
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PD-L1-targeted microbubbles loaded with docetaxel produce a synergistic effect for the treatment of lung cancer under ultrasound irradiation†
Tiankuan
Li‡
 a,
Zhongqian
Hu‡
b,
Chao
Wang
a,
Jian
Yang
c,
Chuhui
Zeng
a,
Rui
Fan
a and
Jinhe
Guo
*a
a,
Zhongqian
Hu‡
b,
Chao
Wang
a,
Jian
Yang
c,
Chuhui
Zeng
a,
Rui
Fan
a and
Jinhe
Guo
*a
aCenter of Interventional Radiology and Vascular Surgery, Department of Radiology, Zhongda Hospital, Medical School, Southeast University, Nanjing, China. E-mail: jinheguo@sina.com
bDepartment of Ultrasound, Zhongda Hospital, Medical School, Southeast University, Nanjing, China
cJiangsu Key Laboratory of Molecular and Functional Imaging, Department of Radiology, Zhongda Hospital, Medical School, Southeast University, Nanjing, China
First published on 23rd December 2019
Abstract
Immunotherapy is gradually becoming as important as traditional therapy in the treatment of cancer, but adverse drug reactions limit patient benefits from PD1/PD-L1 checkpoint inhibitor drugs in the treatment of non-small cell lung cancer (NSCLC). As a chemotherapeutic drug for NSCLC, docetaxel (DTX) can synergize with PD1/PD-L1 checkpoint inhibitors but increase haematoxicity and neurotoxicity. Herein, anti-PD-L1 monoclonal antibody (mAb)-conjugated and docetaxel-loaded multifunctional lipid-shelled microbubbles (PDMs), which were designed with biologically safe phospholipids to produce synergistic antitumour effects, reduced the incidence of side effects and promoted therapeutic effects under ultrasound (US) irradiation. The PDMs were prepared by the acoustic-vibration method and then conjugated with an anti-PD-L1 mAb. The material features of the microbubbles and their cytotoxic effects, cellular apoptosis and cell cycle inhibition were studied. A subcutaneous tumour model was established to test the drug concentration-dependent and antitumour effects of the PDMs combined with US irradiation, and an orthotopic lung tumour model simultaneously confirmed the antitumour effect of this synergistic treatment. The PDMs achieved higher cellular uptake than free DTX, especially when combined with US irradiation. The PDMs combined with US irradiation also induced an increased rate of cellular apoptosis and an elevated G2-M arrest rate in cancer cells, which was positively correlated with PD-L1 expression. An in vivo study showed that synergistic treatment had relatively strong effects on tumour growth inhibition, increased survival time and decreased adverse effect rates. Our study possibly provides a well-controlled design for immunotherapy and chemotherapy and has promising potential for clinical application in NSCLC treatment.
Introduction
Lung cancer is the type of cancer with the highest morbidity and mortality worldwide,1 and NSCLC is the main type of lung cancer.2 Over the past few years, the most exciting antitumour drug class has been PD1/PD-L1 checkpoint inhibitors, which can counteract the abilities of tumour cells to suppress the immune system and promote self-tolerance.3–5 Due to its broad application, immunotherapy has become as valid as chemotherapy for advanced NSCLC.6,7 Clinical evidence has shown that the combination of a PD1/PD-L1 checkpoint inhibitor with a chemotherapeutic drug can achieve better therapeutic effects than monotherapy.8,9 However, in addition to the cardiotoxicity of PD1/PD-L1 checkpoint inhibitors,10,11 this combination of free drugs also aggravates haematotoxicity, hepatotoxicity, and neurotoxicity.12,13 Therefore, an appropriate drug delivery system is needed to reduce the adverse events of these two drugs.Drug delivery systems based on microbubbles are good contrast agents and drug carriers and have the advantages of satisfactory chemical stability, biodegradability, low toxicity, and easy chemical modification.14 The common components of the microbubble shell are albumin, phospholipids, and macromolecular polymers. Clinical studies have shown that lung surfactant microbubbles increase lipophilic drug payloads for ultrasound (US)-targeted delivery.15 The smaller size of microbubbles allows extravasation from blood vessels into surrounding tissues, improving stability and giving longer residence times in the systemic circulation.16 Therefore, lipid-shelled microbubbles might be an appropriate drug delivery system for the combination of a PD1/PD-L1 checkpoint inhibitor and docetaxel (DTX).
As one of the most common noninvasive physical radiation sources, US plays an important role in clinical diagnosis and therapy. Lung US as an emerging theranostic modality exhibits non-ionizing properties, high local resolution, real-time imaging, and low cost.17 Clinical trials have confirmed the efficacy and safety of pulmonary US irradiation treatment combined with corresponding drugs.18,19 The cavitation and sonoporation effects are generally believed to contribute to the therapeutic effect of US irradiation, which can ensure specialized targeted delivery of proteins, genes, exosomes or traditional chemotherapeutic drugs.20–22 Hence, we hypothesized that immunochemotherapeutic phospholipid microbubbles combined with US irradiation might enhance the efficacy and reduce the adverse effects of the combination of a PD1/PD-L1 inhibitor and DTX.
To verify our hypothesis, we designed a multifunctional microbubble system in which the membrane was DTX loaded and then anti-PD-L1 monoclonal antibody (mAb)-modified (PDMs). The anti-PD-L1 mAb could block the immunosuppressive PD1/PD-L1 pathway, while the PDMs directly kill tumour cells via DTX. Moreover, US irradiation was used to rupture the PDMs to further increase drug concentrations in the tumour. The cavitation and sonoporation effects improve the ability of drugs to enter the interstitial space and tumour cells. Our study showed that the PDMs showed excellent antitumour activity both in vitro and in vivo. Fluorescence microscopy and flow cytometry showed that PDM treatment led to increased DTX uptake, which enhanced cellular apoptosis and cell cycle inhibition. The distribution of DTX and PD-L1 in tumour tissues was also observed by in vivo fluorescence imaging. Our results revealed that the PDMs could not only increase the effect of chemotherapy but also enhance the antitumour effect by promoting the proliferation of CD4+ T and CD8+ T cells and decreasing the levels of the cytokines VEGF and TGF-β. All these outcomes indicated that the PDMs had good antitumour efficacy.
Results and discussion
Characterization of immunochemotherapeutic microbubbles
The synthesis process of the PD-L1-mediated immunochemotherapeutic microbubbles is summarized in Fig. 1A. A schematic diagram showing the US irradiation-enhanced drug cytotoxicity and immune-activating mechanism of the microbubbles is presented in Fig. 1B. The round shape of the microbubbles was manifested by a scanning electron micrograph (Fig. 2A). The sizes of the microbubbles are shown in Table S1;† the size of the blank microbubble (BM) was initially 607.6 ± 22.7 nm, the polydispersity index (PDI) was 0.234 ± 0.018 and the zeta potential was −3.91 ± 0.861 mV. The size of the microbubbles later increased to 629.1 ± 19.8 nm, with a PDI of 0.259 ± 0.026 and zeta potential of −8.98 ± 0.693 mV after loading DTX. The size further increased to 666.4 ± 35.9 nm, with a PDI of 0.298 ± 0.036 and zeta potential of −9.69 ± 0.964 mV after coupling to an anti-PD-L1 mAb. These increases were concluded to be the result of successful synthesis of the antibody. The dispersions of the microbubbles are shown in Fig. 2B. The change in size could be a sign of successful binding of the antibody to the surface of the microbubbles. The encapsulation and loading efficiency of DTX in the DTX-loaded microbubble (DM) were 59.21 ± 1.97% and 4.75 ± 0.65%, respectively, and in the PDMs they were 57.34 ± 2.61% and 4.45 ± 0.91%, respectively (Table S2†).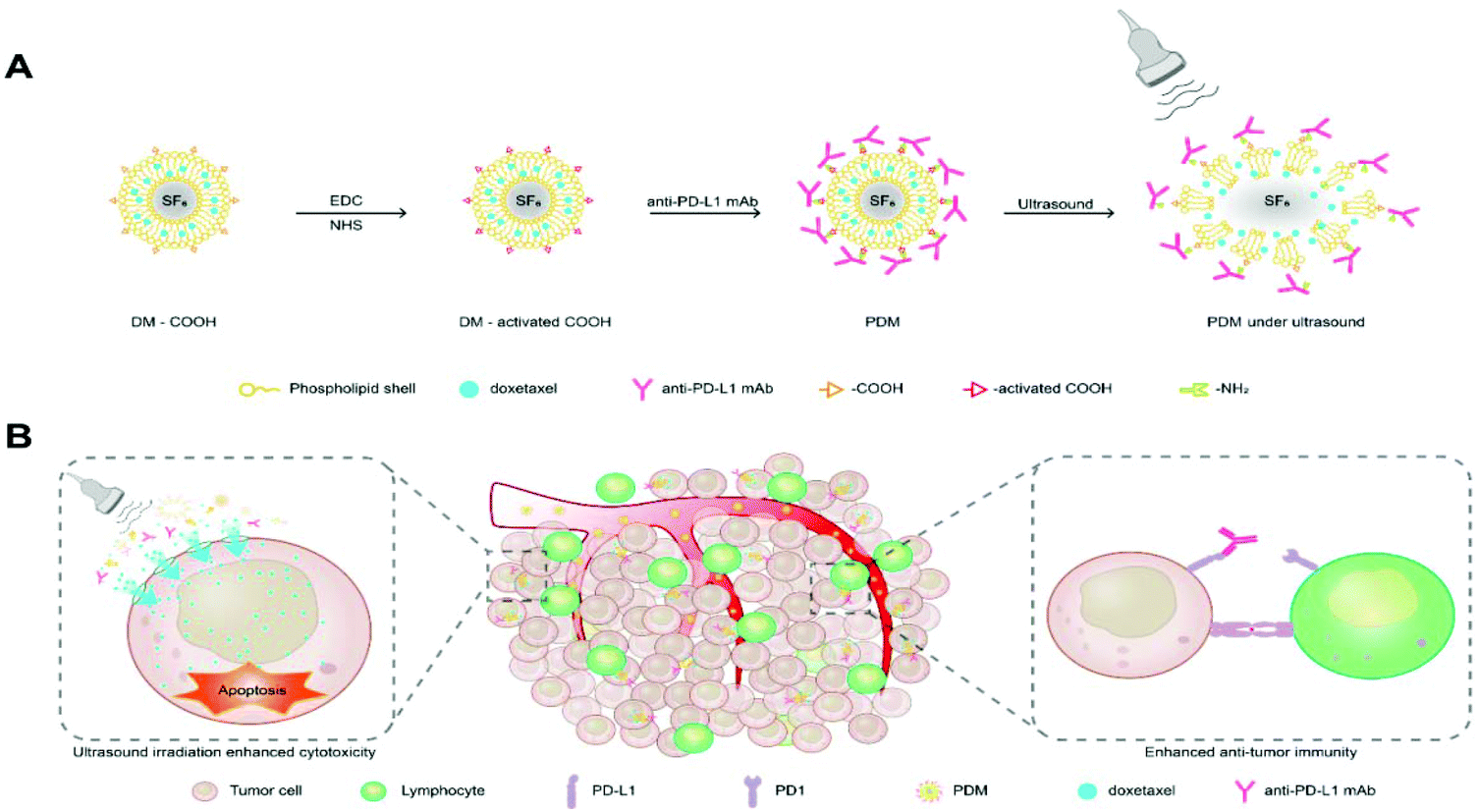 | ||
| Fig. 1 Novel microbubbles combining DTX antitumour activity with PD-L1 blockade in the context of ultrasound irradiation. (A) Schematic diagram showing the preparation of DTX and anti-PD-L1 mAb-co-loaded microbubbles. (B) Scheme of synergistic chemotherapy and immunotherapy under ultrasound irradiation for tumour treatment. | ||
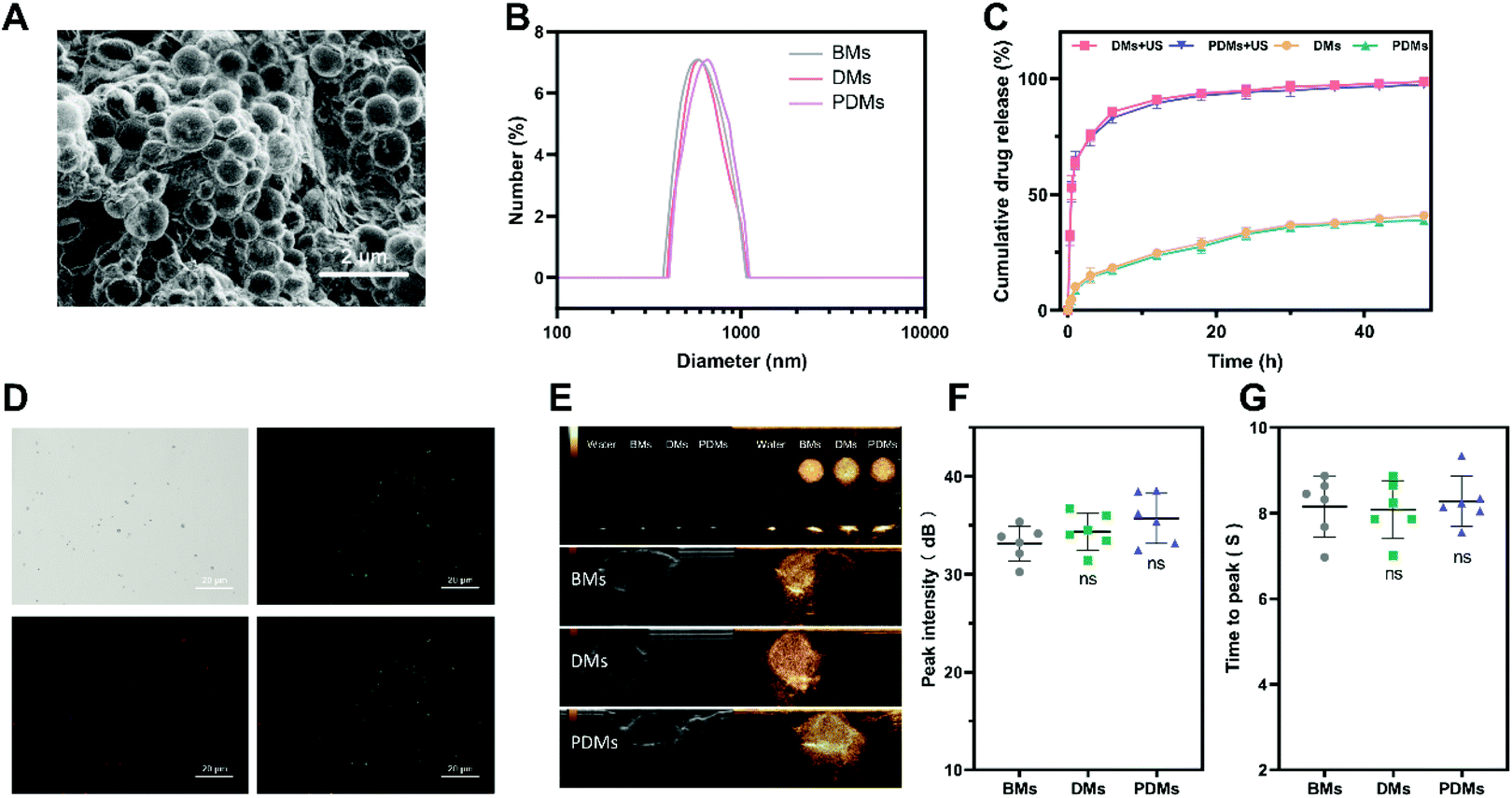 | ||
| Fig. 2 Characterization of docetaxel and anti-PD-L1 mAb-co-loaded microbubbles. (A) SEM image of PDMs, scale bar = 2 μm. (B) Particle size distributions. (C) In vitro cumulative drug release by different bubbles with or without US irradiation. (D) CLSM images of docetaxel and CY5-labelled anti-PD-L1 mAb-co-loaded microbubbles with a shell containing FITC, scale bar = 20 μm. (E) In vitro and in vivo contrast effects of microbubbles. (F) Peak intensity of in vivo imaging of microbubbles. (G) Time to peak in vivo imaging of microbubbles. The data in 2F and 2G are expressed as the mean ± s.d.; n = 6 independent samples, analysed using one-way ANOVA with the Newman–Keuls post hoc test. ns indicates P > 0.05 compared with the BM group. BMs (blank microbubbles), DMs (docetaxel-loaded microbubbles), PDMs (docetaxel and anti-PD-L1 mAb-co-loaded microbubbles), and US (ultrasound irradiation). | ||
The profiles of the release of DTX from the microbubbles with/without US irradiation were examined to assess the effects of sonication on DTX release. As shown in Fig. 2C, US irradiation significantly promoted drug release. CY5-labelled anti-PD-L1 mAb conjugation with FITC-labelled DTX microbubbles was observed by laser scanning confocal microscopy (LSCM) (Fig. 2D). The microbubbles consistently maintained a round shape and were bound with a fluorescein-labelled antibody.
The BMs, DMs, and PDMs had similar contrast imaging capabilities in vitro and in vivo (Fig. 2E); the peak intensity and time to peak were not significantly different among the microbubbles (Fig. 2F and G). In addition to the expression on a number of cancer cell lineages, PD-L1 was also expressed on non-parenchymal cells and non-haematopoietic lineages.5 The distribution of PD-L1 in other organs might prevent the enhancing effect of the microbubbles from being obvious over a short period of time.
We also investigated whether BMs, DMs, and PDMs can induce haemolysis (Fig. S1A and B†), and the results showed that none of the formulations was haemolytic. The toxicity study showed that no weight loss was observed, and serum biochemical parameters were within the corresponding reference ranges (Fig. S2A–E†). HE staining showed that the free combo group resulted in thickened alveolar walls and lymphocyte infiltration in normal lung tissue. Our findings illustrate that the immunochemotherapeutic microbubbles are biologically safe and lung protective.
Microbubble cytotoxicity in vitro
The PD-L1 expression levels of one type of mouse cell (LLC) and three types of human cells (NCI-H460, NCI-H1299, and A549) were tested by flow cytometry. As shown in Fig. 3A and B, the expression of LLC cells was similar to that of NCI-H460 cells, while NCI-H1299 cells had the highest expression and A549 cells had the lowest. Free DTX and DMs had similar cytotoxicity in the four tumour cell lines. PDMs had higher efficacy because of the conjugated anti-PD-L1 mAb, and this efficacy was increased by US irradiation (Fig. 3C–F). The results demonstrated that both anti-PD-L1 mAb targeting and US irradiation could enhance the cytotoxicity of DTX to the target tumour tissues.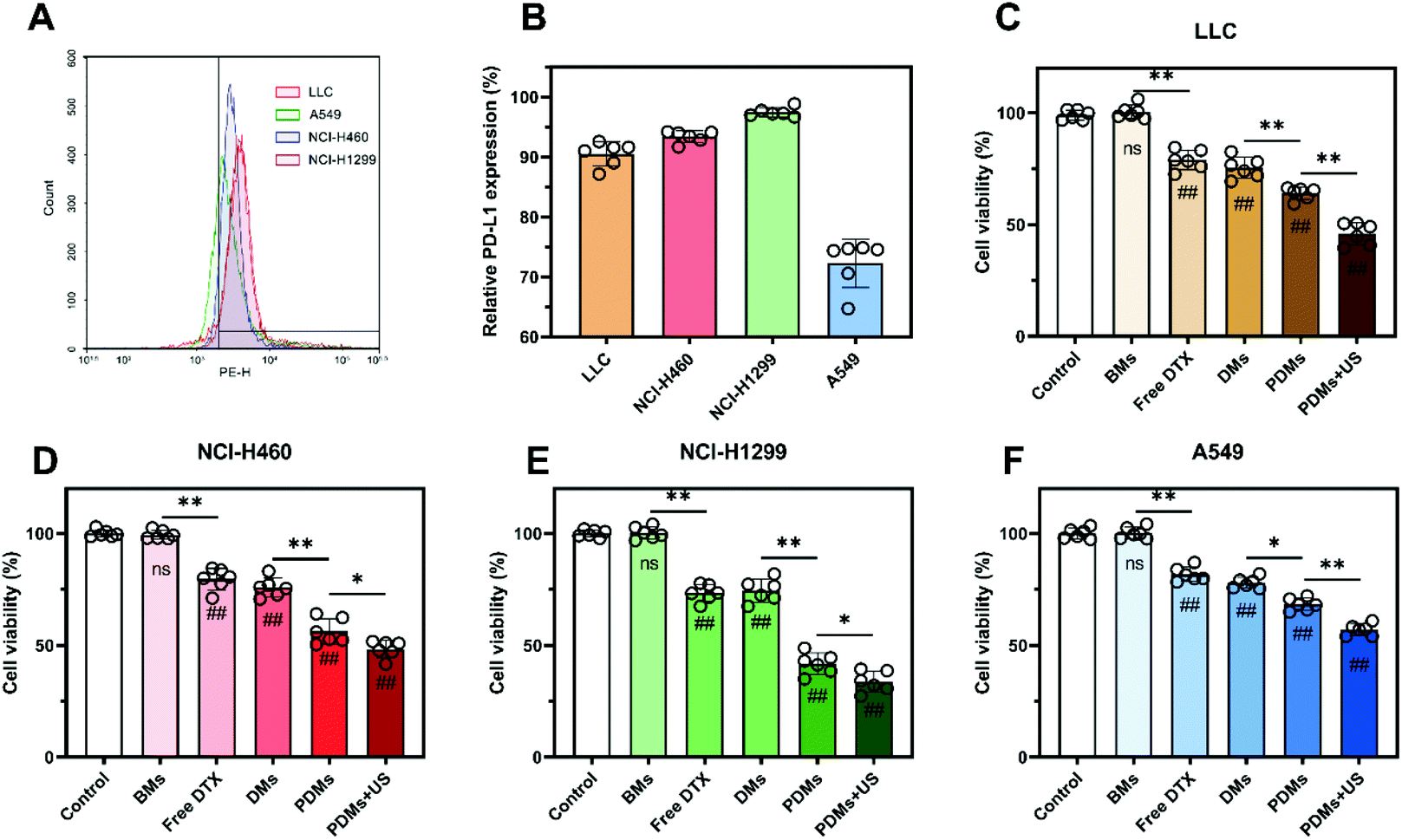 | ||
| Fig. 3 PD-L1 expression of LLC, A549, NCI-H460, and NCI-H1299 cells and the toxicity of microbubbles to tumour cells. (A) Flow cytometry was used to assess the PD-L1 expression profiles of cells. (B) LLC, NCI-H460, and NCI-1299 cells showed relatively high PD-L1 expression, while A549 cells showed the lowest PD-L1 expression. (C–F) A CCK-8 assay was used to determine the cell viabilities of tumour cells incubated with different formulations for 24 h. The results in B–F are expressed as the mean ± s.d.; n = 6 independent samples, analysed using one-way ANOVA with the Newman–Keuls post hoc test. #P < 0.05 and ##P < 0.01 compared with the control group (without microbubbles); *P < 0.05 and **P < 0.01 for comparisons among groups. | ||
In vitro enhancement of cellular drug uptake mediated by PD-L1
To further assess the relationship between the targeting efficiency of PDMs and affinity of DTX for LLC cells in vitro, the cellular uptake of DTX in different formulations was determined by fluorescence imaging and flow cytometry. C6 to indicate drug uptake was contained in microbubbles at a concentration equimolar to DTX. As shown in Fig. 4A and D, CLSM images indicated elevated intracellular drug uptake of C6-PDMs, especially under US irradiation. The flow cytometry results were also consistent with the fluorescence imaging results (Fig. S4†). Therefore, PD-L1-mediated internalization was more efficient than passive diffusion and nonspecific targeting. US irradiation could promote drug uptake, which might account for the increased cellular toxicity of PDMs when combined with US irradiation.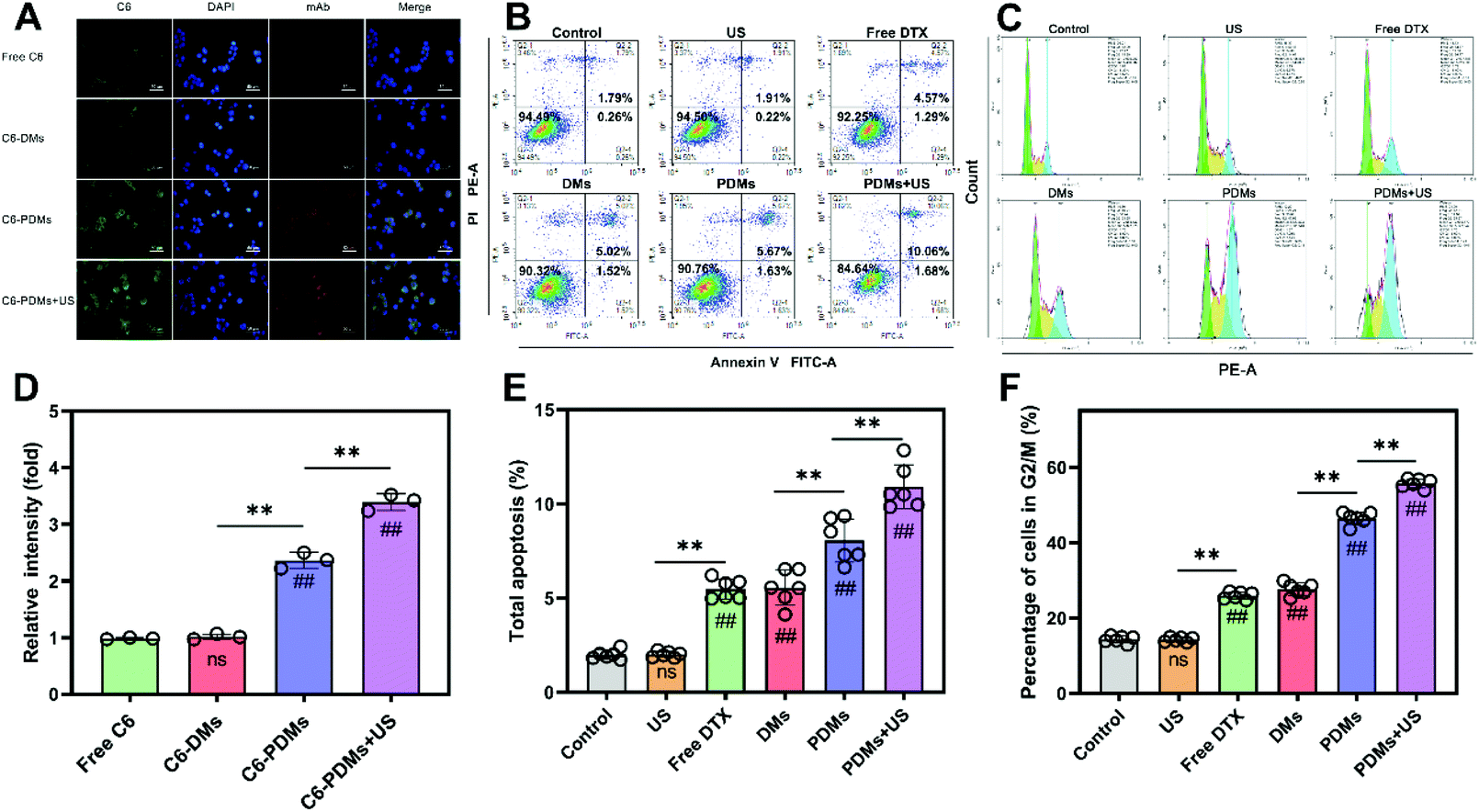 | ||
| Fig. 4 Cellular uptake, apoptosis, and cell cycle inhibition of LLC cells given different treatments. (A and D) Fluorescence images obtained after a four-hour incubation with free C6, C6-DMs, C6-PDMs, or C6-PDMs with 30 seconds of ultrasound irradiation; DAPI, blue; C6, green; and a goat anti-rat secondary antibody, red; scale bar = 50 μm; n = 3. The data were processed using ImageJ software. (B and E) Cell apoptosis study of LLC cells after incubation. The cells were treated with ultrasound irradiation, free DTX, DMs, PDMs, or PDMs + US, and the total apoptosis rate was analysed. (C and F) Cell cycle inhibition study of LLC cells treated with ultrasound irradiation, free DTX, DMs, PDMs, or PDMs with ultrasound irradiation after incubation. Data are shown as the mean ± s.d.; n = 6 independent samples, analysed using one-way ANOVA with the Newman–Keuls post hoc test. #P < 0.05 and ##P < 0.01 compared with the control group (without microbubbles), *P < 0.05 and **P < 0.01 for comparisons among groups. C6 (coumarin-6), C6-DMs (docetaxel and coumarin-6-co-loaded microbubbles), and C6-PDMs (coumarin-6, docetaxel and anti-PD-L1 mAb-co-loaded microbubbles). | ||
Apoptosis induction and cell cycle inhibition in LLC cells in vitro
The cell apoptosis study for assessing different formulations utilized the Annexin V-FITC/PI method to further explore tumour killing. LLC cells were treated with US irradiation, free DTX, DMs, PDMs, or PDMs + US. After 24 h of incubation, the total apoptosis rate was analysed by flow cytometry. As shown in Fig. 4B and E, PDMs + US induced the highest apoptosis rate, free DTX and DMs produced similar apoptosis rates, and US irradiation monotherapy had no influence on apoptosis.For cell cycle analysis, the PI/RNase method was employed and the remaining steps were similar to the abovementioned method. In agreement with the cytotoxicity, cellular uptake, and apoptosis studies, the cell cycle analysis showed that cell cycle inhibition increased in response to different formulations (Fig. 4C and F). Hence, using the aforementioned data, it can be concluded that the PDMs combined with US irradiation increased drug uptake, inhibited the cell cycle, promoted apoptosis, and enhanced drug toxicity to the cells.
Tumour resistance to the paclitaxel family has been a problem troubling clinicians for a long time until studies on transcriptional and post-transcriptional mechanisms revealed that the paclitaxel family induces the expression of PD-L1 immunosuppressive molecules through the mitogen-activated protein kinase (MAPK) pathway.23 The evidence suggests that patients may benefit from a synergy of docetaxel and a PD-1/PD-L1 checkpoint inhibitor, while PD-L1 is expressed in various tumor cells.24
Enriched bio-distribution in melanoma tumour tissue mediated by PD-L1 and US targeting in vivo
Therapeutic efficacy and adverse effects could be influenced by the distribution and accumulation of various formulations in tumours and vital organs. DiR, a lipophilic fluorescent probe, was used to mark microbubbles directly. Free DiR, DiR-DMs, or DiR-PDMs were injected via the tail vein. DiR-PDMs combined with 10-minute US irradiation were also investigated. The in vivo bio-distributions of different formulations were monitored using an IVIS Spectrum system. Compared with free DiR and DiR-DMs, DiR-PDMs exhibited an enhanced signal in tumour tissues in vivo and ex vivo, and this signal intensity was further enhanced under US irradiation (Fig. 5A). The signal of DiR-PDMs combined with US irradiation was found to be the strongest at the first hour. Among the groups, the DiR-PDM group maintained the highest signal level. The DiR-PDMs showed a good targeting effect, and the signal reached a peak at 12 h. The DiR-DMs were more enriched in tumours than free DiR, which consistently exhibited a low signal intensity (Fig. 5B). | ||
| Fig. 5 IVIS images of mice treated with different formulations. (A) Fluorescence images of mice-bearing LLC melanoma at 1, 6, 12, and 24 h after free DiR, DiR-DM, or DiR-PDM treatment with/without ultrasound irradiation. Tumours and major organs were harvested after the last imaging session. (B) Statistical analysis of the superficial ROI average radiant efficiency. (C) Statistical analysis of the average radiant efficiency of the organs and tumours. Data are shown as the mean ± s.d.; n = 4 biologically independent mice, analysed using one-way ANOVA with the Newman–Keuls post hoc test. *P < 0.05 and **P < 0.01 among groups. | ||
To further observe the distribution of formulations in organs after administration, tumour-bearing mice were sacrificed after the last imaging time point, and the tumours and all the vital organs (the heart, lungs, spleen, kidneys, and liver) were harvested for the following experiments. The ex vivo experimental results showed increased accumulation of DiR in tumour tissue (Fig. 5A). In addition, quantitative region of interest analysis (Fig. 5C) showed that PDMs combined with US irradiation exhibited the highest fluorescence efficiency, which might portend enhanced therapeutic effects. As predicted, US irradiation promoted rapid drug release in tumours, and the active targeting by the anti-PD-L1 mAb on the surface of PDMs promoted drug accumulation. A comparison of the distributions of different drugs in major metabolic organs showed that the distribution of free drug in the liver was distinctly lower than that of the microbubbles. It is deduced that increasing the time between injections might reduce liver injury, and this was confirmed by the biotoxicity experiment.
Due to the side effects of immunochemotherapy, a variety of materials have been used as adjuncts.25–28 The targeted drug delivery system of ultrasonic microbubbles, coupled with an antibody that targets a corresponding antigen overexpressed on tumour cells, provides a promising strategy for reducing the severe adverse effects associated with chemotherapeutic drugs.29,30 The fabricated PDMs could effectively target the tumour tissue and thus reduce off-target toxicity. PD-L1 is highly expressed on the surface of various tumour cells and helps tumour cells evade antitumour immunity. Previous studies have indicated that anti-PD-L1 mAb-coupled drug delivery systems can target tumour cells31 and activate the immune system,32 and this approach can synergize with chemotherapy. Therefore, it can be concluded that PDMs combined with US irradiation exhibited an increased drug concentration and extended drug duration in tumours and were therefore beneficial for therapeutic effects of increased strength and duration.
Inhibited tumour growth in a subcutaneous tumour model after treatments with different microbubble formulations
A subcutaneous tumour model was established to evaluate the antitumour effects of various formulations of microbubbles in vivo. When the tumour volume reached 200 mm3, normal saline, free DTX, DMs, a free-drug combination, or PDMs were injected via the tail vein; PDMs combined with US irradiation were also investigated. Every 3 days, the mice were weighed, the tumour volumes were measured, and blood was obtained to assess liver and kidney functions. After all the treatments, the mice were sacrificed for subsequent histopathological study of the tumours and vital organs to evaluate their efficacy and safety.As shown in Fig. 6B–D, the PDMs combined with US irradiation group had the highest survival rate through the termination of the experiment, and this group showed the best inhibition of tumour growth. The PDMs alone showed a better therapeutic effect than the free-drug combination and ranked second. In addition, the efficacy of synergistic therapy was more obvious than that of chemotherapy. The DMs produced slightly improved therapeutic effects on the tumour volume inhibition and survival rate. The difference in body weights was not obvious among the groups.
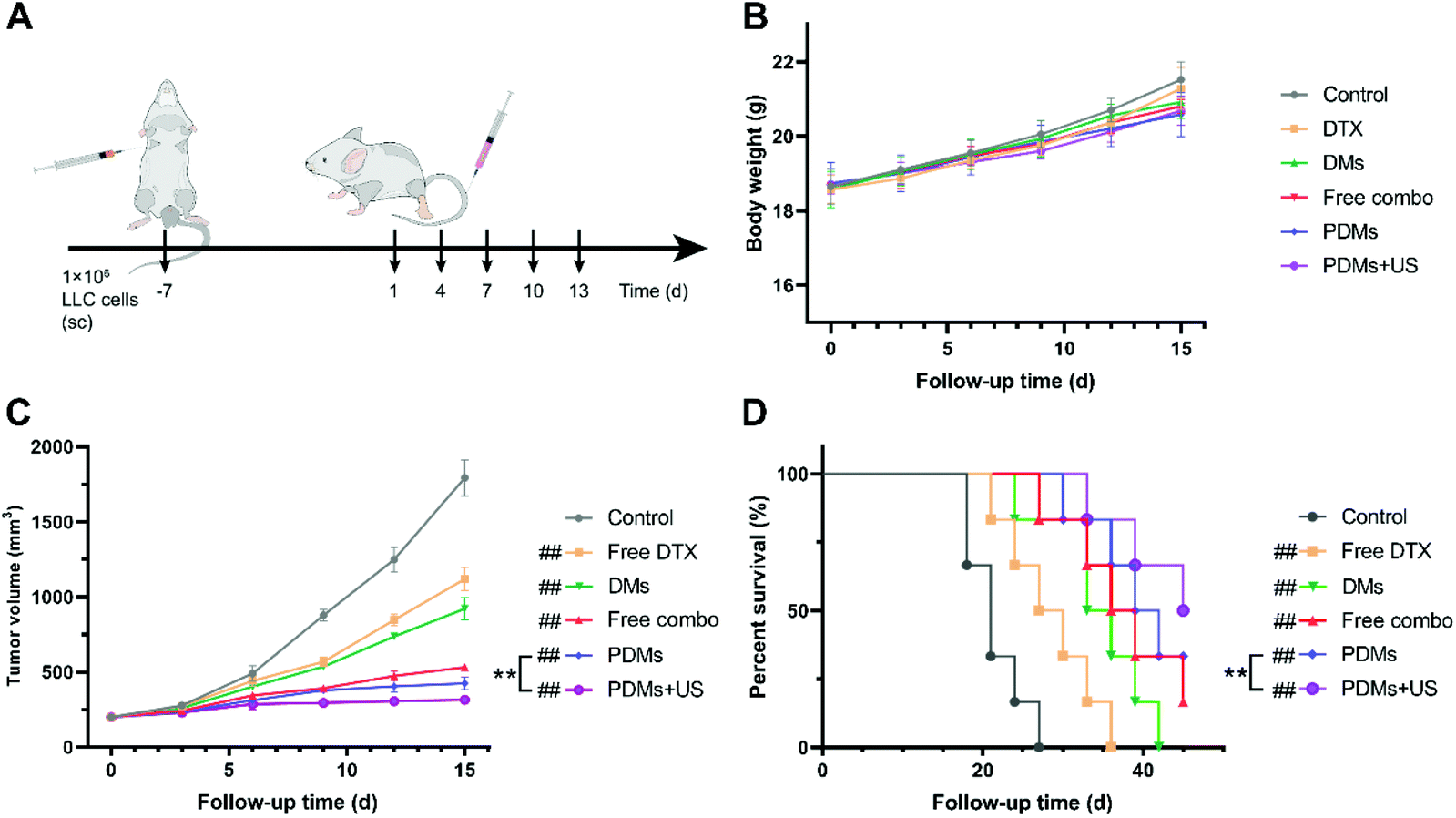 | ||
| Fig. 6 Treatment with PDMs and ultrasound irradiation enhanced tumour inhibition in an LLC subcutaneous tumour model. (A) C57BL/6 mice were subcutaneously injected with LLC cells on day 7. When the tumour volume reached 200 mm3, the mice received different therapeutic formulations. (B) Body weight, (C) tumour volume, and (D) survival rate were analysed. The data in B and C are expressed as the mean ± s.d.; n = 6 biologically independent animals. #P < 0.05 and ##P < 0.01 compared with the control group (normal saline); *P < 0.05 and **P < 0.01 between groups compared using a paired two-way Student's t-test. The log-rank test followed by Tukey's post hoc test was performed to determine the statistical significance in D; n = 6 biologically independent animals. #P < 0.05 and ##P < 0.01 compared with the control group; *P < 0.05 and **P < 0.01 compared with the control group (normal saline). | ||
Apoptosis and proliferation in tumours were analysed using the TUNEL assay and CD31 and Ki67 immunohistochemistry (Fig. 7A–F). The data showed that the PDMs combined with US irradiation group had the highest apoptosis rate and lowest proliferation level. The trend in the data was in accordance with the tumour growth observations. The results for cleaved caspase-3, cleaved caspase-8, and cleaved caspase-9 in western blot assays also revealed that the PDMs combined with US irradiation group had the highest apoptosis rate (Fig. 8A–D).
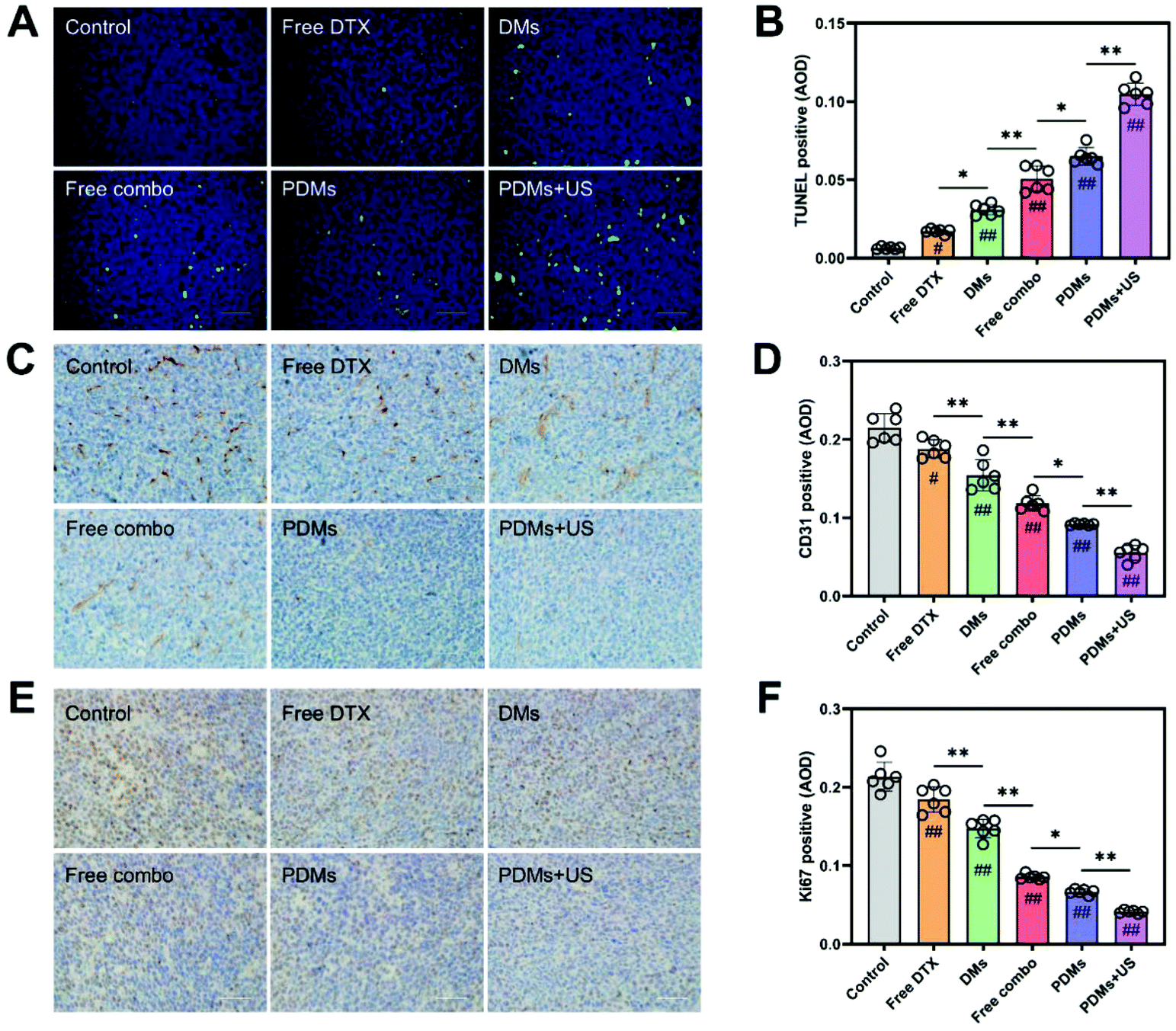 | ||
| Fig. 7 TUNEL staining and immunohistochemical staining for CD31 and Ki67 in tumour sections. (A) TUNEL-stained merged image (apoptotic cells are shown in green) and statistical analysis of (B) the positive rate of TUNEL staining. Immunohistochemical staining for (C) CD31 and (E) Ki67 in tumour sections; angiogenic cells and proliferating cells are shown in brown. Scale bar = 200 μm. Statistical analysis of the positive rates for (D) CD31 and (F) Ki67 staining. Data were processed using ImageJ software and are expressed as mean ± s.d.; n = 6, analysed using one-way ANOVA with the Newman–Keuls post hoc test. #P < 0.05 and ##P < 0.01 compared with the control group (without microbubbles); *P < 0.05 and **P < 0.01 among groups. | ||
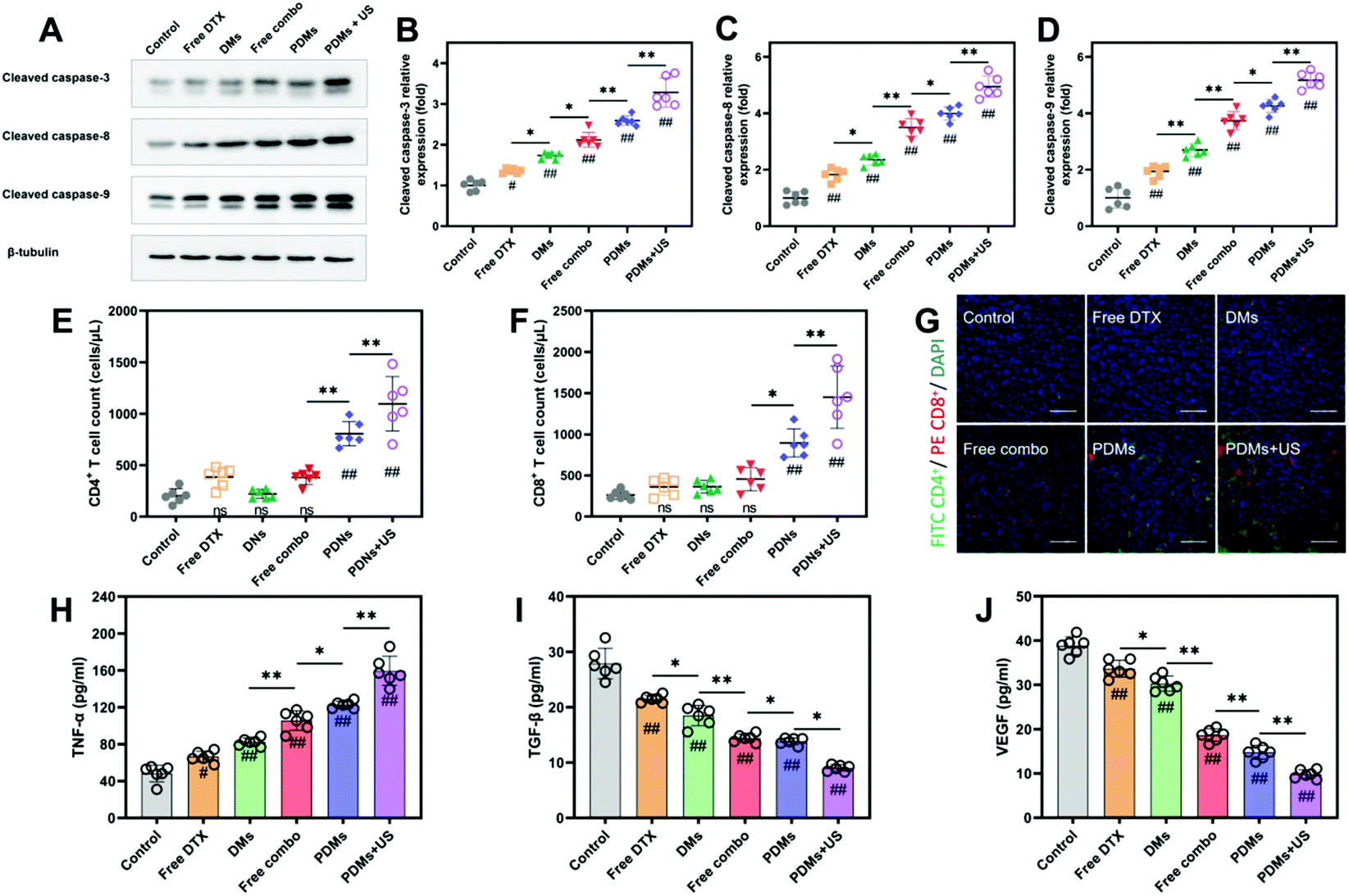 | ||
| Fig. 8 Changes in western blot assays, TIL levels and cytokine levels at 15 days after the first treatment. (A–D) Western blot data for apoptosis-related proteins showed the expression of proapoptotic proteins. Data were processed using ImageJ software. (E and F) Flow cytometry analysis determined the numbers of CD4+ and CD8+ TILs that increased after treatments, and counts of each subset were gated from CD45+ lymphocytes. (G) Fluorescence micrographs of tumour sections showing CD4+ (red) and CD8+ (green) TILs and DAPI staining (blue); scale bar = 200 μm. The levels of cytokines, including TNF-α (H), TGF-β (I) and VEGF (J), in tissue culture supernatants were determined. PDMs + US significantly increased TIL numbers and improved pro-inflammatory cytokine levels. The results are expressed as mean ± s.d.; n = 6 biologically independent mice, analysed by one-way ANOVA with the Newman–Keuls post hoc test. #P < 0.05 and ##P < 0.01 compared with the control group (normal saline); *P < 0.05 and **P < 0.01 among groups. | ||
It has been verified that US irradiation can enhance checkpoint inhibitor therapy.33 US irradiation combined with immunotherapeutic nanomaterials has been studied in colorectal cancer,33 B-cell lymphoma,34 and breast cancer.35 US irradiation combined with microbubble therapy for local lesions has many advantages. The cavitation effect caused by microbubble rupturing under US irradiation can achieve high drug enrichment.18,36 The sonoporation effect promotes drug uptake and enhances the delivery of small and large molecules.37,38 Moreover, studies of US irradiation-enhanced microbubble tumour treatments indicated that this approach could induce rapid vascular damage and shut down blood flow.36 Our results demonstrated that PDMs combined with US irradiation can produce a strong antitumour effect.
Immune activation and cytokine production alleviation by microbubbles
To study the infiltration of immune cells into the tumour site after treatment, tumor-infiltrating lymphocytes (TILs) were harvested from tumours and analysed by immunofluorescence and flow cytometry on day 15 of the experimental process. The flow cytometry results also showed that CD8+ and CD4+ cell infiltration was approximately 4-fold greater in the PDMs combined with US irradiation group than in the control group (Fig. 8E and F). Immunofluorescence staining revealed that the tumours from the PDMs combined with US irradiation group were remarkably infiltrated by both CD8+ and CD4+ T cells, while untreated tumours exhibited limited infiltration (Fig. 8G).We also observed that the level of TNF-α, which induces tumour cell apoptosis, was increased in the culture supernatants of tumour tissues after synergistic treatment (Fig. 8H). The immunosuppressive functions of TGF-β are correlated with malignancy in many cancers, and the level of TGF-β was downregulated in response to the synergistic treatment (Fig. 8I). The expression of TNF-α and TGF-β after treatment with different formulations indicated that the synergistic treatment resulted in the best tumour suppression. In addition, reduced levels of VEGF were observed (Fig. 8J), which plays critical roles in angiogenesis, tumour growth and metastasis cancer. VEGF is also an important index for evaluating the growth of solid tumours. The PDMs combined with US irradiation produced the lowest VEGF expression; this result at the molecular level suggested that synergistic treatments could limit tumour growth.
Further antitumour efficacy in an orthotopic tumour model
We established orthotopic tumour models by injecting LLC cells into Matrigel under X-ray guidance (Fig. 9A and Fig. S6†). When the tumour volume reached 60 mm3, all the interventions were investigated. Increases in tumour volumes were obviously inhibited by PDMs combined with US irradiation therapy at the last follow-up CT scan (Fig. 9B and C), while the tumours in the other groups occupied almost the entire right lung. The synergistic therapy group also had a better health status and longer survival time than the other groups (Fig. 9D and E). The body weight declined gradually in all the groups except for the PDMs combined with US irradiation group, and this phenomenon was different from that observed in the subcutaneous tumour model. The possible reason is that although LLC cells have low invasiveness, pleural cavity occupation can affect cardiopulmonary function and blood oxygen levels, thereby affecting the health status of the mice. | ||
| Fig. 9 Different therapeutic formulations were tested in an orthotopic LLC tumour model. (A) Twenty microliters (containing 2 × 106 LLC cells with Matrigel) were injected into the right lung of C57BL/6 mice under X-ray guidance on day 5. When the tumour volume reached 60 mm3, the mice received different therapeutic formulations. Follow-up analyses (B) CT scanning results, (C) tumour volume, (D) body weight, and (E) survival rate. The data in C and D are expressed as mean ± s.d.; n = 6 biologically independent mice. #P < 0.05 and ##P < 0.01 compared with the control group (normal saline); *P < 0.05 and **P < 0.01 between groups using a paired two-way Student's t-test. The log-rank test was performed followed by Tukey's post hoc test to determine the statistical significance in E; n = 6 biologically independent mice. #P < 0.05 and ##P < 0.01 compared with the control group (normal saline); *P < 0.05 and **P < 0.01 between groups. | ||
Since the lungs are air-filled cavities, lung diseases are often traditionally considered difficult to be treated with US irradiation. However, clinical studies have shown that US irradiation has obvious therapeutic efficacy in the treatment of lung diseases18,19 due to its ability to noninvasively mediate precise drug delivery. Here, the results of the orthotopic tumour experiment indicated that PDMs combined with US irradiation showed good antitumour performance not only in subcutaneous tumour treatment but also in thoracic tumour treatment in mice.
Hence, US irradiation combined with microbubbles would be a promising noninvasive treatment, especially for lesions invading the chest wall. Patients with peripheral lung cancer are the ones most likely to benefit from this synergistic treatment. With the help of endoscopic and endobronchial US irradiation, this treatment may have a therapeutic effect on mediastinal lesions. The therapeutic efficacy of deep lung tumour therapy should be further explored. To our knowledge, this study is the first report on treating lung tumours with nanotechnology-mediated immunochemotherapeutic microbubbles combined with US irradiation.
Experimental
Materials
The anti-PD-L1 mAb was purchased from BioXCell, Inc. (Lebanon, NH, USA). DTX was purchased from J & K Scientific Co., Ltd (Beijing, China). 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dipalmitoyl-sn-glycero-3-phosphate (DPPA), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000] (DSPE-PEG-COOH) were purchased from A.V.T. Pharmaceutical Co., Ltd (Shanghai, China). DSPE-PEG-FITC (molecular weight (MW): 2000) was purchased from Xi'an Ruixi Biological Technology Co., Ltd. Coumarin-6 (C6) was purchased from Yuanye Biotech, Inc. (Shanghai, China). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) was purchased from Aladdin Reagent Co., Ltd (Shanghai, China). N-Hydroxysuccinimide (NHS) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) were purchased from Adamas-beta Inc. (Shanghai, China).Cell counting kit-8 (CCK-8) assay and Annexin V-FITC/propidium iodide (PI) apoptosis detection, cell cycle detection, and VEGF, TNF-α, and TGF-β enzyme-linked immunosorbent assay (ELISA) kits were purchased from KeyGEN BioTECH Corp., Ltd (Nanjing, China). Western blotting antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Flow cytometry antibodies were purchased from eBioscience (Thermo Fisher Scientific, MA, USA).
Cell lines
LLC, A549, NCI-H460 [H460], and NCI-H1299 cells were kindly provided by Stem Cell Bank, Chinese Academy of Sciences. LLC cells were cultured in DMEM, A549 cells were cultured in F-12 K medium, and NCI-H460 [H460] and NCI-H1299 cells were cultured in RPMI-1640 medium. All the media were supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. The cells were incubated at 37 °C with 5% CO2.Synthesis of microbubbles
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DPPA:DSPE:DTX in a weight ratio of 5.0 mg:0.7 mg:0.5 mg:1.0 mg were dissolved in a solution of 6.0 mL of chloroform and 4.0 mL of methanol and then evenly ultrasonically mixed. The solvent was evaporated in the flask at 0.1 MPa and 42 °C to obtain a thin lipid film. When the residual chloroform evaporated at room temperature, the film was eluted with PBS. After the solution was concentrated to 1.5 mL, the dissolved gas in the solution was removed by ultrasonic clearing. Before starting the sonicator at 100 W and 4 °C for 10 min, we placed the probe approximately 3 mm below the liquid level and used SF6 to completely remove air. After separation by a dispersion and differential centrifugation method, relatively uniform DMs were obtained, and free DTX was washed away.
:DPPA:DSPE:DTX in a weight ratio of 5.0 mg:0.7 mg:0.5 mg:1.0 mg were dissolved in a solution of 6.0 mL of chloroform and 4.0 mL of methanol and then evenly ultrasonically mixed. The solvent was evaporated in the flask at 0.1 MPa and 42 °C to obtain a thin lipid film. When the residual chloroform evaporated at room temperature, the film was eluted with PBS. After the solution was concentrated to 1.5 mL, the dissolved gas in the solution was removed by ultrasonic clearing. Before starting the sonicator at 100 W and 4 °C for 10 min, we placed the probe approximately 3 mm below the liquid level and used SF6 to completely remove air. After separation by a dispersion and differential centrifugation method, relatively uniform DMs were obtained, and free DTX was washed away.
Characterization of the microbubbles
The particle sizes, size distributions, and zeta potentials of blank microbubbles (BMs), DMs, and PDMs were measured by a dynamic light scattering method using a ZetaPlus instrument (Brookhaven Instruments Corporation, NY, USA). The morphological characterization of the PDMs was performed with a scanning electron microscope (UltraPlus, Zeiss, Japan) after 200 μL of the sample was drip-dried on silicon slices at room temperature for 48 h.The drug loading efficiency and in vitro DTX release behaviours of microbubbles were detected by high-performance liquid chromatography (HPLC) with an Agilent 1200 HPLC system (Agilent, USA). We used acetonitrile/water (volume ratio of 1:1) as the mobile phase and Phenomenex C18 (4.6 mm × 250 mm, 5 μm) to separate the samples. The samples were dissolved in the mobile phase under US irradiation, and then the AUC was detected at 232 nm at 1 mL min−1 and 30 °C. According to our standard sample curve, we determined the concentration of DTX in each sample. The entrapment efficiency (EE%) and drug loading of DTX (DL%) were determined by HPLC and calculated using the following equations:
| EE% = Wencapsulated DTX/Wtotal feeding DTX × 100%; |
| DL% = Wencapsulated DTX/Wtotal amount of microbubbles × 100%. |
To investigate drug release behaviours, microbubbles and PBS were enclosed in dialysis bags (MWCO, 8000–14000 Da) with shaking in 2 L of PBS at 100 rpm and 37 °C with/without US irradiation (2.0 W cm−2, 1 MHz, duty cycle: 50% for 5 minutes). After 1 mL of dialysate water was removed from the container at predetermined intervals and stored at 4 °C for analysis, the same volume of PBS was used to replenish the sampled mixture. The release amount was measured by the same HPLC method described above. Three independent samples from each group were tested and analysed.
LSCM was used to show the conjugation of a CY5-labelled anti-PD-L1 mAb with FITC-loaded microbubbles. The anti-PD-L1 mAb was labelled with CY5 according to the operation manual of a Lightning-Link Rapid Cyanine Dye 5 Kit (Expedeon, Heidelberg, Germany). DMs-FITC were prepared according to a method similar to the one described above.
To investigate the haemolytic effect of microbubbles, 50 μL of fresh anticoagulant-treated rabbit blood was mixed with (−) normal saline, BMs (w/v, 1 mg mL−1), DMs (w/v, 1 mg mL−1), PDMs (w/v, 1 mg mL−1), or (+) pure water, and the samples were incubated at 37 °C for 60 minutes before centrifugation at 2000 rpm for 5 minutes. Then, the optical density (OD) of each supernatant solution was measured at 540 nm using a microplate reader (Thermo Scientific Multiskan FC, Thermo Fisher Scientific, MA, USA).
Toxicities of different microbubble formulations
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Southeast University and approved by the Animal Ethics Committee of Southeast University. C57BL/6 mice (6–8 weeks, n = 20) were randomly assigned to five groups for the toxicity test: the control group (normal saline), free DTX group, DM group, combination of free DTX and anti-PD-L1 mAb group, and PDM group. The drug was injected every three days for a total of five times as scheduled at a DTX dose of 2 μg g−1. During the experimental schedule, the body weight of each animal was measured every three days, while blood biochemical indexes were evaluated weekly. The serum biochemical parameters including aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), and creatinine levels were measured using a Hitachi 7020 automatic biochemical analyser.Evaluation of contrast-enhanced US imaging
Solutions of BMs, DMs, and PDMs were ascertained by the freeze-drying method. Next, 1.0 mL of microbubbles (w/v, 1.0 mg mL−1) or normal saline (as a control) was added to the holes in an agarose mould. One hundred microliters of BMs, DMs, or PDMs (w/v, 1 mg mL−1) was intravenously injected into subcutaneous tumour model mice (n = 3) to evaluate the in vivo contrast imaging capability. Contrast-enhanced US images were obtained with an ultrasonic machine (Voluson E8, GE, USA) and analysed using QontraXt V3.06 (AMID-qX3, Bracco, Italy).Influence of microbubbles on cell viability in vitro
The PD-L1 antigen expression of LLC, NCI-H460, NCI-H1299, and A549 cells was tested with an anti-CD274 (PD-L1, B7-H1) mAb (MIH5) labelled with PE (eBioscience, Thermo Fisher Scientific) using a flow cytometer (NovoCyte3000, ACEA Biosciences, Inc., CA, USA). All the analyses were performed using NovoExpress1.3.0 software.The cytotoxicities of different microbubbles were measured using the CCK-8 method according to the manufacturer's protocol (KeyGEN BioTECH). LLC, NCI-H460, NCI-H1299, and A549 cells were seeded in 96-well plates with the corresponding incubation medium at a density of 5000 cells per well and incubated overnight. Then, 100 μL of free DTX, BMs, DMs, PDMs, PDMs + US (US irradiation: 0.2 W cm−2, 1 MHz, duty cycle: 50% for 30 seconds after PDM addition) or complete medium (control group) was added into each well so that the final concentration of DTX was 2 μg mL−1. After the cells were incubated for 24 h at 37 °C under a 5% CO2 atmosphere, the supernatant was removed, and the cells were rinsed carefully with PBS twice, followed by the addition of the RPMI 1640 medium (100 μL) and CCK-8 (10 μL) for 1 h. The OD was measured at 450 nm using the microplate reader. The relative cell viability (RCV) (%) was calculated using the equation RCV (%) = ODtest/ODcontrol × 100%.
Evaluation of specific DTX cellular uptake mediated by PD-L1
C6 was added to microbubbles to analyse cellular drug uptake. LLC cells were seeded in six-well plates with the corresponding incubation medium at a density of 5 × 105 cells per well. After incubating overnight, the cells were washed twice with PBS and treated with free C6, C6-DMs, C6-PDMs, or C6-PDMs + US at 37 °C for 2 h. US irradiation was implemented as described above. The cells were washed with PBS three times and fixed with 4% cold paraformaldehyde for 15 min. The cells were washed with PBS three times, and then a goat anti-rabbit IgG (H+L) secondary antibody conjugated with CY5 (Thermo Fisher Scientific) was added and incubated at room temperature for 1 h. After the secondary antibody was removed by washing two times, DAPI was added to the wells and incubated for 10 min. The cells were visualized with a fluorescence microscope (IX71, Olympus Corp., Tokyo, Japan). Then, we detached the cells with a trypsin solution and quantified the uptake efficiency using the flow cytometer.Apoptosis assay and cell cycle analysis in vitro
Apoptosis induction was detected with the flow cytometer according to the manufacturer's protocol for the Annexin V-FITC/PI Reagent (KeyGEN BioTECH). LLC cells were seeded in six-well plates at a density of 1 × 105 per well. After incubating overnight, the cells were treated with complete medium (control), US, DTX, DMs, PDMs, or PDMs + US. Twenty-four hours later, the cells were detached with 100 μL of EDTA-free trypsin, stained and washed twice with PBS. Five hundred microliters of binding buffer was used to suspend the cells, 5 μL of Annexin V-FITC and 5 μL of PI were added and mixed, and the mixture was incubated for 15 min before analysis by flow cytometry.For cell cycle analysis, a PI/RNase Cell Cycle Kit (KeyGEN BioTECH) was used according to the manufacturer's protocol. LLC cells were seeded in six-well plates at a density of 1 × 105 per well and incubated overnight. Then, the cells were starved in complete medium (2% FBS) for 24 h before treatment with complete medium (control), US, DTX, DMs, PDMs, or PDMs + US. Twenty-four hours later, the cells were collected, fixed in 75% ethanol at 4 °C overnight, washed twice with cold PBS, and stained with PI/RNase working buffer at room temperature for 30 min. Then, the cells were detected by flow cytometry.
Subcutaneous and orthotopic tumour models established with LLC cells
C57BL/6 mice (6–8 weeks) without black spots on their skin were used to establish subcutaneous tumour models. Each mouse was subcutaneously injected with 100 μL of LLC cells (1 × 107 mL−1 in PBS) in the right flank. The size of the orthotopic tumour was measured every other day and calculated using the following formula: Volume = (Length × Width2)/2. Approximately 7 days later, when the tumour volume reached the predetermined size of 200 mm3, all the model mice were randomly divided into groups.Orthotopic tumour models were established by injecting 20 μL of cell suspension into the right lung under imaging with an X-ray machine (WeMed Medical Equipment Co., Ltd, Beijing, China). The cell concentration was 1 × 107 mL−1, and the cells were mixed with basement membrane matrices (Matrigel, BD Biosciences, NJ, USA). The injection device was constructed with a needle from a U-40 insulin injector (B. Braun, Melsungen AG, Germany) and an injector body from a 50 μL microsyringe (Gaoge Industrial and Trading Co., Ltd, Shanghai, China). The orthotopic tumour size was monitored with a Hiscan XM Micro CT platform (Suzhou Hiscan Information Technology Co., Ltd, Suzhou, China). Approximately 5 days later, when the tumour volume reached the predetermined size of 60 mm3, all the model mice were randomly divided into groups.
Assay of bio-distributions in mice treated with different microbubbles
Tumour-bearing mice were intravenously injected with free DiR, DiR-DMs, DiR-PDMs, or DiR-PDMs + US (n = 4 each group). Then, real-time images were acquired at different time points (1 h, 3 h, 6 h, 12 h, and 24 h) using the IVIS Spectrum system (Caliper Life Sciences, PerkinElmer Inc., MA, USA) after the mice were anaesthetized with isoflurane. Tumour tissue and the heart, lungs, liver, spleen, and kidneys were harvested for ex vivo imaging after the last imaging time point. The parameters for US irradiation were 2.0 W cm−2, 1 MHz, and a duty cycle 50% for five minutes, which were repeatedly used in the following in vivo experiments. The images were analysed using Living Image 4.1 software (Caliper LifeSciences, PerkinElmer Inc., MA, USA).In vivo antitumour study
To evaluate the antitumour efficacies of microbubbles, subcutaneous model mice were randomly divided into six groups (n = 6 each group) and intravenously injected with 100 μL of PBS, free DTX, free combination (DTX + anti-PD-L1 mAb), DMs, PDMs, or PDMs + US at a DTX dose of 2 μg g−1. Treatments were performed every 3 days. The body weight and tumour volume were measured every 3 days to observe antitumour efficacy and toxicity. All of the tumours, vital organs (the heart, liver, spleen, lungs, and kidneys), and blood were harvested. Tumour tissues were divided into three parts and processed for analyses: a real-time tissue homogenate for flow cytometry, a mass frozen at −80 °C for ELISA and western blot analyses, and a 4% (w/v) paraformaldehyde-fixed mass for sectioning were prepared.Tumour-infiltrating lymphocyte (TIL) suspensions were obtained by the mechanical dissociation and filtration method and stained with fluorochrome-conjugated mAbs specific for the cell surface markers CD4 (PE-CY5) and CD8a (PE), and an anti-rat IgG2a kappa antibody was used as an isotype control. The cells were analysed with the NovoCyte3000 flow cytometer using NovoExpress 1.3.0 software. In addition, immunofluorescence staining was used for double validation.
The antitumour efficacies of the microbubbles in the orthotopic tumour models were evaluated by CT scan, body weight, and survival rate statistics.
Statistical analysis
All statistical analyses were performed using SPSS 21.0 software and GraphPad Prism 8 software. Data are expressed as mean ± standard deviation unless otherwise noted. An unpaired two-tailed t-test was used to compare between two groups. When comparing multiple groups, one-way ANOVA with the Newman–Keuls post hoc test was performed. Kaplan–Meier survival curves were analysed using the log-rank test with the Tukey post hoc test. Differences were considered statistically significant when P < 0.05. Statistical significance was noted as follows: #P < 0.05 and ##P < 0.01 compared with the control group, and *P < 0.05 and **P < 0.01 compared between groups.Conclusion
Here, we developed anti-PD-L1 mAb-conjugated and DTX-loaded multifunctional lipid-shelled microbubbles and verified that the anti-PD-L1 mAb and US irradiation could promote DTX uptake. We validated that a checkpoint inhibitor could be integrated into a therapeutic microbubble to produce a combination with synergistic effects. We also proved that US irradiation-mediated immunochemotherapeutic microbubble therapy could be used to treat lung cancer. This immunochemotherapeutic microbubble approach illustrates a successful treatment strategy that can be extended to other combinations based on clinically approved antibodies (e.g., anti-CTLA-4, anti-41-BB, or anti-TIM-3 antibodies) or traditional chemotherapeutic drugs (e.g. doxorubicin, pemetrexed, or gemcitabine). The synergistic therapy of US combined with immunochemotherapeutic microbubbles might be a promising treatment for a variety of malignancies, potentially leading to the development of a new approach for combination immunotherapy.Conflicts of interest
The authors have declared that no competing interest exists.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 81671795, 81971628, and 81971716). We are grateful to Jiangsu Key Laboratory of Molecular and Functional Imaging for assistance with materials synthesis, materials characterization, and biological integrated experiments.References
- R. L. Siegel, K. D. Miller and A. Jemal, CA Cancer J. Clin., 2019, 69, 7–34 CrossRef PubMed.
- R. Alanni, J. Hou, H. Azzawi and Y. Xiang, IET Syst. Biol., 2019, 13, 129–135 CrossRef PubMed.
- J. J. Havel, D. Chowell and T. A. Chan, Nat. Rev. Cancer, 2019, 19, 133–150 CrossRef CAS PubMed.
- S. L. Topalian, J. M. Taube, R. A. Anders and D. M. Pardoll, Nat. Rev. Cancer, 2016, 16, 275–287 CrossRef CAS PubMed.
- A. H. Sharpe and K. E. Pauken, Nat. Rev. Immunol., 2018, 18, 153–167 CrossRef CAS PubMed.
- R. S. Herbst, P. Baas, D. W. Kim, E. Felip, J. L. Perez-Gracia, J. Y. Han, J. Molina, J. H. Kim, C. D. Arvis, M. J. Ahn, M. Majem, M. J. Fidler, G. de Castro Jr., M. Garrido, G. M. Lubiniecki, Y. Shentu, E. Im, M. Dolled-Filhart and E. B. Garon, Lancet, 2016, 387, 1540–1550 CrossRef CAS.
- National Comprehensive Cancer Network(NCCN) Guidelines with NCCN Evidence Blocks, Version 5. 2019, https://www.nccn.org/professionals/physician_gls/default.aspx, (accessed September 2019).
- M. Nagasaka and S. M. Gadgeel, Expert Rev. Anticancer Ther., 2018, 18, 63–70 CrossRef CAS PubMed.
- C. Schmidt, Nature, 2017, 552, S67–S69 CrossRef CAS PubMed.
- J. J. Moslehi, J. E. Salem, J. A. Sosman, B. Lebrun-Vignes and D. B. Johnson, Lancet, 2018, 391, 933 CrossRef.
- S. Baxi, A. Yang, R. L. Gennarelli, N. Khan, Z. Wang, L. Boyce and D. Korenstein, Br. Med. J., 2018, 360, k793 CrossRef PubMed.
- G. Quintero Aldana, M. Salgado, S. Candamio, J. C. Méndez, M. Jorge, M. Reboredo, L. Vázquez Tuñas, C. Romero, M. Covela, A. Fernández Montes, M. Carmona, Y. Vidal Insua and R. López, Clin. Transl. Oncol., 2019 DOI:10.1007/s12094-019-02151-6.
- S. Kanda, K. Goto, H. Shiraishi, E. Kubo, A. Tanaka, H. Utsumi, K. Sunami, S. Kitazono, H. Mizugaki, H. Horinouchi, Y. Fujiwara, H. Nokihara, N. Yamamoto, H. Hozumi and T. Tamura, Ann. Oncol., 2016, 27, 2242–2250 CrossRef CAS PubMed.
- N. Li, L. Han and H. Jing, Exp. Ther. Med., 2017, 14, 3768–3773 CrossRef CAS PubMed.
- S. R. Sirsi, C. Fung, S. Garg, M. Y. Tianning, P. A. Mountford and M. A. Borden, Theranostics, 2013, 3, 409–419 CrossRef CAS PubMed.
- R. Cavalli, M. Soster and M. Argenziano, Ther. Delivery, 2016, 7, 117–138 CrossRef CAS PubMed.
- M. Gutierrez, M. Tardella, L. Rodriguez, J. Mendoza, D. Clavijo-Cornejo, A. Garcia and C. Bertolazzi, Radiol. Med., 2019, 124, 989–999 CrossRef PubMed.
- M. G. Sugiyama, V. Mintsopoulos, H. Raheel, N. M. Goldenberg, J. E. Batt, L. Brochard, W. M. Kuebler, H. Leong-Poi, R. Karshafian and W. L. Lee, Am. J. Respir. Crit. Care Med., 2018, 198, 404–408 CrossRef PubMed.
- B. Lu, L. Sun, X. Yan, Z. Ai and J. Xu, Med. Oncol., 2015, 32, 345 CrossRef PubMed.
- S. Xenariou, U. Griesenbach, H. D. Liang, J. Zhu, R. Farley, L. Somerton, C. Singh, P. K. Jeffery, S. Ferrari, R. K. Scheule, S. H. Cheng, D. M. Geddes, M. Blomley and E. W. Alton, Gene Ther., 2007, 14, 768–774 CrossRef CAS PubMed.
- C. M. Schoellhammer, G. Y. Lauwers, J. A. Goettel, M. A. Oberli, C. Cleveland, J. Y. Park, D. Minahan, Y. Chen, D. G. Anderson, A. Jaklenec, S. B. Snapper, R. Langer and G. Traverso, Gastroenterology, 2017, 152, 1151–1160 CrossRef CAS PubMed.
- W. Sun, Z. Li, X. Zhou, G. Yang and L. Yuan, Drug Delivery, 2019, 26, 45–50 CrossRef CAS PubMed.
- W. Gong, Q. Song, X. Lu, W. Gong, J. Zhao, P. Min and X. Yi, J. Chemother., 2011, 23, 295–299 CrossRef CAS PubMed.
- S. C. Liang, Y. E. Latchman, J. E. Buhlmann, M. F. Tomczak, B. H. Horwitz, G. J. Freeman and A. H. Sharpe, Eur. J. Immunol., 2003, 33, 2706–2716 CrossRef CAS PubMed.
- C. S. Chiang, Y. J. Lin, R. Lee, Y. H. Lai, H. W. Cheng, C. H. Hsieh, W. C. Shyu and S. Y. Chen, Nat. Nanotechnol., 2018, 13, 746–754 CrossRef CAS PubMed.
- W. J. Wan, C. X. Qu, Y. J. Zhou, L. Zhang, M. T. Chen, Y. Liu, B. G. You, F. Li, D. D. Wang and X. N. Zhang, Int. J. Pharm., 2019, 566, 731–744 CrossRef CAS PubMed.
- R. Kuai, W. Yuan, S. Son, J. Nam, Y. Xu, Y. Fan, A. Schwendeman and J. J. Moon, Sci. Adv., 2018, 4, eaao1736 CrossRef PubMed.
- Z. S. Dunn, J. Mac and P. Wang, Biomaterials, 2019, 217, 119265 CrossRef CAS PubMed.
- E. C. Abenojar, P. Nittayacharn, A. C. de Leon, R. Perera, Y. Wang, I. Bederman and A. A. Exner, Langmuir, 2019, 35, 10192–10202 CrossRef CAS PubMed.
- S. Zhou, S. Zheng, Y. Shan, L. Li, X. Zhang and C. Wang, Oncol. Rep., 2016, 35, 801–808 CrossRef CAS PubMed.
- S. Xu, F. Cui, D. Huang, D. Zhang, A. Zhu, X. Sun, Y. Cao, S. Ding, Y. Wang, E. Gao and F. Zhang, Int. J. Nanomed., 2019, 14, 17–32 CrossRef CAS PubMed.
- Z. Gu, Q. Wang, Y. Shi, Y. Huang, J. Zhang, X. Zhang and G. Lin, J. Controlled Release, 2018, 286, 369–380 CrossRef CAS PubMed.
- S. Bulner, A. Prodeus, J. Gariepy, K. Hynynen and D. E. Goertz, Ultrasound Med. Biol., 2019, 45, 500–512 CrossRef PubMed.
- S. Zheng, D. Song, X. Jin, H. Zhang, M. Aldarouish, Y. Chen and C. Wang, Nanomedicine, 2018, 13, 297–311 CrossRef CAS PubMed.
- W. Sun, Y. Du, X. Liang, C. Yu, J. Fang, W. Lu, X. Guo, J. Tian, Y. Jin and J. Zheng, Biomaterials, 2019, 217, 119264 CrossRef CAS PubMed.
- D. E. Goertz, M. Todorova, O. Mortazavi, V. Agache, B. Chen, R. Karshafian and K. Hynynen, PLoS One, 2012, 7, e52307 CrossRef CAS PubMed.
- H. Yu, Z. Lin, L. Xu, D. Liu and Y. Shen, Ultrasonics, 2015, 61, 136–144 CrossRef CAS PubMed.
- B. L. Helfield, X. Chen, B. Qin, S. C. Watkins and F. S. Villanueva, Ultrasound Med. Biol., 2017, 43, 2678–2689 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental results and methods are depicted in the ESI Table S1: Characteristics of different microbubbles. Table S2: Encapsulation efficiency and drug-loading efficiency of microbubbles. Fig. S1: Haemolysis tests of BMs, DMs, and PDMs. Fig. S2: Mouse body weight (A) as well as AST (B), ALT (C), creatinine (D) and blood urea nitrogen (E) levels were monitored over the course of treatment. Fig. S3: HE assessment of the heart, liver, lungs, spleen, and kidneys. Fig. S4: Flow cytometry assay of C6 uptake by LLC cells treated with different formulations. ESI Fig. S5: Flow cytometry analysis of CD4+ and CD8+ TIL numbers after treatments. Fig. S6: LLC cells with Matrigel were injected into the right lung of C57BL/6 mice under X-ray guidance to establish an orthotopic tumour model. See DOI: 10.1039/c9bm01575b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |