3 mRNA. Also, remaining HA in Has2-KD skin might be a tightly integrated portion and/or enzymatically modified type which was not affected by HA turnover after Has2-KD. The fact that this model resulted in a variety of reduced HA concentrations in skin opened the opportunity to investigate HA impact on tumor growth in vivo in dependence on HA concentration.
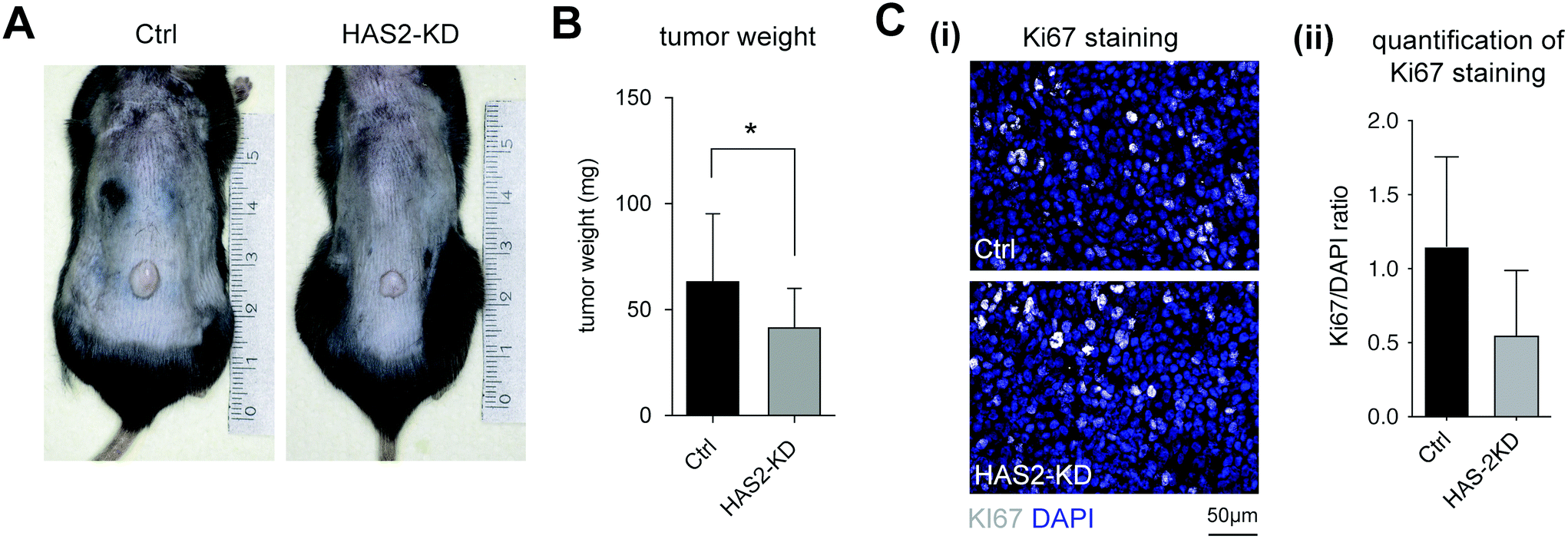 | ||
| Fig. 2 Suppressed growth of melanoma cells in Has2-KD mice. (A) Representative image of melanoma tumor in vivo. (B) Quantitative analysis of tumor weight from Ctrl and Has2-KD mouse after 16 days of injection. (C) Proliferation activity in the tumor was analyzed by fluorescence microscopy using DAPI (blue) and Ki67 antibody (white) staining. (C(i)) Representative image of the staining (scale bar: 50 μm) and (C(ii)) quantitative analysis of the Ki67 staining normalized against DAPI staining (data are represented as mean ± SD; * – significance level of p < 0.05). | ||
3.2. Biomimetic tissue models in vitro
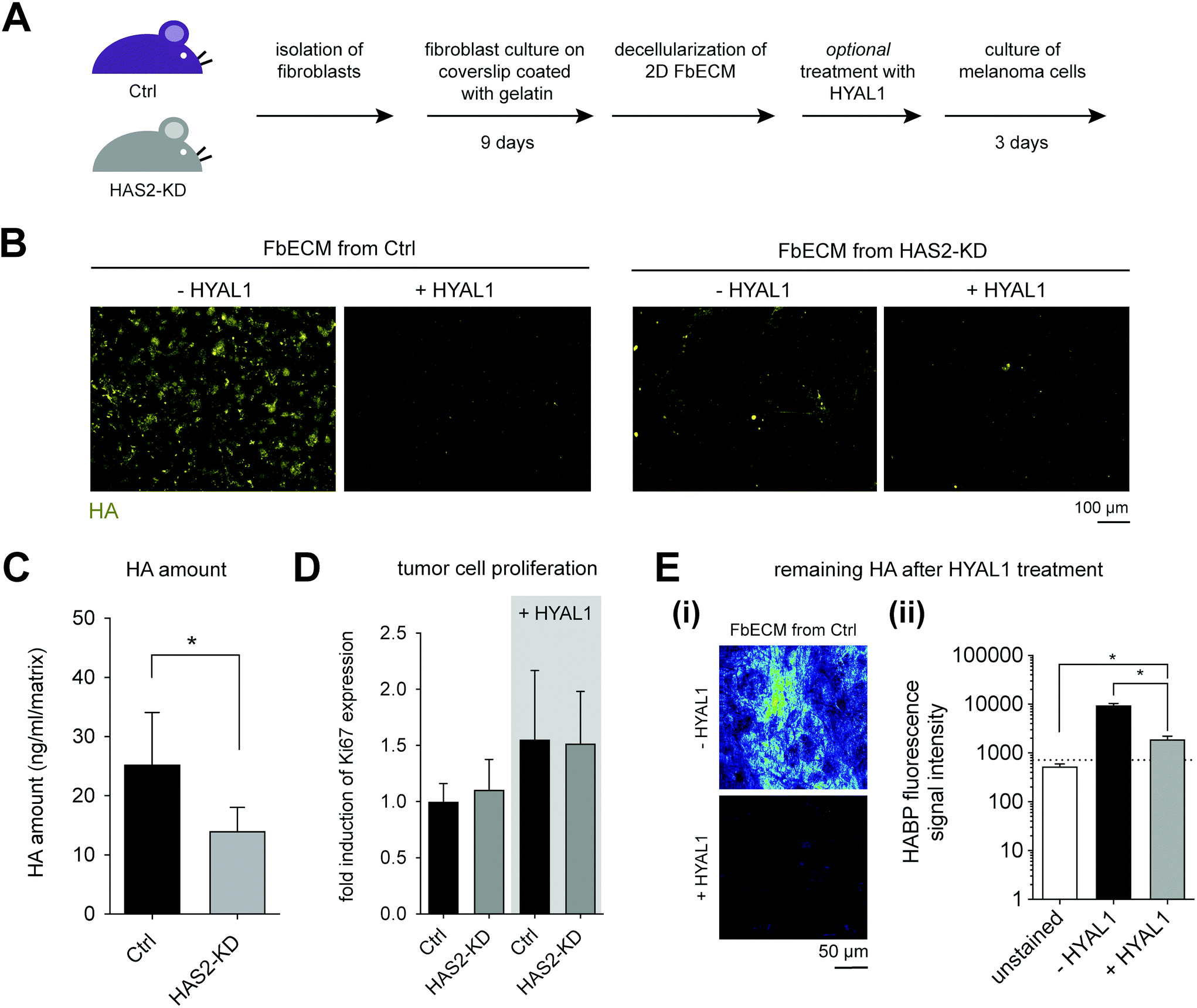 | ||
| Fig. 3 HYAL1 digested FbECM matrices promote proliferation of melanoma cells. (A) Schematic illustration of the experimental workflow. (B) Representative images of FbECMs reconstituted using Ctrl and Has2-KD fibroblasts. Matrices were treated with or without HYAL1 and analyzed by cLSM using HABP staining. (C) Quantitative analysis of HA amount in FbECM using commercial HA-ELISA kit. (D) Proliferation analysis of melanoma cells by means of Ki67 expression. (E) Analysis of remaining HA after HYAL1 treatment by cLSM using HABP staining. (E(i)) Representative image of FbECM from Ctrl. Color of the images is shown as heatmap. (E(ii)) Quantitative analysis of HABP fluorescence signal intensity. The dot line represents the fluorescence signal of FbECM stained with secondary antibody alone (data are represented as mean ± SD; * – significance level of p < 0.05). | ||
To study the impact of HA on melanoma cell proliferation, B78D14 cells were cultivated on decellularized FbECM for 5 days. The gene expression of Ki67 was quantified to compare proliferative capability of B78D14 cells grown on different FbECM. Additionally, we used HYAL1 treated FbECM samples, as HA negative control due to the enzymatic degradation of existing HA by HYAL1. As shown in Fig. 3D, Ctrl and Has2-KD FbECM demonstrated no differences in Ki67 expression. No remaining HA could be measured using HA-ELISA kit after HYAL1 digestion. However, low signal intensity was still detectable with HAPB using cLSM (Fig. 3E-ii). This finding suggests remaining HA residues after treatment with HYAL1. Interestingly, cells cultivated on HYAL1-treated FbECMs of both Ctrl and Has2-KD fibroblasts showed an increase in proliferation (Fig. 3D). Hence, it is suggested that the residuals of low molecular HA might cause an increased proliferation of melanoma cells. It is interesting to note, that HYAL1 is known to be highly expressed in tumor microenvironments and it is suspected to contribute to tumor aggressiveness.34,52
Decreased HA deposition in Has2-KD FbECM was shown to have no impact on melanoma cell proliferation, but HYAL1 treatment of both FbECM types led to upregulated proliferation. However, the matrix organization and HA deposition by fibroblasts are not homogenous, as depicted in Fig. 3B, which limits the comparison of melanoma behavior on FbECMs. Enhanced proteolysis of the FbECM by melanoma cells could be excluded by analyzing B78D14 melanoma cells growing on FbECM by mRNA expression microarray: none of the major MMPs was significantly regulated in these melanoma cells (data not shown). Additionally, we may not exclude that decrease of HA content in FbECM might lead also to changes in the deposition of collagens or fibronectin. However, microarray analysis of skin from Ctrl vs. Has2-KD mice did not reveal significant changes of mRNA levels for collagens I and III and fibronectin (data not shown). Furthermore, FbECM is a very thin ECM of approximately 3 to 7 μm, which did not allow cell invasion studies and measurement of mechanical properties.
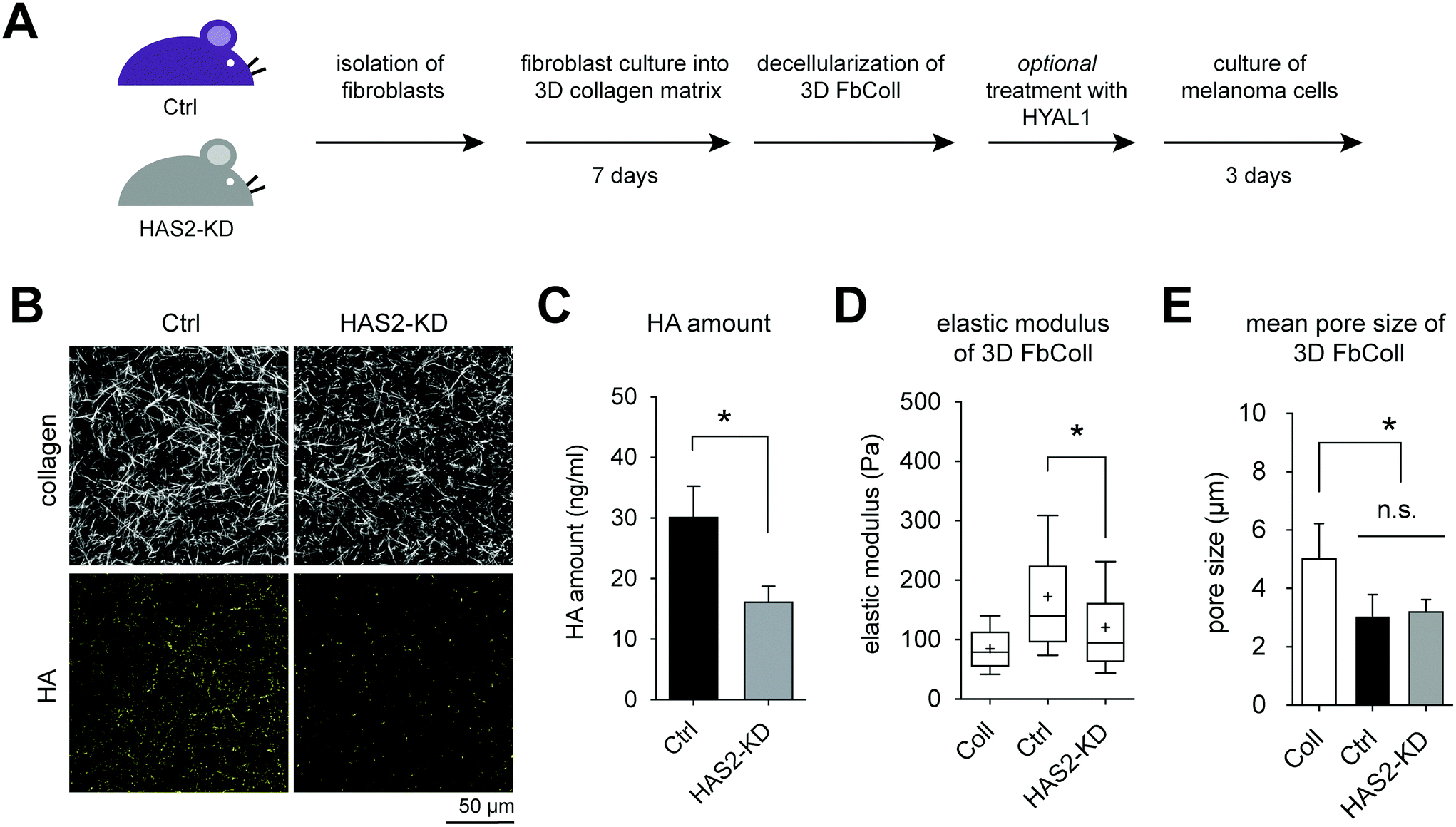 | ||
| Fig. 4 Characterization of FbColl matrices. (A) Schematic illustration of the experimental workflow. (B) Representative images of FbColl reconstituted using Ctrl and Has2-KD fibroblasts. Collagen and HA were analyzed by cLSM using TAMRA-SE staining and HABP staining, respectively. (C) Quantitative analysis of HA amount in FbColl using commercial HA-ELISA kit. Quantitative analysis of (D) elastic modulus and (E) pore size of FbColl matrices (data are represented as mean ± SD; * – significance level of p < 0.05). | ||
Analogous to the FbECM, GFP-expressing B78D14 melanoma cells were cultured for 5 days on decellularized FbColl matrices with and without HYAL1 treatment. Additionally, non-decellularized matrices were used for co-culture with fibroblasts. Cells were analyzed regarding their invasiveness and proliferative capacity. Fig. 5A shows an xz-plot of B78D14 cells in FbColl matrices. Proliferating cells were quantified by counting EdU positive cells (Fig. 5B). Representative images of EdU assay are shown in ESI Fig. S6.† There was no difference between Has2-KD and Ctrl FbColl matrices, both in co-culture as well as in decellularized matrices. The only significant difference was an increased proliferation of melanoma cells due to HYAL1 treatment of matrices. This happened in both Ctrl and Has2-KD matrices. However, for Ctrl-HYAL1-treated matrices the effect was stronger than for Has2-KD-HYAL1-treated matrices. Again, these findings showed that reduced deposition of fibroblast-derived HA did not play a crucial role in melanoma cell proliferation. However, HA degradation did, even in a concentration dependent manner, since Has2-KD matrices had less HA incorporated and therefore less degraded HA. A similar influence was observed on the percentage of invaded cells and maximum invasion depth, where co-culture and/or reduced HA-deposition did not change melanoma cell behavior (30% invaded cells and maximum invasion distance of 30 μm), but HYAL1 treatment did (Fig. 5C and D). Here, an even higher impact of total amount of HA before HYAL-treatment was observed, as Has2-KD-HYAL1-treated matrices had both less invading cells and smaller invasion distance (65% and 90 μm for Ctrl compared to 45% and 65 μm for Has2-KD).
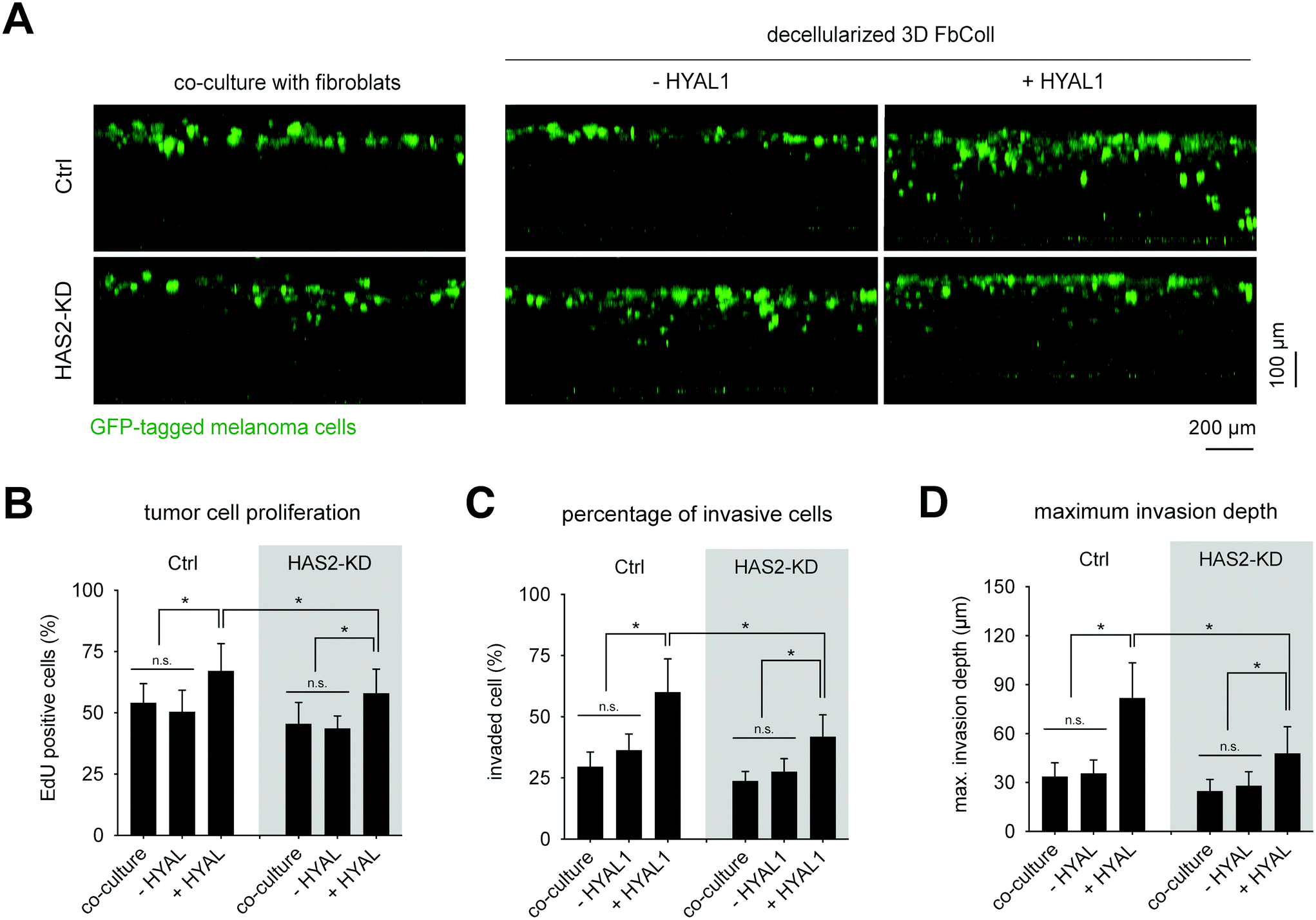 | ||
| Fig. 5 HYAL1 treated FbECM matrices enhance proliferation and invasion of melanoma cells. (A) Representative images of xz-view of GFP expressing melanoma cells invasion into 3D FbColl matrices (B) and proliferation analysis of melanoma cells by means of EdU incorporation. EdU positive cells were analyzed throughout the matrix thickness by counting cells using home-built MATLAB script. (C) Quantitative analysis of percentage of invasive cells and (D) maximum invasive distance of melanoma cells. Cells found >20 μm beneath the matrix surface were counted as invasive cells. The maximum invasive distance was defined as the distance crossed by 10% of all cells (data are represented as mean ± SD; * – significance level of p < 0.05). | ||
In sum, HYAL1-treatment of matrices led to increased proliferation in both, FbECM and FbColl, accompanied by a decrease in cell invasion in the 3D FbColl matrices. Based on these findings we hypothesized that digested, small fragments of HA (in the range of <30 kDa) still remained in FbECM and FbColl, since HYAL1 degrade HA into HA fragments in the range of 20 kDa53 to 66 kDa.54 Furthermore, it has to be recognized, that the small HA fragments cannot be detected with the HA-ELISA kit used here or with HA-binding proteins.55 Since Ctrl matrices contained higher amounts of HA than Has2-KD matrices, it can be assumed that more LMW-HA fragments were present after HYAL treatment, and in turn, could have led to an increased response of melanoma cells.
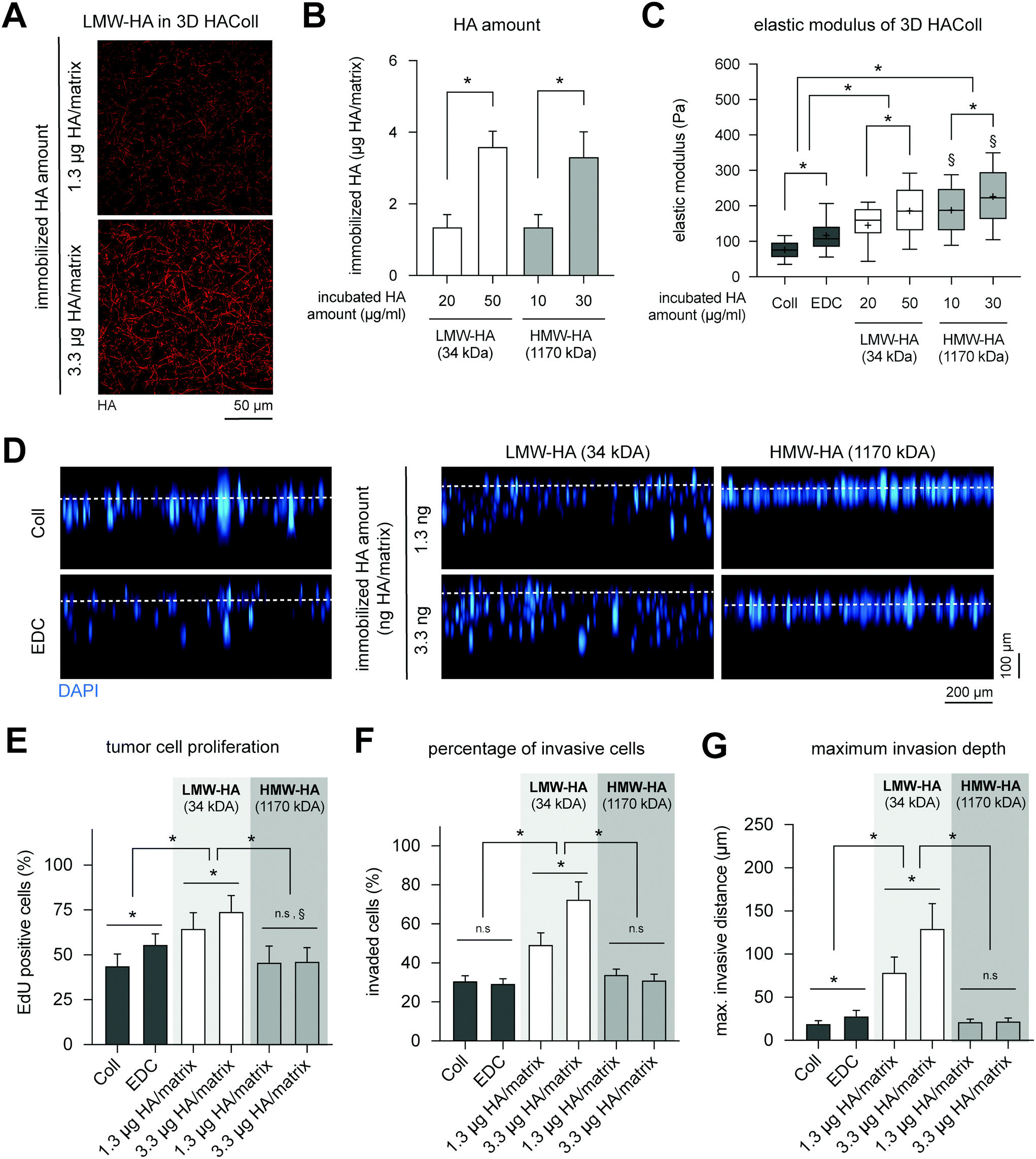 | ||
| Fig. 6 LMW-HA immobilized 3D collagen matrices modulate proliferation and invasion of melanoma cell in HA concentration dependent manner. (A) Representative image of LMW-HA immobilized 3D collagen matrices and the quantitative analysis of (B) HA immobilized amount (C) elastic modulus. (D) Representative images of xz-view of DAPI-stained melanoma cells invasion into 3D HAColl matrices. (E) Proliferation analysis of melanoma cells by means of EdU incorporation. EdU positive cells were analyzed throughout the full matrix thickness by counting cells using home-built MATLAB script. (F) Quantitative analysis of percentage of invasive cells and (G) maximum invasive distance of melanoma cells. Cells found >20 μm beneath the matrix surface were counted as invasive cells. The maximum invasive distance was defined as the distance crossed by 10% of all cells (data are represented as mean ± SD; * – significance level of p < 0.05). | ||
Culturing of B78D14 cells on HAColl matrices showed that melanoma cell proliferation and invasion is dependent on molecular weight of presented HA (Fig. 6E). Cell proliferation was found to be enhanced in a concentration-dependent manner for LMW-HA, but not for HMW-HA-functionalized matrices (Fig. 6B). Similar to cell proliferation, a higher invasion, both percentage of invaded cells and maximum invasion distance, was observed in a concentration-dependent manner for LMW-HA functionalized matrices (Fig. 6D, F and G). Again, HMW-HA did not affect invasion of melanoma cells, as previously reported for other human melanoma cell lines and in vitro models.11 It has to be noted that controls of EDC crosslinking without HA immobilization showed a slight increase in proliferation and invasion in comparison to pure Coll, however, to a much lower extent as LMW-HA immobilization. This is in line with previous reports that matrix stiffness and LMW-HA, but not HMW-HA, co-jointly regulate proliferation of melanoma cells, as also reported in an animal model.56
In sum, only LMW-HA was able to increase proliferation and invasion of melanoma cells in a concentration-dependent manner, whereas HMW-HA did not affect both cell functions. This result, together with the effects seen in FbECM and FbColl matrices caused by HYAL1 treatment suggests that HMW-HA might have a homeostatic effect on the surrounding cells, whereas LMW-HA tend to trigger signal transduction in cells in terms of proliferation and invasion.57,58
3.3. Discussion: biomimetic models reveal alternative explanation for in vivo observation
An increase of HA, especially via overexpression of HAS2, showed tumor driving properties via different mechanisms.49,59–61 Our findings fall in line with those reports, since the knockdown of Has2 led to decreased primary tumor mass. It can be assumed that knockdown of the main HMW-HA producing enzyme Has2 and only the smaller amount of existing HMW-HA would result in smaller tumors. But taken the in vitro models into account (FbColl and HAColl), we could show that exposure to matrix-incorporated HMW-HA did not lead to increased proliferation or migration of B78D14 melanoma cells, even when increasing HMW-HA amounts.This led to the question why decreased expression of HMW-HA in vivo would lead to reduced tumor weight. An alternative explanation could be that lower expression of HMW-HA is accompanied by less enzymatic catabolism to LMW-HA. Since some tumors are known to have high expression of HYALs62–64 and B78D14 cells also produces a moderate amount of hyaluronidase (see ESI Fig. S7†), constant HYAL expression can lead to digestion of HMW-HA in the tumor microenvironment accompanied by activating effects on tumor cells. A reduction of HMW-HA amount, e.g. by Has2 knockdown, would also decrease those activating effects, resulting in reduced tumor progression. However, this hypothesis could not be easily proved in an in vivo model because of its complexity. Furthermore, knocking down of genes in animal models can cause changes in expression of other ECM components e.g. versican and fibronectin,65 which might affect tumor progression and development.66
Using biomimetic matrices, we finally could show that such an alternative explanation of an HA-dependent melanoma activation could be in place. We showed that both, HYAL1-treated matrices and LMW-HA functionalized matrices, enhanced proliferative and invasive capabilities of melanoma cells, which fit into the hypothesis of LMW-HA-induced tumor progression. Moreover, the results is well correlated to our previous report demonstrated that LMW-HA enhanced proliferation and invasion of BRO human melanoma cells, which was attributed to strong CD44-LMW-HA interactions.11
4. Conclusion
Overall our study found a correlation of Has2-knockdown dependent melanoma tumor growth in vivo and melanoma cell proliferation and invasion in three biomimetic matrix models of HA presentation of varied complexity. We demonstrated that the usage of engineered and well-defined cell culture models can help to better understand the complex behavior of melanoma growth in vivo. Moreover, our findings suggest that HA expression and its degradation by hyaluronidases in the tumor microenvironment trigger tumor progression.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
The authors acknowledge the support of grants from Deutsche Forschungsgemeinschaft (DFG, grants: AN276/6-1 to UA and PO713/9-1 to TP). The usage of the BioImaging Core Facility of the Faculty of Life Science of Leipzig University, supported by a grant from Deutsche Forschungsgemeinschaft INST 268/293-1 FUGG to Tilo Pompe, is gratefully acknowledged.References
- M. De Palma, D. Biziato and T. V. Petrova, Nat. Rev. Cancer, 2017, 17, 457–474 CrossRef CAS PubMed.
- J. Sapudom and T. Pompe, Biomater. Sci., 2018, 6, 2009–2024 RSC.
- D. Nikitovic, M. Tzardi, A. Berdiaki, A. Tsatsakis and G. N. Tzanakakis, Front. Immunol., 2015, 6, 169 Search PubMed.
- R. H. Tammi, A. Kultti, V. M. Kosma, R. Pirinen, P. Auvinen and M. I. Tammi, Semin. Cancer Biol., 2008, 18, 288–295 CrossRef CAS PubMed.
- T. D. Camenisch, A. P. Spicer, T. Brehm-Gibson, J. Biesterfeldt, M. L. Augustine, A. Calabro, S. Kubalak, S. E. Klewer and J. A. McDonald, J. Clin. Invest., 2000, 106, 349–360 CrossRef CAS PubMed.
- A. J. Day and C. A. De La Motte, Trends Immunol., 2005, 26, 637–643 CrossRef CAS PubMed.
- K. M. Bullard, M. T. Longaker and H. P. Lorenz, World J. Surg., 2003, 27, 54–61 CrossRef PubMed.
- U. Anderegg, J. C. Simon and M. Averbeck, Exp. Dermatol., 2014, 23, 295–303 CrossRef CAS PubMed.
- N. Itano, L. Zhuo and K. Kimata, Cancer Sci., 2008, 99, 1720–1725 CrossRef CAS PubMed.
- A. Willenberg, A. Saalbach, J. C. Simon and U. Anderegg, J. Invest. Dermatol., 2012, 132, 385–393 CrossRef CAS PubMed.
- J. Sapudom, F. Ullm, S. Martin, L. Kalbitzer, J. Naab, S. Möller, M. Schnabelrauch, U. Anderegg, S. Schmidt and T. Pompe, Acta Biomater., 2017, 50, 259–270 CrossRef CAS PubMed.
- D. Vigetti, M. Viola, E. Karousou, G. De Luca and A. Passi, Matrix Biol., 2014, 35, 8–13 CrossRef CAS PubMed.
- M. Viola, D. Vigetti, A. Genasetti, M. Rizzi, E. Karousou, P. Moretto, M. Clerici, B. Bartolini, F. Pallotti, G. De Luca and A. Passi, Connect. Tissue Res., 2008, 49, 111–114 CrossRef CAS PubMed.
- K. Fuchs, A. Hippe, A. Schmaus, B. Homey, J. P. Sleeman and V. Orian-Rousseau, Cell Death Dis., 2013, 4, e819–e819 CrossRef CAS PubMed.
- A. E. Rizzardi, R. I. Vogel, J. S. Koopmeiners, C. L. Forster, L. O. Marston, N. K. Rosener, N. Akentieva, M. A. Price, G. J. Metzger, C. A. Warlick, J. C. Henriksen, E. A. Turley, J. B. McCarthy and S. C. Schmechel, Cancer, 2014, 120, 1800–1809 CrossRef CAS PubMed.
- L. Soltés, R. Mendichi, G. Kogan, J. Schiller, M. Stankovska and J. Arnhold, Biomacromolecules, 2006, 7, 659–668 CrossRef PubMed.
- V. Deguine, M. Menasche, P. Ferrari, L. Fraisse, Y. Pouliquen and L. Robert, Int. J. Biol. Macromol., 1998, 22, 17–22 CrossRef CAS PubMed.
- N. Itano and K. Kimata, IUBMB Life, 2002, 1, 195–199 CrossRef PubMed.
- A. B. Csoka, G. I. Frost and R. Stern, Matrix Biol., 2001, 20, 499–508 CrossRef CAS PubMed.
- R. Stern, A. Asari and K. Sugahara, Eur. J. Cell Biol., 2006, 85, 699–715 CrossRef CAS PubMed.
- A. Nagaoka, H. Yoshida, S. Nakamura, T. Morikawa, K. Kawabata, M. Kobayashi, S. Sakai, Y. Takahashi, Y. Okada and S. Inoue, J. Biol. Chem., 2015, 290, 30910–30923 CrossRef CAS PubMed.
- S. P. Fink, L. L. Myeroff, R. Kariv, P. Platzer, B. Xin, D. Mikkola, E. Lawrence, N. Morris, A. Nosrati, J. K. V. Willson, J. Willis, M. Veigl, J. S. Barnholtz-Sloan, Z. Wang and S. D. Markowitz, Oncotarget, 2015, 6, 30500–30515 CrossRef PubMed.
- H. Yamamoto, Y. Tobisawa, T. Inubushi, F. Irie, C. Ohyama and Y. Yamaguchi, J. Biol. Chem., 2017, 292, 7304–7313 CrossRef CAS PubMed.
- V. B. Lokeshwar, W. H. Cerwinka, T. Isoyama and B. L. Lokeshwar, Cancer Res., 2005, 65, 7782–7789 CrossRef CAS PubMed.
- S. Matou-Nasri, J. Gaffney, S. Kumar and M. Slevin, Int. J. Oncol., 2009, 35, 761–773 CAS.
- C. Fieber, P. Baumann, R. Vallon, C. Termeer, J. C. Simon, M. Hofmann, P. Angel, P. Herrlich and J. P. Sleeman, J. Cell Sci., 2003, 117, 359–367 CrossRef PubMed.
- V. Voelcker, C. Gebhardt, M. Averbeck, A. Saalbach, V. Wolf, F. Weih, J. Sleeman, U. Anderegg and J. Simon, Exp. Dermatol., 2008, 17, 100–107 CrossRef CAS PubMed.
- A. G. Bharadwaj, J. L. Kovar, E. Loughman, C. Elowsky, G. G. Oakley and M. A. Simpson, Am. J. Pathol., 2009, 174, 1027–1036 CrossRef.
- S. Dang, Y. Peng, L. Ye, Y. Wang, Z. Qian, Y. Chen, X. Wang, Y. Lin, X. Zhang, X. Sun, Q. Wu, Y. Cheng, H. Nie, M. Jin and H. Xu, Clin. Dev. Immunol., 2013, 2013, 1–11 Search PubMed.
- L. Y. W. Bourguignon, G. Wong, C. A. Earle and W. Xia, Cytoskeleton, 2011, 68, 671–693 CrossRef CAS PubMed.
- M. S. Karbownik and J. Z. Nowak, Pharmacol. Rep., 2013, 65, 1056–1074 CrossRef CAS PubMed.
- J. M. Song, K. Molla, A. Anandharaj, I. Cornax, M. G. O'Sullivan, A. R. Kirtane, J. Panyam and F. Kassie, Oncotarget, 2017, 8, 26927–26940 Search PubMed.
- A. Schmaus, S. Klusmeier, M. Rothley, A. Dimmler, B. Sipos, G. Faller, W. Thiele, H. Allgayer, P. Hohenberger, S. Post and J. P. Sleeman, Br. J. Cancer, 2014, 111, 1–9 CrossRef PubMed.
- A. Schmaus and J. P. Sleeman, Glycobiology, 2014, 25, 258–268 CrossRef PubMed.
- Y. Yamaguchi, H. Yamamoto, Y. Tobisawa and F. Irie, Matrix Biol., 2019, 78–79, 139–146 CrossRef CAS PubMed.
- J. Sapudom and T. Pompe, Biomater. Sci., 2018, 6, 2009–2024 RSC.
- J. Sapudom, S. Rubner, S. Martin, T. Kurth, S. Riedel, C. T. Mierke and T. Pompe, Biomaterials, 2015, 52, 367–375 CrossRef CAS.
- J. Sapudom, L. Kalbitzer, X. Wu, S. Martin, K. Kroy and T. Pompe, Biomaterials, 2019, 193, 47–57 CrossRef CAS PubMed.
- A. Nyga, U. Cheema and M. Loizidou, J. Cell Commun. Signal., 2011, 5, 239–248 CrossRef PubMed.
- A. W. Holle, J. L. Young and J. P. Spatz, Adv. Drug Delivery Rev., 2016, 97, 270–279 CrossRef CAS PubMed.
- M. D. Amatangelo, D. E. Bassi, A. J. Klein-Szanto and E. Cukierman, Am. J. Pathol., 2005, 167, 475–488 CrossRef CAS PubMed.
- K. Franke, J. Sapudom, L. Kalbitzer, U. Anderegg and T. Pompe, Acta Biomater., 2014, 10, 2693–2702 CrossRef CAS PubMed.
- L. Kalbitzer, K. Franke, S. Möller, M. Schnabelrauch and T. Pompe, J. Mater. Chem. B, 2015, 3, 8902–8910 RSC.
- J. Sapudom, L. Kalbitzer, X. Wu, S. Martin, K. Kroy and T. Pompe, Biomaterials, 2019, 193, 47–57 CrossRef CAS PubMed.
- B. P. Toole, Glycobiology, 2002, 12, 37R–42R CrossRef CAS PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS PubMed.
- C. Voutouri and T. Stylianopoulos, PLoS One, 2018, 13, e0193801 CrossRef PubMed.
- S. T. Kreger and S. L. Voytik-Harbin, Matrix Biol., 2009, 28, 336–346 CrossRef CAS PubMed.
- B. Bernert, H. Porsch and P. Heldin, J. Biol. Chem., 2011, 286, 42349–42359 CrossRef CAS PubMed.
- L. Udabage, G. R. Brownlee, S. K. Nilsson and T. J. Brown, Exp. Cell Res., 2005, 310, 205–217 CrossRef CAS PubMed.
- Y. Li, L. Li, T. J. Brown and P. Heldin, Int. J. Cancer, 2007, 120, 2557–2567 CrossRef CAS PubMed.
- C. J. Whatcott, H. Han, R. G. Posner, G. Hostetter and D. D. Von Hoff, Cancer Discovery, 2011, 1, 291–296 CrossRef CAS PubMed.
- Extracellular Sugar-Based Biopolymers Matrices, ed. E. Cohen and H. Merzendorfer, Springer International Publishing, Cham, 2019, vol. 12 Search PubMed.
- A. Schmaus, J. Bauer and J. P. Sleeman, Cancer Metastasis Rev., 2014, 33, 1059–1079 CrossRef CAS.
- S. Haserodt, A. Metin and R. A. Dweik, Glycobiology, 2011, 21, 175–183 CrossRef CAS PubMed.
- X. Li, H. M. Shepard, J. A. Cowell, C. Zhao, R. J. Osgood, S. Rosengren, B. Blouw, S. A. Garrovillo, M. D. Pagel, C. J. Whatcott, H. Han, D. D. Von Hoff, D. M. Taverna, M. J. LaBarre, D. C. Maneval and C. B. Thompson, Clin. Cancer Res., 2018, 24, 4798–4807 CrossRef CAS PubMed.
- J. M. Cyphert, C. S. Trempus and S. Garantziotis, Int. J. Cell Biol., 2015, 2015, 1–8 CrossRef PubMed.
- J. Monslow, P. Govindaraju and E. Puré, Front. Immunol., 2015, 6, 231 Search PubMed.
- P. Li, T. Xiang, H. Li, Q. Li, B. Yang, J. Huang, X. Zhang, Y. Shi, J. Tan and G. Ren, Int. J. Clin. Exp. Pathol., 2015, 8, 12101–12114 CAS.
- H. Okuda, A. Kobayashi, B. Xia, M. Watabe, S. K. Pai, S. Hirota, F. Xing, W. Liu, P. R. Pandey, K. Fukuda, V. Modur, A. Ghosh, A. Wilber and K. Watabe, Cancer Res., 2012, 72, 537–547 CrossRef CAS PubMed.
- Z. Zhang, D. Tao, P. Zhang, X. Liu, Y. Zhang, J. Cheng, H. Yuan, L. Liu and H. Jiang, J. Exp. Clin. Cancer Res., 2016, 35, 181 CrossRef PubMed.
- E. J. Franzmann, G. L. Schroeder, W. J. Goodwin, D. T. Weed, P. Fisher and V. B. Lokeshwar, Int. J. Cancer, 2003, 106, 438–445 CrossRef CAS.
- J. X. Tan, X. Y. Wang, X. L. Su, H. Y. Li, Y. Shi, L. Wang and G. S. Ren, PLoS One, 2011, 6, e22836 CrossRef CAS PubMed.
- M. W. Kramer, R. Golshani, A. S. Merseburger, J. Knapp, A. Garcia, J. Hennenlotter, R. C. Duncan, M. S. Soloway, M. Jorda, M. A. Kuczyk, A. Stenzl and V. B. Lokeshwar, Eur. Urol., 2010, 57, 86–94 CrossRef CAS PubMed.
- K. E. Keller, Y. Y. Sun, J. A. Vranka, L. Hayashi and T. S. Acott, PLoS One, 2012, 7, e48523 CrossRef CAS PubMed.
- N. Kobayashi, S. Miyoshi, T. Mikami, H. Koyama, M. Kitazawa, M. Takeoka, K. Sano, J. Amano, Z. Isogai, S. Niida, K. Oguri, M. Okayama, J. A. McDonald, K. Kimata, S. Taniguchi and N. Itano, Cancer Res., 2010, 70, 7073–7083 CrossRef CAS PubMed.
- J. C. Becker, M. H. Andersen, D. Schrama and P. Thor Straten, Cancer Immunol. Immunother., 2013, 62, 1137–1148 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01636h |
| ‡ Equally contributed to this work |
| This journal is © The Royal Society of Chemistry 2020 |