
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust alginate/hyaluronic acid thiol–yne click-hydrogel scaffolds with superior mechanical performance and stability for load-bearing soft tissue engineering†
Maria M.
Pérez-Madrigal
 *a,
Joshua E.
Shaw
b,
Maria C.
Arno
a,
Judith A.
Hoyland
bc,
Stephen M.
Richardson
b and
Andrew P.
Dove
*a
*a,
Joshua E.
Shaw
b,
Maria C.
Arno
a,
Judith A.
Hoyland
bc,
Stephen M.
Richardson
b and
Andrew P.
Dove
*a
aSchool of Chemistry, University of Birmingham Edgbaston, Birmingham, B15 2TT, UK. E-mail: a.dove@bham.ac.uk
bDivision of Cell Matrix Biology and Regenerative Medicine, School of Biological Sciences, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, University of Manchester, Manchester M13 9PT, UK
cNIHR Manchester Biomedical Research Centre, Central Manchester Foundation Trust, Manchester Academic Health Science Centre, Manchester, UK
First published on 6th November 2019
Abstract
Hydrogels based on hyaluronic acid (HA) exhibit great potential as tissue engineering (TE) scaffolds as a consequence of their unique biological features. Herein, we examine how the advantages of two natural polymers (i.e. HA and alginate) are combined with the efficiency and rapid nature of the thiol–yne click chemistry reaction to obtain biocompatible matrices with tailored properties. Our injectable click-hydrogels revealed excellent mechanical performance, long-term stability, high cytocompatibility and adequate stiffness for the targeted application. This simple approach yielded HA hydrogels with characteristics that make them suitable for applications as 3D scaffolds to support and promote soft tissue regeneration.
Hyaluronic acid (HA), an anionic, hydrophilic, nonsulfated glycosaminoglycan, is a key component of articular cartilage which is responsible for bearing compressive loads and providing lubrication, as well as playing a major role in biological signalling interactions.1 Among other processes, HA promotes cell migration and proliferation and facilitates the remodelling of the extracellular matrix (ECM) when interacting with the receptor CD44.2,3 Moreover, this polysaccharide has been reported as anti-inflammatory and chondroprotective (HA attenuates joint damage in arthritis),4 while recent evidence demonstrates its role in many processes involved in stem cell biology.5,6 In addition to its unique biological functions, HA is a natural, accessible polysaccharide, which diminishes any safety and commercially related concerns. Indeed, the US Food and Drug Administration (FDA) approved to market HA intra-articular products intended for treatment of knee osteoarthritis as medical devices. HA also degrades efficiently through enzymatic routes in the human body via hyaluronidase (HAse), which allows for its controlled clearance.7 Together, all these features make HA attractive as a substrate for customized biomedical devices,8 such as patch-type transdermal platforms for vaccination9 or chronic wound healing,10 3D hydrogel scaffolds or drug delivery systems,11–13 as well as hydrogel models with spatiotemporal control of both stiffness and viscoelastic cell-instructive cues.14
Polysaccharides in general satisfy several key properties as 3D cell scaffolds (i.e. affordability, structural support, and promotion of cell attachment, proliferation, and differentiation).15 Among those, alginate spontaneously forms gels in the presence of divalent cations through electrostatic interactions.16 In contrast, HA often requires chemical modifications to introduce functional groups that are then able to cross-link the HA-based network (e.g. methacrylates,17 thiols,18 enzymes,19,20 amino acids (lysine21 or tyramine22), gallol conjugates,23 and Schiff's bases,24,25 among others26,27). In addition, supramolecular assembly of guest–host pairs produces physically-crosslinked HA hydrogels.28–30 For instance, a recent work reported the preparation of nano-micelle crosslinked methacrylated HA hydrogels with low swelling for potential cartilage repair.31 Similarly, the bioclick reaction toolbox32 (i.e. reactions that are quick, highly selective, versatile, high yielding, and take place in the presence of cells and/or biomacromolecules) has been explored to prepare HA-based hydrogels with the aim of applying one-step, aqueous-based, no-catalyst required cross-linking methods.33–36 However, despite the benefits obtained by these “click” synthetic routes, a series of unmet requirements needs to be addressed for load-bearing soft tissue regeneration, namely: (i) the mechanical performance under compressive loading requires at least compressive strength in the order of hundreds of kPa and a stiffness of ca. 24 kPa;37 (ii) adequate swelling and degradation to enable long-term application; and (iii) their administration through minimally-invasive procedures (by injection). Indeed, injectable hydrogels represent attractive options for cartilage and bone tissue engineering (TE) approaches.38
Recently, we have reported the application of the nucleophilic thiol–yne addition reaction39 to prepare poly(ethylene glycol) (PEG) hydrogels that display outstanding mechanical performance,40,41 non-swellable properties42 and the versatility to incorporate alginate-based networks, which renders them self-healing and stretchable,43 under physiological conditions (i.e. pH 7.4 at 37 °C). However, although this metal-free biorthogonal click reaction is highly suitable for hydrogel synthesis because of its efficiency and rapid nature,39 we have only applied it to obtain PEG-based hydrogels, which intrinsically lack cell adhesion capability and display low serum protein absorption. As reviewed by Means et al.,44 hydrogels that simultaneously mimic the hydration, strength, and stiffness of soft and load-bearing tissues have the potential to be used in a much broader range of biomedical applications. Therefore, herein, we envisaged that the thiol–yne click-reaction provided a straightforward and suitable approach, with few synthetic steps, to prepare robust click-hydrogels based on polysaccharides. Indeed, only when these three elements (i.e. HA, alginate, and thiol–yne click chemistry) are exploited simultaneously, does this powerful mechanism yield hydrogels with superior mechanical performance while fulfilling the requirements needed for our specific biological application, i.e. cartilage regeneration.
To investigate our strategy, polymer precursors were prepared bearing thiol and alkyne functionalities. Firstly, HA was modified with thiol moieties (HA-SH) following, in part, a literature procedure.19,20 As a result, HA-SH (Fig. 1A) was obtained in adequate yield (82% ± 8.7%) with a high degree of modification (39.7% ± 1.8% of the disaccharide units were functionalized with the thiol group, as determined by 1H NMR spectroscopy, Fig. S1, ESI†). All ten repetitions of this protocol displayed similar values, which highlights the robustness of this method and its reproducibility. Moreover, our modification procedure ensures that the resulting compound will undergo efficient chemical cross-linking (gelation) to produce click-hydrogels without compromising the inherent cell signaling capability of HA, which involves complex multivalent binding events between a minimum sequence of three HA disaccharide units and the cell receptor CD44.45,46 Secondly, alkyne-functionalized PEG precursors with different architecture (i.e. 2-arm, 3-arm, or 4-arm) were prepared as cross-linking units for the click-hydrogel network. The preparation of these compounds, which relies on highly efficient Fischer esterification, yielded materials with conversion values higher than 91% (Fig. S2–S5, Table S1†).
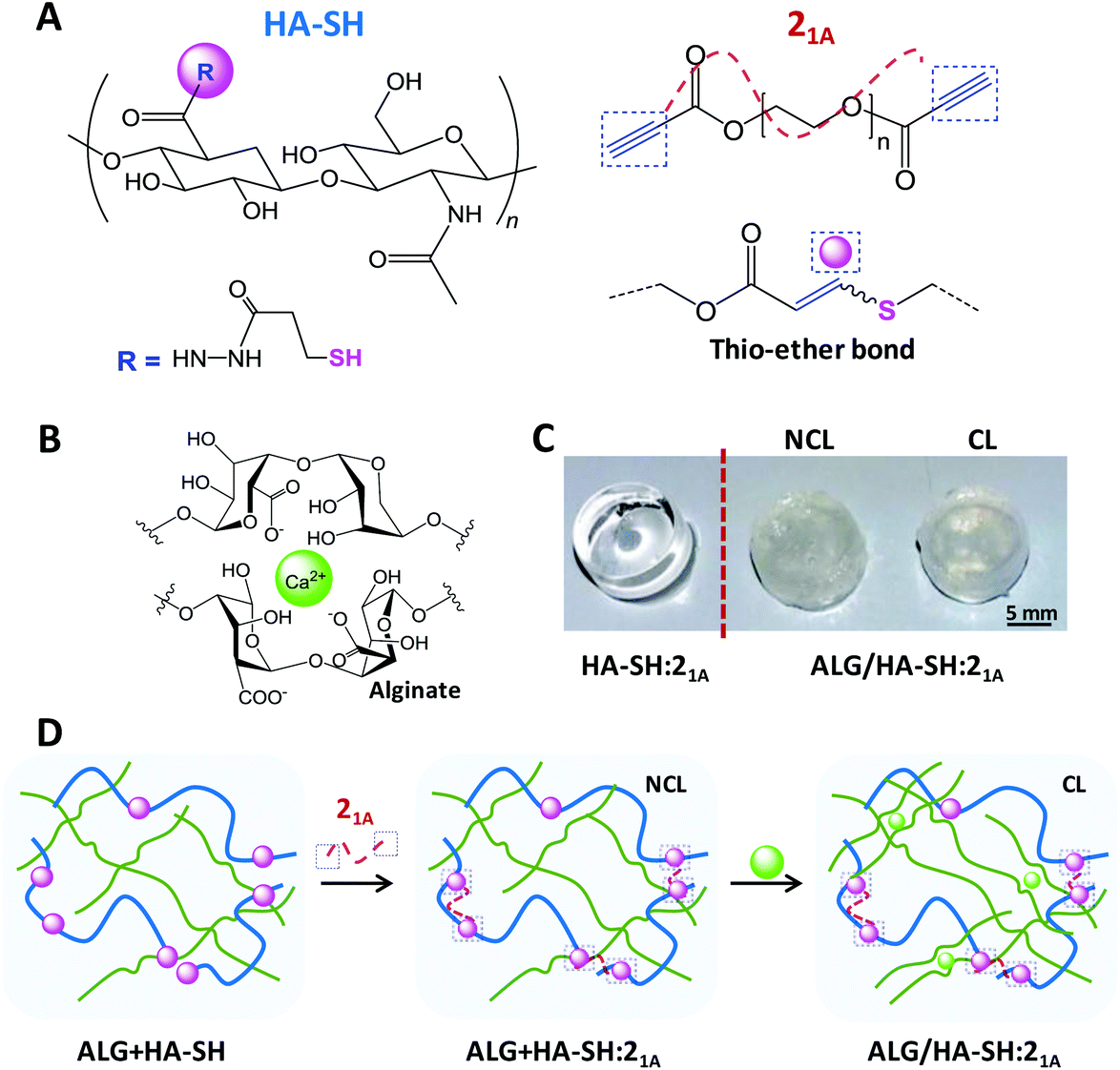 | ||
Fig. 1 Preparation of ALG/HA-SH:21A click-hydrogels. Schematics illustrating the composition and cross-linking of the dense HA-SH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :yne network (A) and the alginate-based network (B). (C) Photographs of as prepared click-hydrogels: HA-SH:21A (no alginate in the composition) and ALG/HA-SH:21A (NCL = not cross-linked with Ca2+; CL = cross-linked with Ca2+). (D) Schematics illustrating the preparation steps of ALG/HA-SH:21A click-hydrogels. :yne network (A) and the alginate-based network (B). (C) Photographs of as prepared click-hydrogels: HA-SH:21A (no alginate in the composition) and ALG/HA-SH:21A (NCL = not cross-linked with Ca2+; CL = cross-linked with Ca2+). (D) Schematics illustrating the preparation steps of ALG/HA-SH:21A click-hydrogels. | ||
We carefully designed our HA-SH:alkyne click-hydrogels to meet the general technical requirements of cell-laden scaffolds for a 3D culture configuration, as recently established by Diekjürgen and Grainger.15 Accordingly, several parameters, which included the alkyne-terminated PEG cross-linker (i.e. 21A, 31A, or 42A), the polymer content (i.e. 1.5, 2.5, 5, or 10 wt%) and the weight ratio between HA-SH and the alkyne-terminated PEG (i.e. 50:50 or 75:25), were optimized to ensure (i) an enhanced residence time of the hydrogel network (natural HA has a short half-life of only a few days in subcutaneous implant47) and (ii) the homogeneous distribution of encapsulated cells within the hydrogels. A detailed description of the optimization process, as well as additional data, are provided in the ESI (section 2.2 – Table S2 and S3, Scheme S1, and Fig. S6 and S7†).
Optically transparent HA-SH:alkyne hydrogels were prepared with a solid content of 2.5 wt% and a HA-SH:alkyne weight ratio of 75:25 using 21A as the alkyne cross-linker in phosphate-buffered saline (PBS) (pH 7.4) at room temperature (23 °C) – these hydrogels are denoted hereafter as HA-SH:21A (Fig. 1). As determined by the vial tilt method, HA-SH:21A hydrogels gelled within 2 minutes (118 ± 5 seconds) – this result was further confirmed by rheological characterization (Fig. S10†). Furthermore, in order to enhance the mechanical performance of the resulting click-hydrogels, we sought to form an interpenetrating polymer network (IPN) by the introduction of a second more flexible network based on alginate physically cross-linked with Ca2+ (Fig. 1B). Alginate was chosen as it can regulate the differentiation of mesenchymal stem cells (MSCs), in particular chondrogenesis.16 Hence, through another optimization process (refer to ESI for further details, section 2.2 – Fig. S8 and S9†), it was decided that alginate (2.8 wt%) would be mixed with the HA-SH before the gelation process, which takes place by adding the alkyne PEG precursor to HA-SH (Fig. 1D) – these hydrogels are denoted hereafter as ALG + HA-SH:21A. When the alginate-based network is included into the system (2.8 wt%), the hydrogel turned opaque (Fig. 1C) and the gelation time of the hydrogel slightly increased to ca. 5 minutes, possibly as a consequence of the alginate chains interfering with the rapid covalent cross-linking of the thiol–yne network (Fig. S11A†).
We next verified the efficiency of the Ca2+ ionic cross-linking of the loose network by rheological characterization (Fig. S11B and C†) and determined to immerse the ALG + HA-SH:21A hydrogels for 10 minutes in the cross-linking solution (i.e. CaCl2 150 mM). The resulting hydrogels with both polymeric networks cross-linked are denoted hereafter as ALG/HA-SH:21A (Fig. 1C and D). Overall, this configuration allows for an injectable system in which the thiol–yne network would be initially cross-linked in situ inside a knee defect, while the alginate-based network would be cross-linked in a second step by injecting the Ca2+ solution, making sure to cover the hydrogel, and thus avoiding the inconveniences of highly invasive implantation techniques.
ALG/HA-SH:21A click-hydrogels displayed a gel faction (GF) of 78 ± 0.9%, which evidences the high efficiency of the cross-linking process. Moreover, their equilibrium water content (EWC, 96 ± 0.40%), an important parameter for the design of tissue engineering scaffolds, reveals a porous network able to retain large amounts of water, as well as favour the necessary diffusion of oxygen, nutrients, and other relevant biomolecules for encapsulated cells to grow and proliferate.
The mechanical performance of the click-hydrogels was assessed by uniaxial compression testing and improved once the non-covalently cross-linked network was introduced into the composition (Fig. S9†). Specifically, the maximum compressive strength increased from 17.5 kPa ± 1.33 kPa up to 1.4 MPa ± 0.55 MPa in the presence of the alginate-based network in the IPN, the strain at break now being ca. 97% (Fig. 2A). Not only did ALG/HA-SH:21A hydrogels sustain higher compressive loads before breaking, they also displayed compressive Young's moduli almost four times higher (67.5 kPa ± 13.9 kPa) than HA-SH:21A (8.8 kPa ± 4.0 kPa). We relate the generic enhancement of the mechanical properties to the synergistic effect between the individual networks. HA-SH:21A materials were extremely brittle as a consequence of the highly cross-linked nature of the dense thiol–yne network which was alone unable to withstand large deformation, typical of covalent mechanisms. These materials in fact started to break at lower strain values, in the range between 50 to 65% (Fig. S9†). In contrast, ALG/HA-SH:21A hydrogels contain both covalent and non-covalent interactions that cooperatively were able to sustain the load, thus protecting the system. Furthermore, ALG/HA-SH:21A hydrogels still retained their excellent mechanical behavior after being immersed in cell culture media (1.8 mM Ca2+) for 21 days provided the swelling factor (SF) is constant, although both the compressive strength (0.66 ± 0.55 MPa) (Fig. 2B) and the Young's modulus (37.1 ± 12.9 kPa) appeared to decrease slightly during this time (Fig. 2C). Most importantly, under physiological conditions, our ALG/HA-SH:21A hydrogels meet the requirements as soft scaffolds for load-bearing soft tissue regeneration since they match the biological stiffness of cartilage (E ca. 24 kPa)37 and are close to the stress that natural cartilage would experience in the human body (i.e. from 5 to 10 MPa).48
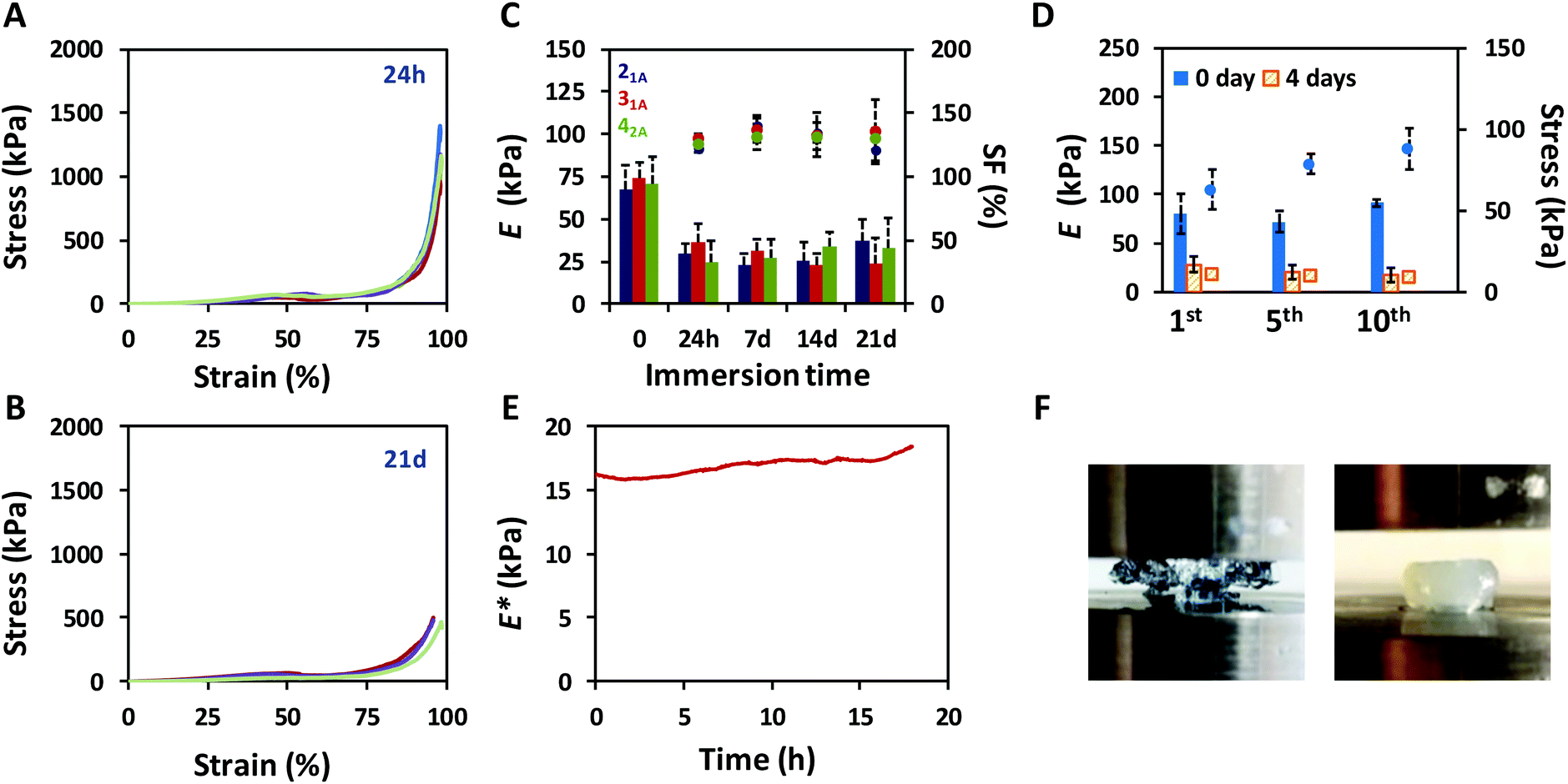 | ||
| Fig. 2 Mechanical properties of ALG/HA-SH:yne click-hydrogels. Representative compressive stress–strain curves recorded for ALG/HA-SH:21A after being immersed in cell culture media at 37 °C for (A) 24 hours (n = 9) and (B) 21 days (n = 7), thus highlighting the reproducibility of the manufacturing process and hydrogel performance. (C) Evolution of the Young's moduli (bars, left axis) displayed by ALG/HA-SH:yne click-hydrogels (n = 10) with immersion time (in cell culture media at 37 °C) and the swelling factor (SF, symbols right axis). (D) Evolution of the Young's moduli (bars, left axis) and compressive stress at 30% strain (symbols, right axis) with the cycle number for ALG/HA-SH:21A click-hydrogels (n = 10) as prepared (0 day) and after being immersed in cell culture media at 37 °C for four days. (E) Evolution of the complex modulus (E*) with time for ALG/HA-SH:21A click-hydrogels immersed in cell culture media at 37 °C. (F) Images of HA-SH:21A (left) and ALG/HA-SH:21A (right) click-hydrogels after compression to 70% of the initial height. As can be observed that HA-SH:21A system is brittle and not able to withstand such compression, being completely destroyed between the compression circular plates, as opposed to ALG/HA-SH:21A, which still retains its mechanical stability. | ||
Cartilage tissue in the knee experiences continuous load-release compression cycles. Hence, to simulate such conditions,49 10 consecutive cyclic compressions to 30% strain were performed to assess the ability of ALG/HA-SH:21A hydrogels to dissipate energy, which is confirmed by the hysteresis between the loading and unloading curves (Fig. S12†). The maximum compressive stress achieved was shown to slightly increase after each cycle (from 63.0 kPa ± 12.4 kPa to 88.1 kPa ± 12.5 kPa for the 1st and 10th, respectively), whereas the Young's moduli did not change significantly (from 80.8 kPa ± 15.5 kPa to 79.1 kPa ± 14.2 kPa for the 1st and 10th, respectively) (Fig. 2D). We believe that small amounts of water are expelled from the hydrogel after each cycle, which increases the polymer content of the system, thus affecting its mechanical performance. This response was also observed with samples that had been immersed in cell culture media for 4 days (Fig. 2D) and did not depend on the swelling of the hydrogel.
At this point, and to determine the mechanical properties of ALG/HA-SH:21A hydrogels in situ when immersed in cell culture media, we carried out dynamic mechanical analysis (DMA) experiments. Specifically, uniaxial compression was performed in stress strain mode with a displacement setting of 10% for 18 h at a frequency of 1 Hz at 37 °C on hydrogels (n = 3) that had equilibrated in cell culture media for four days at 37 °C. The complex modulus (E*), which sums both the storage (E′, elastic response of a material) and the loss (E′′, viscous response of a material) modulus of the mechanical response, remained stable within the time window tested (Fig. 2E), which indicates that the click-hydrogel efficiently retained its initial performance after a continuous load-release compression event. Indeed, E* increased only by 15% after such amount of time (18 h).
Additionally, not only did the alginate-based IPN improve the mechanical performance of the HA-based click-hydrogel, but it also modified the viscoelastic properties, as determined by rheological characterization (Fig. 3). The storage modulus (G′), which accounts for the material's ability to store energy elastically under shear, was monitored by running frequency sweeps (Fig. S13†), and it was found to be 3.0 ± 0.26 kPa and 70 ± 6.9 kPa for HA-SH:21A and ALG/HA-SH:21A, respectively, which indicates that ALG/HA-SH:21A is a much stiffer hydrogel scaffold (Fig. 3A). In line with this, cryo-SEM images also evidenced the stiffer nature of the ALG/HA-SH:21A hydrogels by the smaller size (2.3 ± 0.9 μm in contrast to 27.0 ± 11.0 μm) of the deformations created by the ice crystals when there was a higher polymer content and the presence of a dual polymeric network that resulted from the hydrogel network being stiffer (Fig. 4 and Fig. S14†). Moreover, G′ decreased down to 30 ± 3.4 kPa for ALG/HA-SH:21A after being immersed in cell culture media for four days (Fig. 3A). When submitted to amplitude sweep tests, the high G′ values obtained for ALG/HA-SH:21A hydrogels remained stable during almost the whole amplitude range, only yielding at strain values higher than 4% (Fig. 3B). In comparison to gelatin-based hydrogels cross-linked with the thiol–yne click reaction,50 ALG/HA-SH:21A hydrogels display higher stiffness values and longer stability in aqueous media. Under physiological conditions, our system closely simulates the biomechanics of the cartilage microenvironment, which is of vital importance considering that hydrogel stiffness influences cell viability after encapsulation, as well as increases ECM production.51
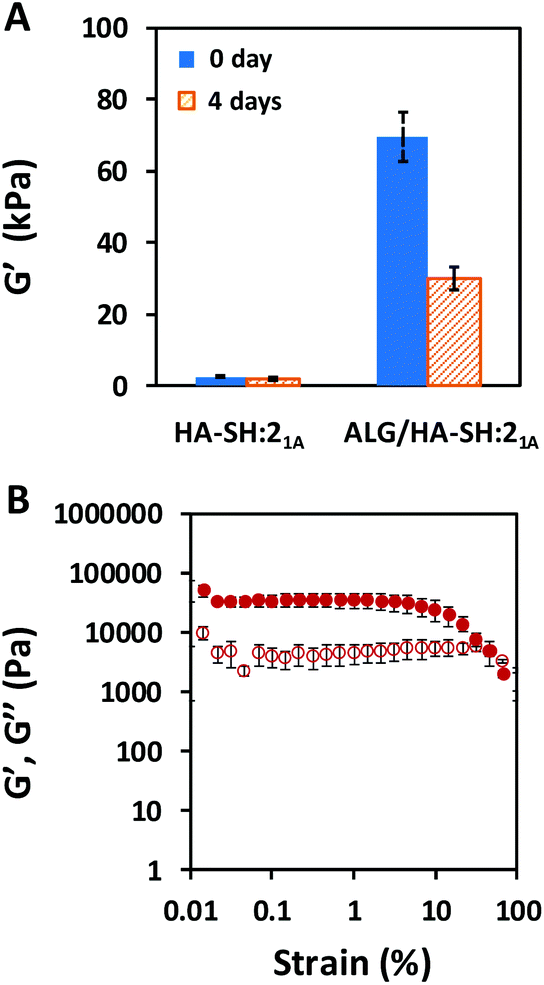 | ||
| Fig. 3 Rheological characterization of ALG/HA-SH:21A click-hydrogels. (A) Storage moduli (G′) mean values determined at 1.27 rad s−1 and 0.5% strain (frequency sweeps) as prepared (0 day) and after being immersed in cell culture media at 37 °C for 4 days. (B) Representative data for ALG/HA-SH:21A recorded under amplitude sweep (at 10 rad s−1) after being immersed in cell culture media at 37 °C for 4 days. Error bars: SD with n = 3. | ||
 | ||
| Fig. 4 Cryo-SEM images taken for HA-SH:21A (left) and ALG/HA-SH:21A (right) click-hydrogels. | ||
Hydrogels generally tend to swell when immersed in aqueous solutions. As the volume increases, their mechanical performance changes, which compromises their application in a biological context. To assess the swelling response of our click-hydrogels, samples were immersed in different aqueous solutions at 37 °C under mild shaking. In PBS, ALG/HA-SH:21A hydrogels swelled up sharply to 198% ± 5.3% after 7 days, before beginning to lose weight, which resulted in an unstable material that was difficult to handle (Fig. S7B†). We ascribed such response to the ion exchange between Ca2+ and the Na+ ions present in the PBS buffer, which ultimately weakens the ion cross-linked network. However, as long as calcium ions are present in the solution, this negative effect is prevented (Fig. S15†). It is important to note that alginate-only hydrogels swelled considerably (ca. 274% ± 11.8%) and were completely dissolved after 11 days of immersion (Fig. S15†) in PBS + 0.1 M Ca2+. In those same conditions, ALG/HA-SH:21A hydrogels retained their stability. Hence, in our ALG/HA-SH:21A hydrogels, the thiol–yne network essentially provides a potential biological function to the system and is required to slow down the degradation of the hydrogel and render a scaffold stable for almost 7 weeks in cell culture media (1.8 mM Ca2+) (Fig. 5A).
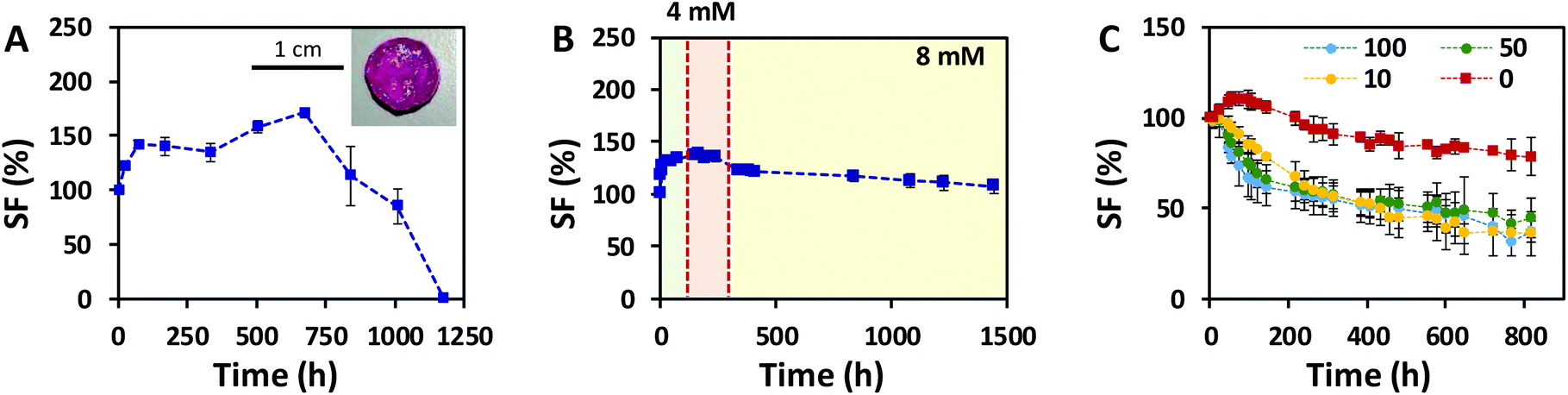 | ||
| Fig. 5 Long-term stability of ALG/HA-SH:21A click-hydrogels at 37 °C in different environments. Swelling factor (SF) values recorded for click-hydrogels immersed in (A) cell culture media with 1.8 mM Ca2+ (inset shows photo of ALG/HA-SH:21A after 14 days of immersion); (B) Ringer's solution with varying concentrations of Ca2+ (2.7 mM, 4 mM, and 8 mM); and (C) Ringer's solution with 8 mM of Ca2+ at various concentrations of hyaluronidase (100 U mL−1, 50 U mL−1, or 10 U mL−1, and 0 U mL−1). Error bars: SD with n = 4. | ||
We next explored the swelling response and long-term stability of ALG/HA-SH:21A hydrogels mimicking as close as possible the Ca2+ concentration of an in vivo situation, where a Ca2+ concentration higher than 10 mM for long periods is considered cytotoxic.52 ALG/HA-SH:21A hydrogels were immersed in Ringer's solution (i.e. 7.2 g L−1 NaCl and 0.37 g L−1 KCl) with an initial concentration of 0.3 g L−1 CaCl2, i.e. 2.7 mM Ca2+. The clinical use of Ringer's solution involves therapeutic procedures, such as arthroscopic lavage in the case of septic arthritis. At the 7th and 14th day of immersion, the Ca2+ concentration was increased up to 0.44 g L−1 CaCl2 (i.e. 4 mM Ca2+) and 0.88 g L−1 CaCl2 (i.e. 8 mM Ca2+), respectively. After 60 days, the SF reached a value of 107.1% ± 6.8%, which allows for an “expansion fit” of the hydrogel to fill the void space in the joint, with the hydrogels retaining their mechanical integrity (Fig. 5B). The balance between the hydrophilic alginate and the hydrophobic thiol–yne cross-linking sites present in the ALG/HA-SH:21A network, as well as the Ca2+ concentration, adequately slowed down the hydrolytic degradation of the system.
Finally, we assessed the swelling response of ALG/HA-SH:21A hydrogels mimicking a situation with enzymatic degradation. HAse was added to a Ringer's solution (8 mM Ca2+) at varying concentrations (i.e. 100 U mL−1, 50 U mL−1, or 10 U mL−1). After 34 days at 37 °C, the mass loss for ALG/HA-SH:21A hydrogels was determined to be 21.4 ± 10.2%, 64.1 ± 11.9%, 55.2 ± 10.8%, and 62.9 ± 5.5% for solutions containing 0, 10, 50, and 100 U mL−1, respectively (Fig. 5C). Within the first 5 days of immersion, the degradation rate varied with the concentration of HAse and it was faster for the more concentrated solution (100 U mL−1). After 8 days in solution, the degradation of the hydrogels followed a similar general trend regardless of the concentration of HAse employed, and, even though only ca. 40% of the weight remained, the mechanical integrity of the click-hydrogels was stable enough to allow for their handling. This behaviour is in good agreement with the fact that the activation center of HA enzymatic biodegradation is found in the carboxylic acid groups in the β-glucoronic acid units,53 which were being blocked as a consequence of the functionalization of HA with the thiol-containing group via EDC coupling. Such biodegradation rate is anticipated to allow the click-hydrogels to degrade while new tissue is forming. Overall, these results represent a major improvement in comparison to previous works33,34,54–56 where HA-based hydrogels subjected to HAse degradation were only stable for shorter periods, between 24 h54 and 14 days.55
Once satisfied with the mechanical performance of our click-hydrogels, we assessed their potential for biomedical applications by evaluating the cytotoxicity of the degradation products in the first instance. To that end, ALG/HA-SH:yne hydrogels were immersed in 5 mL of cell culture media for 35 days at 37 °C under mild shaking. After that time, MC3T3 cells (osteoblast precursor cell line derived from mouse calvaria) were then cultured for 72 h with the incubated cell media, and their metabolic activity was tested with PrestoBlue® (Fig. 6A). The degradation products caused (i.e. condition 1) ca. 20–30% cell death, without any dilution which decreased with the dilution factor regardless of the composition (i.e. the alkyne precursor used). Overall, the degradation products from all three systems displayed some level of cytotoxicity, albeit only minimal; specifically, ALG/HA-SH:21A hydrogels exhibited the highest cell viability, thus further confirming the suitability of this system as a biocompatible cell matrix.
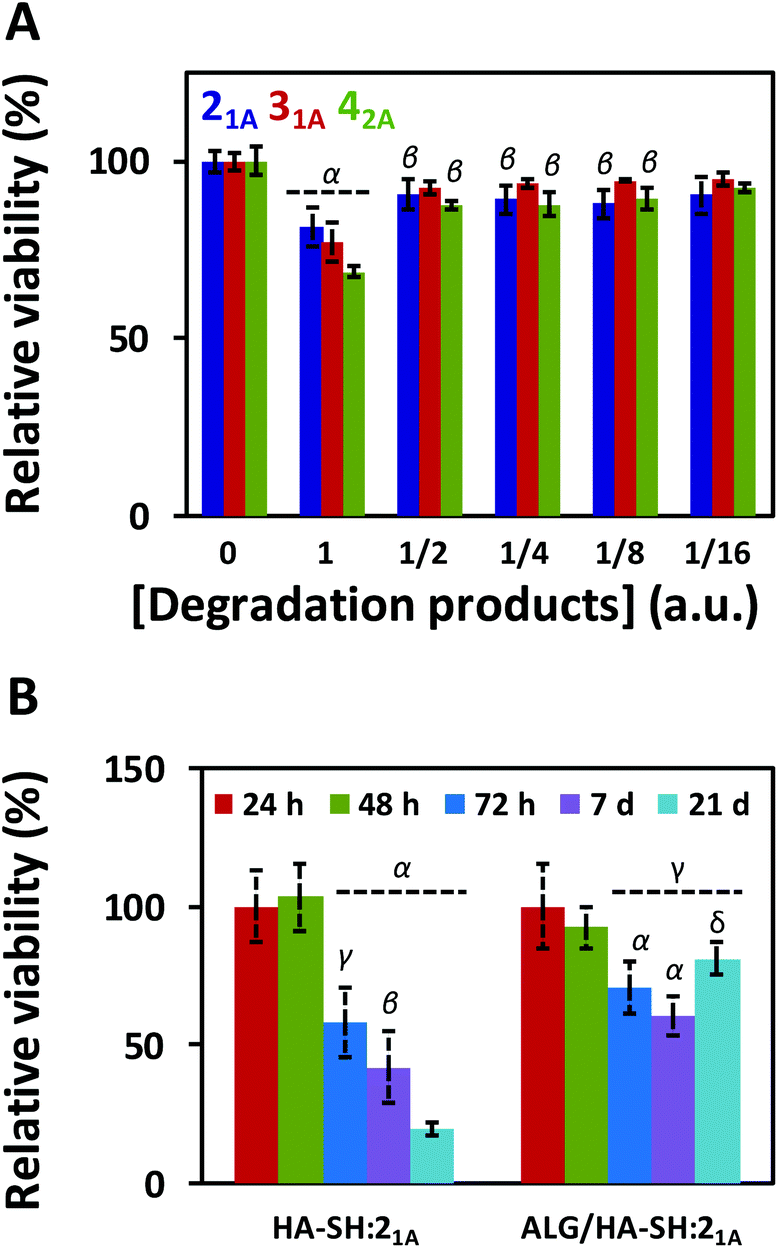 | ||
| Fig. 6 Cytocompatibility of ALG/HA-SH:21A click-hydrogels: (A) Cytotoxicity of the degradation products released from ALG/HA-SH:yne immersed in 5 mL of cell culture media at 37 °C for 35 days (viability in % relative to control). Greek letters on the bars refer to significant differences (p-value < 0.05): α vs. all; and β vs. [ ] = 0 (control). (B) Metabolic activity of cells encapsulated in HA-SH:21A and ALG/HA-SH:21A after different incubation times (24, 48, and 72 h, and 7 and 21 days; viability in % relative to 24 h). Error bars: SD with n = 3. Greek letters on the bars refer to significant differences (p-value < 0.05): α vs. 24 h and 48 h; β vs. ALG/HA-SH:21A at 72 h; γ vs. HA-SH:21A at 21 d; and δ vs. HA-SH:21A at 7 d. | ||
To evaluate the suitability of our click-hydrogels as 3D scaffolds, human MSCs (Y201 hTERT-immortalized human clonal MSCs57) were encapsulated within the ALG/HA-SH:21A hydrogel networks before gelation. Cell viability was assessed with AlamarBlue® metabolic assay (Fig. 6B) and live-dead fluorescent staining (Fig. 7) at different time points. After 21 days of incubation, ALG/HA-SH:21A click-hydrogels exhibited cell viability higher than 81% ± 6.0%, a value that is not significantly different from time 48 h (92% ± 7.3%). Hence, although cell viability does decline slightly at time points 72 h and 7 days, cell growth is being sustained for the whole incubation period (Fig. 6B). This trend is consistent with numerous 3D culture experiments reported in the literature, including studies with alginate gels, where encapsulated cells do not show evidence of significant proliferation but cell viability is maintained with time, thus indicating good cytocompatibility of the 3D scaffolds.22,58,59
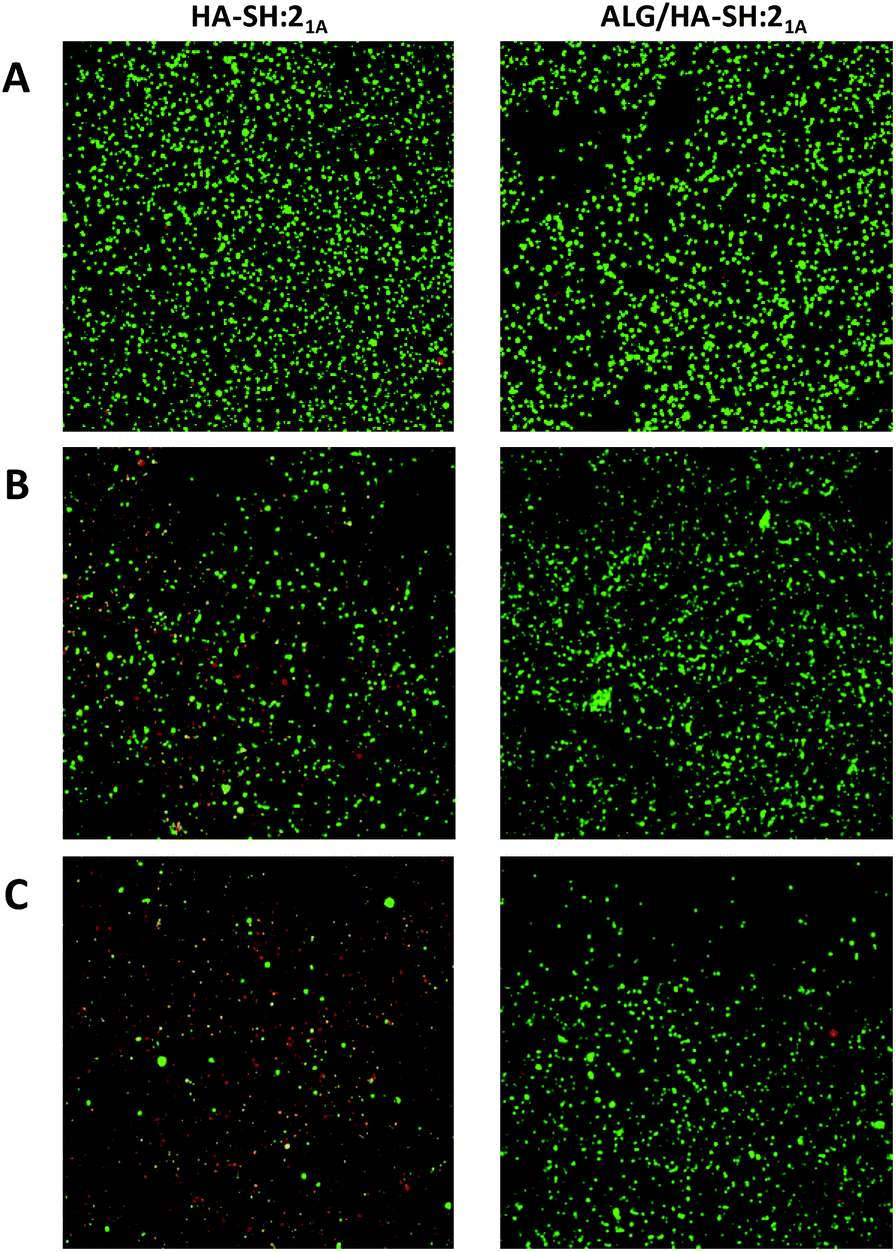 | ||
| Fig. 7 Cytocompatibilty of HA-SH:21A (left column) and ALG/HA-SH:21A (right column) click-hydrogels. Representative live/dead images of 3D encapsulated cells after (A) 24 hours, (B) 7 days, and (C) 21 days of incubation. | ||
In contrast, HA-SH:21A click-hydrogels showed high cytotoxicity with time and, after 21 days, ca. 80% ± 4.2% of the cells were dead (Fig. 6B). In our case, we mainly ascribe the high cytocompatibility of ALG/HA-SH:21A click-hydrogels to the presence of alginate. As previously discussed, we hypothesized that the alginate chains slow down the gelation process of the dense HA-SH:21A network, thereby allowing for a more efficient cross-linking. Consequently, fewer alkyne functional groups remain unreacted, thus reducing the risk of cleavage during hydrolysis, which releases propiolic acid, a highly cytotoxic compound (Fig. S16†).60 Overall, not only does alginate substantially improve the mechanical performance of the click-hydrogels, it also plays an important role in enhancing the biocompatibility of the system.
In conclusion, exploiting adaptable mechanisms and natural polymers guides the design of the next generation of biomaterials. This work demonstrates how combining the advantages of two polysaccharide-based networks and the efficiency and rapid nature of the thiol–yne click chemistry reaction yields hydrogels with superior performance. ALG/HA-SH:21A click-hydrogels displayed excellent mechanical features, sustaining high compressive loads even after being immersed in cell culture media for three weeks, while simultaneously retaining all the key properties required for a soft 3D scaffold, i.e. injectability, long-term stability, adequate stiffness, and cytocompatibility. In our system, such promising performance is only achievable when the three components of the system (i.e. HA, alginate, and thiol–yne click chemistry) are present. Overall, we envisage the biocompatible matrices prepared by this strategy as promising 3D cell culture systems to support and promote soft tissue regeneration. Further studies will be required to demonstrate the utility of ALG/HA-SH:21A click-hydrogels to support chondrogenesis and hence find clinical application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr A. C. Weems is thankfully acknowledged for his assistance during DMA characterization. M. M. P.-M. acknowledges funding from the European Union Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 703415. BBSRC are thanked for their award of a DTP CASE studentship to J. S. (grant number: BBSRC DTP CASE BB/M011208/1). The Bioimaging Facility microscope used in this study was purchased with grants from BBSRC, Wellcome and the University of Manchester Strategic Fund.Notes and references
- D. D. Allison and K. J. Grande-Allen, Tissue Eng., 2006, 12, 2131–2140 CrossRef CAS.
- S. P. Evanko, J. C. Angello and T. N. Wight, Arterioscler., Thromb., Vasc. Biol., 1999, 19, 1004–1013 CrossRef CAS.
- D. Naor, R. V. Sionov and D. Ish-Shalom, Adv. Cancer Res., 1997, 71, 241–319 CrossRef CAS.
- K. Masuko, M. Murata, K. Yudoh, T. Kato and H. Nakamura, Int. J. Gen. Med., 2009, 2, 77–81 CrossRef CAS.
- M. A. Solis, Y.-H. Chen, T. Y. Wong, V. Z. Bittencourt, Y.-C. Lin and L. L. H. Huang, Biochem. Res. Int., 2011, 2012, 346972 Search PubMed.
- R. M. L. Simpson, X. Hong, M. M. Wong, E. Karamariti, S. I. Bhaloo, D. Warren, W. Kong, Y. Hu and Q. Xu, Stem Cells, 2016, 34, 1225–1238 CrossRef CAS.
- Q. Feng, S. Lin, K. Zhang, C. Dong, T. Wu, H. Huang, X. Yan, L. Zhang, G. Li and L. Bian, Acta Biomater., 2017, 53, 329–342 CrossRef CAS PubMed.
- H. Kim, H. Jeong, S. Han, S. Beack, B. W. Hwang, M. Shin, S. S. Oh and S. K. Hahn, Biomaterials, 2017, 123, 155–171 CrossRef CAS.
- K. S. Kim, H. Kim, Y. Park, W. H. Kong, S. W. Lee, S. J. J. Kwok, S. K. Hahn and S. H. Yun, Adv. Funct. Mater., 2016, 26, 2512–2522 CrossRef CAS.
- Y. S. Kim, D. K. Sung, W. H. Kong, H. Kim and S. K. Hahn, Biomater. Sci., 2018, 6, 1020–1030 RSC.
- C. B. Highley, G. D. Prestwich and J. A. Burdick, Curr. Opin. Biotechnol, 2016, 40, 35–40 CrossRef CAS.
- E. J. Oh, K. Park, K. S. Kim, J. Kim, J.-A. Yang, J.-H. Kong, M. Y. Lee, A. S. Hoffman and S. K. Hahn, J. Controlled Release, 2010, 141, 2–12 CrossRef CAS PubMed.
- W. H. Kong, D. K. Sung, H. Kim, J.-A. Yang, N. Ieronimakis, K. S. Kim, J. Lee, D.-H. Kim, S. H. Yun and S. K. Hahn, Biomaterials, 2016, 81, 93–103 CrossRef CAS.
- E. Hui, K. I. Gimeno, G. Guan and S. R. Caliari, Biomacromolecules, 2019, 20, 4126–4134 CrossRef CAS.
- D. Diekjürgen and D. W. Grainger, Biomaterials, 2017, 141, 96–115 CrossRef.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458–465 CrossRef CAS.
- X. Zhang, P. Sun, L. Huangshan, B.-H. Hu and P. B. Messersmith, Chem. Commun., 2015, 51, 9662–9665 RSC.
- N. Broguiere, L. Isenmann and M. Zenobi-Wong, Biomaterials, 2016, 99, 47–55 CrossRef CAS PubMed.
- N. Broguiere, E. Cavalli, G. M. Salzmann, L. A. Applegate and M. Zenobi-Wong, ACS Biomater. Sci. Eng., 2016, 2, 2176–2184 CrossRef CAS.
- A. La Gatta, G. Ricci, A. Stellavato, M. Cammarota, R. Filosa, A. Papa, A. D'Agostino, M. Portaccio, I. Delfino, M. De Rosa and C. Schiraldi, Int. J. Biol. Macromol., 2017, 103, 978–989 CrossRef CAS.
- E. Jooybar, M. J. Abdekhodaie, M. Alvi, A. Mousavi, M. Karperien and P. J. Dijkstra, Acta Biomater., 2019, 83, 233–244 CrossRef CAS PubMed.
- M. Shin and H. Lee, Chem. Mater., 2017, 29, 8211–8220 CrossRef CAS.
- T. Hozumi, T. Kageyama, S. Ohta, J. Fukuda and T. Ito, Biomacromolecules, 2018, 19, 288–297 CrossRef CAS.
- R. Zhang, X. Li, K. He, X. Sheng, S. Deng, Y. Shen, G. Chang and X. Ye, Polym. Adv. Technol., 2017, 28, 1759–1763 CrossRef CAS.
- E. Larrañeta, M. Henry, N. J. Irwin, J. Trotter, A. A. Perminova and R. F. Donnelly, Carbohydr. Polym., 2018, 181, 1194–1205 CrossRef.
- R. Egbu, S. Brocchini, P. T. Khaw and S. Awwad, Eur. J. Pharm. Biopharm., 2018, 124, 95–103 CrossRef CAS.
- C. B. Rodell, A. L. Kaminski and J. A. Burdick, Biomacromolecules, 2013, 14, 4125–4134 CrossRef CAS.
- C. B. Rodell, J. W. MacArthur Jr., S. M. Dorsey, R. J. Wade, L. L. Wang, Y. J. Woo and J. A. Burdick, Adv. Funct. Mater., 2015, 25, 636–644 CrossRef CAS.
- L. Chen, X. Zhao, Y. Lin, Z. Su and Q. Wang, Polym. Chem., 2014, 5, 6754–6760 RSC.
- P. Ren, H. Zhang, Z. Dai, F. Ren, Y. Wu, R. Hou, Y. Zhu and J. Fu, J. Mater. Chem. B, 2019, 7, 5490–5501 RSC.
- K. S. Anseth and H.-A. Klok, Biomacromolecules, 2016, 17, 1–3 CrossRef CAS PubMed.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Biomacromolecules, 2011, 12, 824–830 CrossRef CAS PubMed.
- A. Takahashi, Y. Suzuki, T. Suhara, K. Omichi, A. Shimizu, K. Hasegawa, N. Kokudo, S. Ohta and T. Ito, Biomacromolecules, 2013, 14, 3581–3588 CrossRef CAS.
- S.-S. Han, H. Y. Yoon, J. Y. Yhee, M. O. Cho, H.-E. Shim, J.-E. Jeong, D.-E. Lee, K. Kim, H. Guim, J. H. Lee, K. M. Huh and S.-W. Kang, Polym. Chem., 2018, 9, 20–27 RSC.
- M. Fan, Y. Ma, J. Mao, Z. Zhang and H. Tan, Acta Biomater., 2015, 20, 60–68 CrossRef CAS PubMed.
- F. Guilak, L. G. Alexopoulos, M. A. Haider, H. P. Ting-Beall and L. A. Setton, Ann. Biomed. Eng., 2005, 33, 1312–1318 CrossRef PubMed.
- M. Liu, X. Zeng, C. Ma, H. Yi, Z. Ali, X. Mou, S. Li, Y. Deng and N. He, Bone Res., 2017, 5, 17014 CrossRef CAS PubMed.
- V. X. Truong and A. P. Dove, Angew. Chem., Int. Ed., 2013, 52, 4132–4136 CrossRef CAS.
- L. J. Macdougall, V. X. Truong and A. P. Dove, ACS Macro Lett., 2017, 6, 93–97 CrossRef CAS.
- V. X. Truong, M. P. Ablett, S. M. Richardson, J. A. Hoyland and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 1618–1622 CrossRef CAS.
- L. J. Macdougall, M. M. Pérez-Madrigal, M. C. Arno and A. P. Dove, Biomacromolecules, 2018, 19, 1378–1388 CrossRef CAS.
- L. J. Macdougall, M. M. Pérez-Madrigal, J. E. Shaw, M. Inam, J. A. Hoyland, R. O'Reilly, S. M. Richardson and A. P. Dove, Biomater. Sci., 2018, 6, 2932–2937 RSC.
- A. K. Means and M. A. Grunlan, ACS Macro Lett., 2019, 8, 705–713 CrossRef.
- J. Lesley, V. C. Hascall, M. Tammi and R. Hyman, J. Biol. Chem., 2000, 275, 26967–26975 CAS.
- M. Morra, Biomacromolecules, 2005, 6, 1205–1223 CrossRef CAS.
- B. A. Buhren, H. Schrumpf, N.-P. Hoff, E. Bölke, S. Hilton and P. A. Gerber, Eur. J. Med. Res., 2016, 21, 5 CrossRef.
- J. Wang, F. Zhang, W. P. Tsang, C. Wan and C. Wu, Biomaterials, 2017, 120, 11–21 CrossRef CAS.
- J. H. Lai and M. E. Levenston, Osteoarthr. Cartilage, 2010, 18, 1291–1299 CrossRef CAS.
- V. X. Truong, K. M. Tsang and J. S. Forsythe, Biomacromolecules, 2017, 18, 757–766 CrossRef CAS.
- L. V. Thomas, V. G. Rahul and P. D. Nair, Int. J. Biol. Macromol., 2017, 104, 1925–1935 CrossRef CAS.
- L. Q. Wan, J. Jiang, D. E. Arnold, X. E. Guo, H. H. Lu and V. C. Mow, Cell. Mol. Bioeng., 2008, 1, 93–102 CrossRef CAS.
- S. P. Zhong, D. Campoccia, P. J. Doherty, R. L. Williams, L. Benedetti and D. F. Williams, Biomaterials, 1994, 15, 359–365 CrossRef CAS.
- A. Borzacchiello, L. Russo, B. M. Malle, K. Schwach-Abdellaoui and L. Ambrosio, BioMed Res. Int., 2015, 2015, 871218 Search PubMed.
- T. Segura, B. C. Anderson, P. H. Chung, R. E. Webber, K. R. Shull and L. D. Shea, Biomaterials, 2005, 26, 359–371 CrossRef CAS.
- K. Xu, F. Lee, S. Gao, M.-H. Tan and M. Kurisawa, J. Controlled Release, 2015, 216, 47–55 CrossRef CAS.
- S. James, J. Fox, F. Afsari, J. Lee, S. Clough, C. Knight, J. Ashmore, P. Ashton, O. Preham, M. Hoogduijn, R. A. R. Ponzoni, Y. Hancock, M. Coles and P. Genever, Stem Cell Rep., 2015, 4, 1004–1015 CrossRef CAS.
- W. Hassan, Y. Dong and W. Wang, Stem Cell Res. Ther., 2013, 4, 11 CrossRef.
- A. Ghanizadeh Tabriz, C. G. Mills, J. J. Mullins, J. A. Davies and W. Shu, Front. Bioeng. Biotechnol., 2017, 5, 13 Search PubMed.
- L. J. Macdougall, K. L. Wiley, A. M. Kloxin and A. P. Dove, Biomaterials, 2018, 178, 435–447 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01494b |
| This journal is © The Royal Society of Chemistry 2020 |