
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioengineering a pre-vascularized pouch for subsequent islet transplantation using VEGF-loaded polylactide capsules†
Naresh
Kasoju‡
 a,
Alžběta
Pátíková
b,
Edyta
Wawrzynska
a,
Alžběta
Vojtíšková
b,
Tomáš
Sedlačík
a,
Marta
Kumorek
a,
Ognen
Pop-Georgievski
a,
Eva
Sticová
c,
Jan
Kříž
*b and
Dana
Kubies
*a
a,
Alžběta
Pátíková
b,
Edyta
Wawrzynska
a,
Alžběta
Vojtíšková
b,
Tomáš
Sedlačík
a,
Marta
Kumorek
a,
Ognen
Pop-Georgievski
a,
Eva
Sticová
c,
Jan
Kříž
*b and
Dana
Kubies
*a
aInstitute of Macromolecular Chemistry Czech Academy of Sciences, Heyrovsky sq.2, Prague 162 06, Czech Republic. E-mail: kubies@imc.cas.cz
bDepartment of Diabetes, Institute for Clinical and Experimental Medicine, Prague, Czech Republic, Videnska 2, Prague 120 00, Czech Republic. E-mail: jkri@ikem.cz
cDepartment of Pathology, Third Faculty of Medicine, Charles University and Faculty Hospital of Královské Vinohrady, Prague, Czech Republic
First published on 15th November 2019
Abstract
The effectiveness of cell transplantation can be improved by optimization of the transplantation site. For some types of cells that form highly oxygen-demanding tissue, e.g., pancreatic islets, a successful engraftment depends on immediate and sufficient blood supply. This critical point can be avoided when cells are transplanted into a bioengineered pre-vascularized cavity which can be formed using a polymer scaffold. In our study, we tested surface-modified poly(lactide-co-caprolactone) (PLCL) capsular scaffolds containing the pro-angiogenic factor VEGF. After each modification step (i.e., amination and heparinization), the surface properties and morphology of scaffolds were characterized by ATR-FTIR and XPS spectroscopy, and by SEM and AFM. All modifications preserved the gross capsule morphology and maintained the open pore structure. Optimized aminolysis conditions decreased the Mw of PLCL only up to 10% while generating a sufficient number of NH2 groups required for the covalent immobilization of heparin. The heparin layer served as a VEGF reservoir with an in vitro VEGF release for at least four weeks. In vivo studies revealed that to obtain highly vascularized PLCL capsules (a) the optimal VEGF dose for the capsule was 50 μg and (b) the implantation time was four weeks when implanted into the greater omentum of Lewis rats; dense fibrous tissue accompanied by vessels completely infiltrated the scaffold and created sparse granulation tissue within the internal cavity of the capsule. The prepared pre-vascularized pouch enabled the islet graft survival and functioning for at least 50 days after islet transplantation. The proposed construct can be used to create a reliable pre-vascularized pouch for cell transplantation.
1. Introduction
The replacement of failed cells by transplantation is one of the most promising approaches that have been already tested and applied in clinical practice. Over the last several decades the transplantations of parathyroid glands,1 thyroid gland,2 adrenal cells,3 thymus4 and pancreatic islets5 have been introduced and tested under experimental and clinical conditions. In addition, cellular transplantation can be theoretically used for the therapy of haemophilia, liver failure, Parkinson's disease, transversal spinal cord injuries and others.6–9 Outcomes of these procedures still vary widely but it seems likely that the procedure's effectiveness can be considerably improved via optimization of the transplantation site and the procedure itself. The generally accepted requirement for an adequate environment for an engraftment of transplanted cells inspires many groups to pay great attention to the characterization and subsequent development of the optimal transplantation site.Some approaches focused on cell protection against inflammation and more specific rejection using isolation of the graft in microcapsules.10 This method can effectively serve in the case of cells releasing hormones slowly and evenly with very small variations in time. Another approach focused on the creation of microencapsulation using a cell pouch with a fully finished healing in order to protect the cells from inflammation and to concentrate them into a small volume thus allowing the subsequent excision and a detailed examination.11,12 The finished healing provides a hospitable environment but suffers from poor blood perfusion.13 This feature would be detrimental for the graft survival particularly in the case of cells serving as a sensor of the metabolic state in addition to hormone secretion. For example, pancreatic islets are extremely oxygen demanding and even a short time disruption of blood perfusion in combination with a high concentration of glucose can cause their destruction. This crucial limitation known for successful islet transplantation can be improved when the pouch providing an adequate blood perfusion in the time of transplantation is created in advance.
Several device-based approaches, including those based on a groin flow-through chamber model,14,15 have been described to create a transplantation site which provides sufficient blood supply for transplanted islets. Although these approaches may be desirable because they allow immediate access to the existing vasculature, none was transferred to clinical practice likely due to the extent of surgical manipulation needed in order to accommodate a sufficient number of islets. Alternatively, devices based on polymeric porous scaffolds are found to be simple in terms of surgical manipulation, offer an adequate volume for islet loading, provide mechanical graft protection and may be easily retrievable in the case of graft infection.16,17
In our study we have focused on the creation of a pre-vascularized pouch with an adequate inner volume, without considerable inflammation in the time of transplantation and rich in a vascular/capillary network on the internal surface. We believe that such induced pre-vascularization could significantly shorten the “hypoxia” time when islets are dependent on oxygen diffusion. A quick islet reconnection to the host vascular network, resulting in a rapid re-perfusion, improves the subsequent islet graft survival and its function. The concept is presented in Fig. 1a. Based on previously published experiments where the number of transplanted islets was considerably higher than the number used in clinical programs,18,19 we have focused on the preparation of a microporous device, which can direct fibrous tissue penetration and gradually release vascular growth factors stimulating ingrowth of vessels together with connective tissue. However, with this type of device, the scaffold porosity is a key parameter for an optimal recipient tissue response and, subsequently, the transplantation outcome. We have previously shown that in comparison with a typical scaffold usually with an isotropic pore morphology, a scaffold consisting of anisotropic channelled pores showed the desired guided cell infiltration.20,21 The proposed capsule-shaped scaffolds have been fabricated from an FDA approved polylactide-based polymer; the scaffolds possess channelled pores oriented into an internal capsule lumen that could serve as a cavity for subsequent cellular, or specifically islet, transplantation (Fig. 1a and 2).
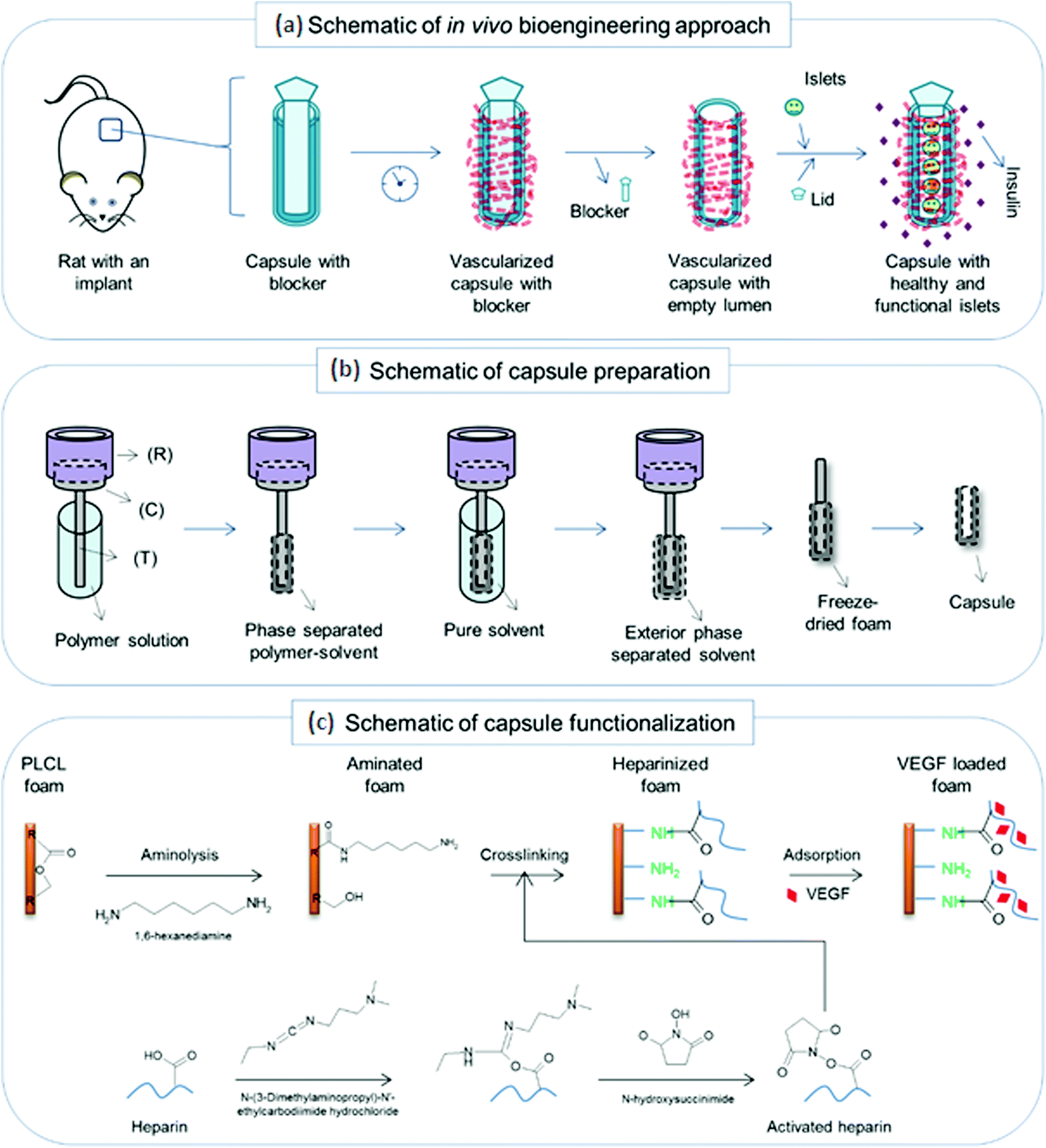 | ||
| Fig. 1 Schematic of the implantation, capsule preparation and functionalization: (a) a schematic of the proposed approach to bioengineer a pre-vascularized chamber for potential cell transplantation using a PLCL capsular scaffold with a Teflon lid. (b) The capsule preparation was performed following a TIPS principle by using a custom-made setup having a reservoir for a dry ice–ethanol mixture (R) that was connected to a conducting block (C) and a template bar (T) (for details see ref. 13). (c) The capsule functionalization was performed in three stages including aminolysis, heparin crosslinking by carbodiimide chemistry and then VEGF loading by physical adsorption. | ||
However, a major drawback of the use of synthetic polymers is the fact that they do not usually provide biochemical cues, e.g., molecules acting as proangiogenic signals that are highly required to trigger an efficient vascularization process in tissue engineering applications.22 Although the natural healing process attempts to vascularize a polymer scaffold, our earlier attempts showed that this process was relatively slow and that the resulting tissue was often mostly fibrotic.20,21 To this end, here we undertook functionalization of a poly(lactide-co-caprolactone) (PLCL) capsule-shaped scaffold with the proangiogenic vascular endothelial growth factor (VEGF) in order to enhance the formation of a microvascular network within the scaffold. The PLCL capsular scaffolds used in this work were prepared using the previously described Dip-TIPS technique (Fig. 1b).21,23 The surface functionalization of the scaffolds was achieved in 3 stages: in the first step, amine groups were introduced onto the scaffold surface without destroying the scaffold's 3D morphology and features; in the second step, a heparin layer was immobilized by a carbodiimide-based covalent crosslinking to the amine groups to obtain a VEGF-cargo with an adequate loading capacity for an efficient vascularization process; finally, VEGF was loaded into the scaffold by physical adsorption to the heparin layer (Fig. 1c). Physico-chemical characterization studies were performed to track the changes in the surface properties of the real three-dimensional scaffold at each stage of the modification, and in vitro VEGF release was evaluated. In vivo evaluation using an inbred Lewis rat model was focused on determining the conditions required for adequate VEGF dosing and the timing of the implantation into the greater omentum to obtain sufficient tissue ingrowth and formation of functional vasculature. Finally, the properly pre-vascularized pouch was tested for Langerhans islet survival using a Lewis rat model.
2. Materials and methods
All materials, chemicals, and reagents were used as received unless stated otherwise. The detailed synthetic procedures, characterization methods, and implantation protocols are provided in the ESI.† In the following, a short summary of the sample preparation, the characterization and implantation methods is presented.2.1 Materials
High molecular weight poly(L-lactide-co-ε-caprolactone) (PLCL, Mw = 316![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and Mn = 120000 g mol−1, 14 wt% of ε-CL) was synthesized according to the protocol described by Kubies et al.24
000 g mol−1 and Mn = 120000 g mol−1, 14 wt% of ε-CL) was synthesized according to the protocol described by Kubies et al.24
2.2 Fabrication of samples
Macroporous 3D samples: PLCL capsule-shaped scaffolds were prepared using a thermally induced phase separation (TIPS) approach according to the protocol reported earlier by our group.20,21,23 The protocol is schematically depicted in Fig. 1b. Briefly, the metal template connected to the cooling platform was dipped into a PLCL solution (5% w/v in 1,4-dioxane). The system was cooled to −80 °C and the phase separation proceeded for 30 s followed by coarsening for 30 s. Then the template with the sample was dipped into 1,4-dioxane for another 30 s to avoid any skin effect, left to coarsen for 30 s and subjected to freeze-drying.Model 2D samples: to properly analyse the surface properties such as topography and the surface elemental composition of PLCL surfaces before and after functionalization, model PLCL films were used instead of the porous scaffolds. The model PLCL films were prepared by the solvent casting procedure from a 10% PLCL solution (w/v in 1,4-dioxane).
2.3 Functionalization of samples
VEGF loading was performed from a VEGF solution in PBS (pH 7.4) supplemented with 0.1% w/v BSA. The model tested concentrations were 0.08 μg VEGF per mg of scaffold (i.e., 2 μg VEGF/scaffold, the sample denoted as heparin/VEGF_2) and 0.4 μg VEGF per mg of scaffold (i.e., 10 μg VEGF/scaffold, the sample denoted as heparin/VEGF_10). The release of VEGF from the scaffolds into PBS supplemented with BSA at 37 °C within four weeks was investigated by the full immersion method.
2.4 Characterization studies
The surface topography was assessed using an atomic force microscope in tapping mode (AFM, Nanoscope IIIa, Digital Instruments.
Surface wettability (CA) was determined by the sessile drop method using a goniometer system (OCA 20, Dataphysics).
Surface chemistry was analysed by X-ray photoelectron spectroscopy (XPS) by Fourier transform infrared spectroscopy in attenuated total reflectance mode (ATR-FTIR).
For SEM, ATR-FTIR and CA analyses the porous scaffolds were used, while for AFM and XPS analyses, the model flat films were used.
To deduce the mode of VEGF release from the scaffolds, the release kinetics data were fitted to four mathematical models, viz., zero-order, first-order, Higuchi and power law models.25–28
2.5 In vivo studies
For the dose-optimization experiment, scaffolds supplemented with 10, 50, or 100 μg VEGF (the samples denoted as heparin/VEGF_10, heparin/VEGF_50 and heparin/VEGF_100) were implanted into the greater omentum of the experimental animals for 2 weeks. For time-optimization experiments, scaffolds loaded with 50 μg VEGF (i.e., heparin/VEGF_50) were implanted for 2, 3, or 4 weeks. After the selected time period, the abdominal cavity was again opened under general anaesthesia, and the scaffold with the surrounding tissues was excised and fixed; the animal was then euthanized.
The scaffolds loaded with 50 μg VEGF (i.e., heparin/VEGF_50) were immersed in platelet enriched plasma and were implanted within the greater omentum of recipients (see paragraph 2.5.2. and the ESI†). In the meantime, insulin pellets were implanted subcutaneously in order to control the blood glucose levels prior to islet transplantation.
Four weeks after the scaffold implantation a cranial midline laparotomy was performed and the scaffold was placed on wet gauze. A small incision in the omentum was performed to allow a Teflon bar removal and islets were then injected into the created cavity; finally, the omental incision and abdominal wall were closed. Two weeks after islet transplantation, insulin pellets were removed.
The blood glucose level was monitored twice a week and body weight once a week for 50 days. Glycemia under 10 mmol l−1 was considered as a sign of a functional graft, and glycemia over 11.1 measured at least three times was considered as a marker of graft failure. An intravenous glucose tolerance test (0.5 g of glucose per kg) was performed 40 days after islet transplantation. At the end of the experiment, each scaffold with the islet graft was harvested, fixed and provided for immunohistological analysis.
The islet structure and the presence of blood capillaries in the graft were evaluated using double-staining with anti-insulin and anti-glucagon antibodies and with anti-CD31 antibody specific for endothelial cells.
3. Results and discussion
3.1 Typical morphological features
The pore properties of a scaffold, both at the macro-scale and micro-scale, greatly influence the fate of a scaffold in cell and tissue engineering; therefore, the morphological characteristics of the prepared 3D microporous PLCL capsules were studied in detail. The capsule lumen has a diameter of 4 mm and a length of 20 mm (Fig. 2a) and can accommodate 250 μl of the cell suspension. The pore size of various sections of the capsule was analysed by SEM. A representative cross-section of the capsule (Fig. 2b) shows the channelled pores and the interconnected character of the pore network; the thickness of the capsule's wall ranged between 600 and 650 μm. The outer surface exhibited pores 50–70 μm in diameter, and the inner surface revealed pores 5–10 μm in diameter (Fig. 2c and d). The capsule possessed a unique radially oriented, channelled, interconnected and anisotropic pore morphology as a result of the Dip TIPS approach that we employed. The use of isotropic porous scaffolds often results in the formation of a necrotic core due to random cell infiltration and associated constraints on nutrient diffusion, whereas anisotropic porous scaffolds inherently promote guided cell infiltration without affecting nutrient diffusion and therefore yield fully functional viable tissue.30 | ||
| Fig. 2 Typical morphological features of PLCL capsules: (a) a digital camera image of the capsule (left) along with a Teflon bar (right) used to block the core of the capsule during implantation. SEM images of the capsule's cross section (b), outer surface (c), and inner surface (d). | ||
3.2 Aminolysis
Polylactide-based surfaces possess only a few carboxyl and hydroxyl groups on the chain ends resulting from the mechanism of lactide polymerization in the presence of tin(II) octoate.31 To introduce functional NH2 groups for covalent immobilization of heparin on the complex surface of the 3D PLCL scaffold, the pristine capsules were subjected to aminolysis. Aminolysis is considered one of the most convenient procedures for surface modifications and can also be used in the surface hydrophilization of polyesters.32,33 Since the reaction takes place only at the polymer surface and does not change the polymer's bulk properties, this procedure is especially favourable for the modification of post-formatted scaffolds and membranes.34–36 Highly reactive amine groups break some of the ester groups of polyester chains and covalently bind to a single carboxyl group.36 Subsequently, the remaining amine end-group can be used for immobilization of various functional biomacromolecules. However, the ester bond cleavage results in a reduction of the molecular weight of the sample. Zhu and co-workers have extensively investigated the functionalization of poly(ε-caprolactone) and poly(lactic acid) films with different diamines.35,37 The group studied the influence of the reaction time, temperature, solvent type, and the length and concentration of the diamine on the mechanical properties of aminolysed membranes. It was found that the reaction of 1,6-hexanediamine (HMDA) proceeds very mildly.We adjusted the aminolysis conditions to achieve minimal degradation of polymer chains and to preserve the morphology of the complex 3D PLCL capsules. The reaction with HMDA was conducted in isopropanol (a non-solvent for PLCL) to avoid any damage to the bulk of the capsule. First-hand visual confirmation of successful amination was obtained by FITC staining followed by CLSM imaging. In agreement with earlier reports,34 we found that pristine PLCL capsules showed no signal, whereas the aminolysed capsules showed intense green fluorescence after staining with FITC (Fig. 3A).
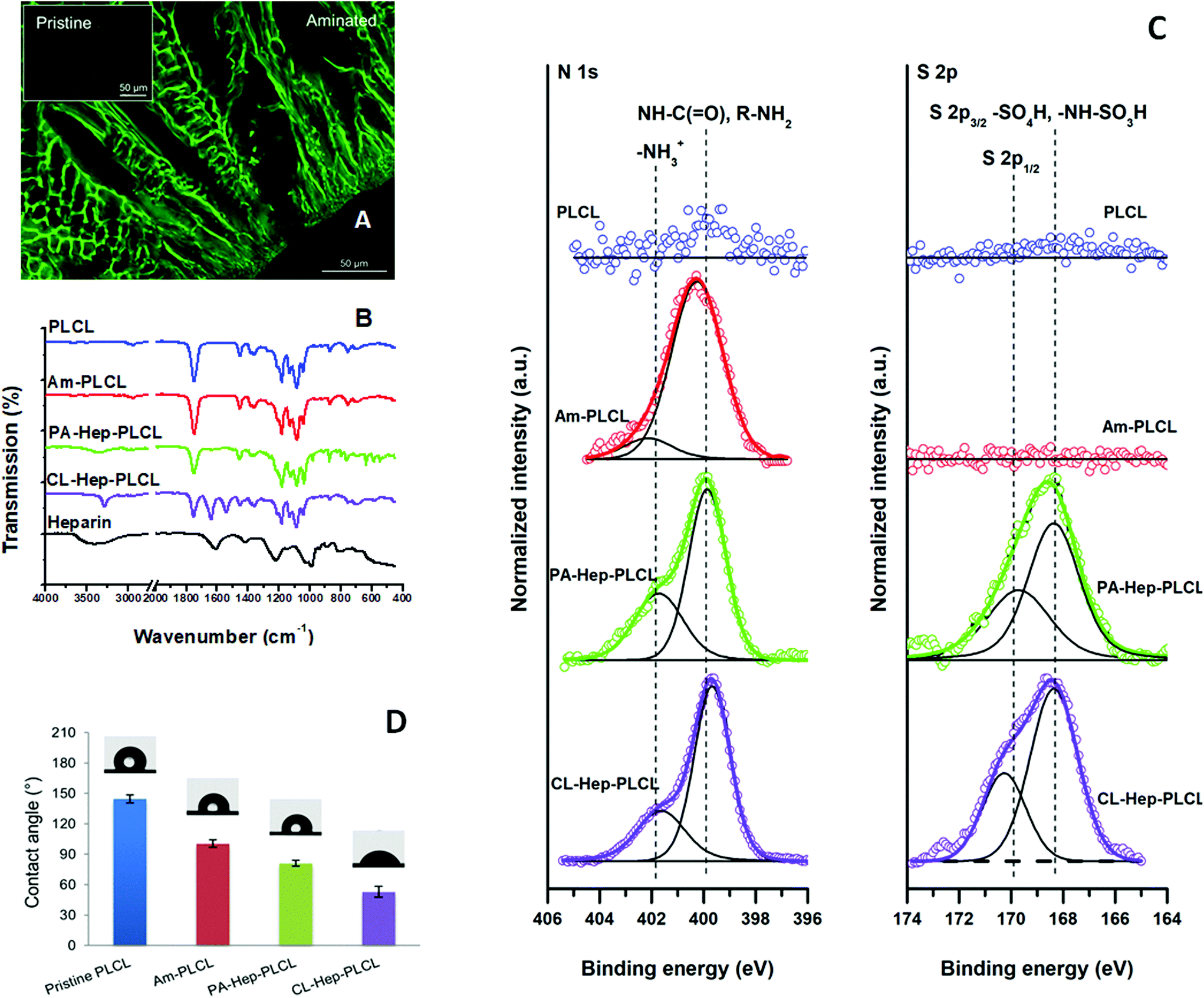 | ||
| Fig. 3 Characterization of PLCL surfaces, i.e., the pristine PLCL, PCLC after aminolysis (Am-PLCL) and PCLC after physically adsorbed (PA-Hep-PLCL) and chemically crosslinked (CL-Hep-PLCL) heparin coatings. (A) Qualitative analysis of amination – confocal microscopy of the FITC-stained Am-PLCL capsule showed a distinct green fluorescence in contrast to the control sample; (B) ATR-FTIR spectra of PLCL, Am-PLCL, PA-Hep-PLCL and CL-Hep-PLCL samples. The spectrum of pure heparin is presented for a comparison; (C) Representative high resolution N 1s and S 2p XPS spectra of the PLCL, Am-PLCL, PA-Hep-PLCL and CL-Hep-PLCL coated model surfaces; (D) Surface wettability after aminolysis (Am-PLCL) and heparinization (PA-Hep-PLCL and CL-Hep-PLCL) evaluated by static water-in-air contact angle measurements. | ||
The chemical composition of pristine and aminated PLCL surfaces was studied by ATR-FTIR and XPS analyses. As presented in Fig. 3B, pristine PLCL showed characteristic peaks associated with ester linkages (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O and long alkyl chain-related) at 1000–1300 cm−1, carbonyl stretching (CO related) at 1750 cm−1 and CH2 stretching (alkyl group-related) at 2945 and 2995 cm−1.38,39 Yuan et al. reported that aminolysis of poly(ε-caprolactone) (PCL) surfaces resulted in additional, tiny peaks at 3321, 1650 and 1550 cm−1; these peaks were attributed to the successful introduction of amine groups.40 However, in our experiments, the FTIR spectra of aminated PLCL did not exhibit detectable peaks of this type, possibly due to the low degree of amination, the limitations of the instrument, or both. The surface composition of the samples was further analysed by XPS (Fig. 3C). The high-resolution C 1s spectrum of pristine PLCL was characterized by aliphatic C—C and C—H contributions at 285.0 eV, C—O contribution at approximately 286.9 ± 0.2 eV and ester C(O)—O contribution at 289.0 ± 0.1 eV. Aminolysis of the PLCL capsules resulted in an increase in the contributions at approximately 286.3 ± 0.2 eV due to the additional presence of C–N moieties that overlap with the initially observed C–O contributions. Consistent with the observations from the C 1s spectrum (ESI Fig. 1†), the high-resolution N 1s spectrum of the aminolysed PLCL capsules was characterized by a dominant amide and amine contribution at 400.0 ± 0.1 eV and a minor charged amine contribution at 401.9 ± 0.2 eV. The obtained results are in agreement with the results reported by Zhu et al.35 The nitrogen content of the PLCL surfaces reached 0.5 atomic % after 60 min of aminolysis irrespective of the reaction temperature. This observation is consistent with previous work on the surface composition of aminolysed PLA and PCL surfaces, in which a nitrogen content in the range of 0.5–1.3 atomic% was reported.33,35,41,42 The surface coverage with amine active species obtained herein should be sufficient for the purpose of further functionalization.33 This assumption was confirmed in subsequent heparin immobilization experiments.
O, C–O and long alkyl chain-related) at 1000–1300 cm−1, carbonyl stretching (CO related) at 1750 cm−1 and CH2 stretching (alkyl group-related) at 2945 and 2995 cm−1.38,39 Yuan et al. reported that aminolysis of poly(ε-caprolactone) (PCL) surfaces resulted in additional, tiny peaks at 3321, 1650 and 1550 cm−1; these peaks were attributed to the successful introduction of amine groups.40 However, in our experiments, the FTIR spectra of aminated PLCL did not exhibit detectable peaks of this type, possibly due to the low degree of amination, the limitations of the instrument, or both. The surface composition of the samples was further analysed by XPS (Fig. 3C). The high-resolution C 1s spectrum of pristine PLCL was characterized by aliphatic C—C and C—H contributions at 285.0 eV, C—O contribution at approximately 286.9 ± 0.2 eV and ester C(O)—O contribution at 289.0 ± 0.1 eV. Aminolysis of the PLCL capsules resulted in an increase in the contributions at approximately 286.3 ± 0.2 eV due to the additional presence of C–N moieties that overlap with the initially observed C–O contributions. Consistent with the observations from the C 1s spectrum (ESI Fig. 1†), the high-resolution N 1s spectrum of the aminolysed PLCL capsules was characterized by a dominant amide and amine contribution at 400.0 ± 0.1 eV and a minor charged amine contribution at 401.9 ± 0.2 eV. The obtained results are in agreement with the results reported by Zhu et al.35 The nitrogen content of the PLCL surfaces reached 0.5 atomic % after 60 min of aminolysis irrespective of the reaction temperature. This observation is consistent with previous work on the surface composition of aminolysed PLA and PCL surfaces, in which a nitrogen content in the range of 0.5–1.3 atomic% was reported.33,35,41,42 The surface coverage with amine active species obtained herein should be sufficient for the purpose of further functionalization.33 This assumption was confirmed in subsequent heparin immobilization experiments.
Proof of successful amination was also sought from contact angle measurements based on the fact that the incorporation of amine groups decreases the hydrophobicity of the capsule surface. As shown in Fig. 3D, the CA of pristine PLCL was 144 ± 4°, thus representing a highly hydrophobic surface. The observed high CA value was not typical of PLA-based flat surfaces and resulted from the porous open structure of the analysed capsule surface. This surface feature was described in more detail in our previous work.23 After aminolysis for 30 min, the CA of the PLCL 3D surfaces decreased significantly to 101 ± 4°. This significant reduction of 30% clearly demonstrates that effective surface hydrophilization occurred due to the presence of the introduced amine groups. Previously, several groups reported an overall reduction in contact angles between 15 and 26% for flat non-porous PCL surfaces aminolysed with an HMDA solution.34,35,43 On the other hand, Croll et al. reported a decrease of only 2% when PLGA surfaces were treated with 0.05 M N-(2aminoethyl)-1,3-propanediamine33 and Sharkawi et al. observed no significant changes in the wettability of HMDA-aminolysed PDLLA films with a thickness of 0.5 mm.42 During aminolysis, some of the ester bonds of the PLCL chains break apart, and HMDA forms amide bonds by nucleophilic substitution. In principle, aminolysis is destructive to the polymer chain; too intensive scission of polymer chains will result in changes in their mechanical properties and thereby in changes in the integrity of the initial 3D scaffold structure that can affect the scaffold's stability and thus its possible practical applications. Therefore, the changes in the material properties after the aminolysis are a matter of serious concern. We analysed the changes in the molecular weight (Mw) and the physical mass of the scaffolds as a function of the duration and temperature of aminolysis treatment (Fig. 4A). There was an increasing trend in the loss of physical mass as a function of time and temperature of aminolysis. Nevertheless, the mass loss was less than 3% under all tested conditions. In contrast, the changes in Mw were more prominent. At 22 °C, the Mw decreased from 6.5 to 20.4% as the duration of aminolysis increased from 5 to 60 min. A similar increasing trend was observed at 40 °C. In addition, the changes in Mw were temperature dependent; after 30 min of aminolysis, the decrease in Mw was 11.4% at 22 °C, whereas it was 28.8% at 40 °C. Only a few groups have investigated the effects of reaction conditions on changes in molecular characteristics. Effects similar to those observed in our study were observed by Zhu et al. during the aminolysis of PCL films in the presence of 5% HMDA at 30 °C for 30 min with a resulting Mw decrease of 11.6%,35 by Hong et al. when PLA microspheres underwent aminolysis in 6% HMDA/n-propanol solution at 60 °C for 60 min, resulting in a mass loss of 30%,43 and by Collins et al. in the case of poly(ethylene terephthalate) (PET) films aminolysed in the presence of 40% aqueous methylamine at 20 °C for 1 h, for which a Mw decrease of 26.8% was reported.44
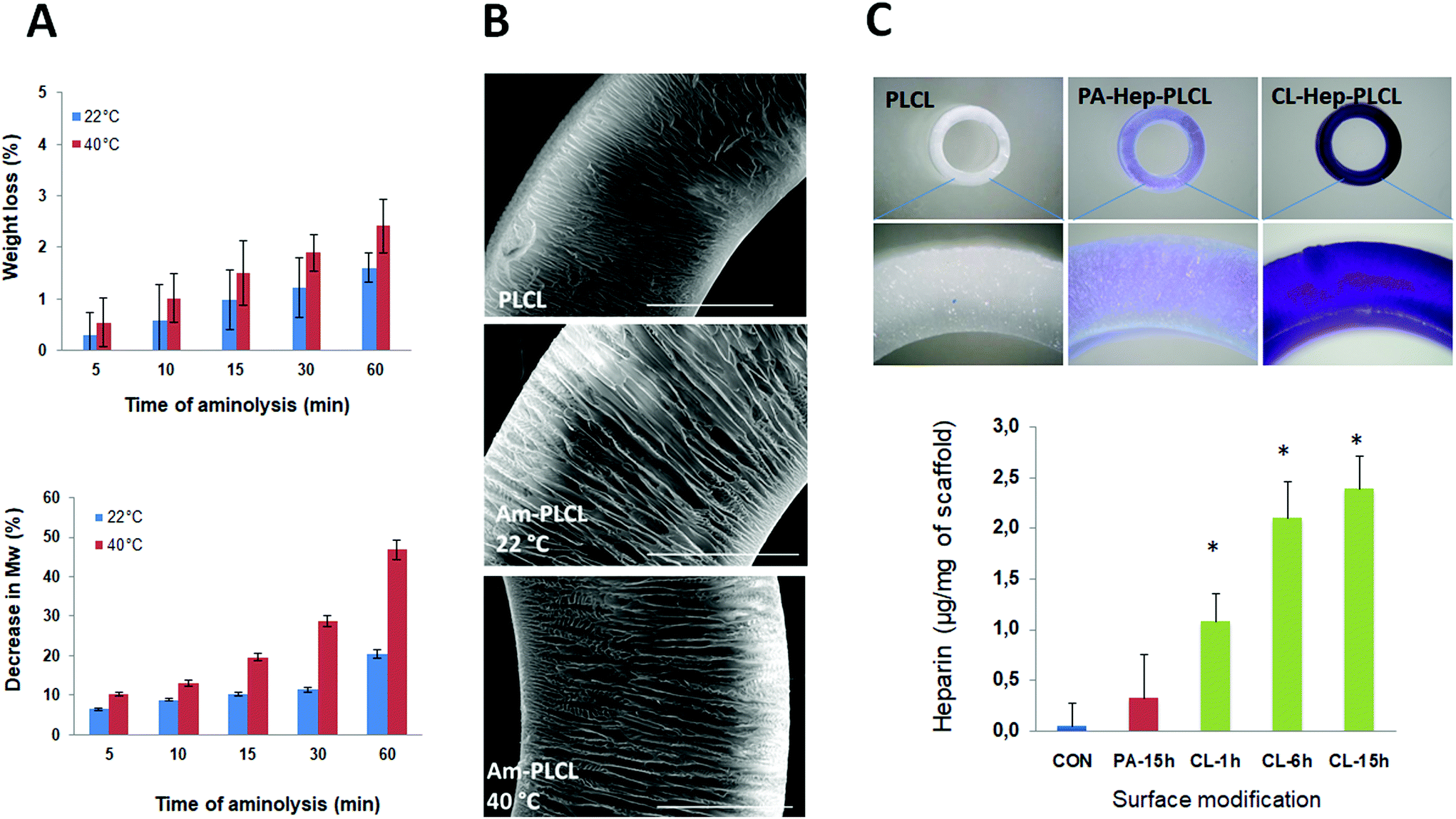 | ||
| Fig. 4 Characterization of PLCL capsules after aminolysis (Am-PLCL) and after physically adsorbed (PA-Hep-PLCL) and chemically crosslinked (CL-Hep-PLCL) heparin coatings. (A) Physical mass and molecular weight changes after aminolysis: the loss in the physical weight caused by aminolysis is negligible while the decrease in the molecular weight is more considerable and reaction time and temperature dependent (B); Morphological changes after aminolysis: SEM images of the pristine capsule and the samples aminolysed at 22 and 40 °C for 30 min, scale bar 500 μm; (C) Qualitative and quantitative analysis of heparinization (toluidine blue assay): stereo zoom microscopy images of the toluidine blue-stained PLCL samples heparinized via physical adsorption (PA) and cross-linking chemistry (CL) showed a distinct purple color in contrast to the control sample. The toluidine blue assay suggested that, in contrast to physically adsorption, the heparin content was higher in the cross-linked samples with a direct correlation with the crosslinking reaction time. *⋯ statistically significant at P < 0.01 versus the control sample. | ||
To obtain information on the overall structure of the aminolyzed capsules, we analysed the samples by SEM. Importantly, there were no visible defects in the gross morphology or in the character of the pore structure before or after aminolysis (ESI Fig. 2† and Fig. 4B). We also tracked the possible changes in the topography of the model solvent-cast PLCL films by AFM (Fig. 5). Notably, the pristine and aminolysed PLCL surfaces exhibited very similar overall topographies with lamellar structures as a result of the crystallization of the PLCL chains during the film processing (Fig. 5A and B). The slight increase in the surface roughness, and subtle changes in the character of the lamellae, can be considered to be a result of the etching procedure. Similar effects of surface etching on the surface topography have been reported by Zhu et al.,34,35 and Yuan et al.41 for aminolyzed PCL substrates and by Drobota et al. for PET substrates.45
 | ||
| Fig. 5 Topographical changes after aminolysis and heparinization of model PLCL films: AFM images and height profiles of the pristine PLCL (a), Am-PLCL (30 min at 22 °C) (b), PA-Hep-PLCL (15 h) (c) and CL-Hep-PLCL (6 h) coated surfaces. Scan size: 1 × 1 μm2. | ||
To summarize, the aminolysis performed at 22 °C for 30 min resulted in sufficient surface modification with an acceptable decrease in the molecular weight while preserving the gross morphology of the modified scaffolds. These conditions were selected for subsequent modification studies with heparin and for the preparation of the samples for in vivo studies.
3.3 Heparinization
To further functionalize the capsule with heparin, a biomacromolecule that has a strong affinity for VEGF and thereby increases its loading amount and structural stability, aminolysed PLCL capsules were incubated with a heparin solution in the presence or absence of crosslinking agents. During physical adsorption (PA), the negatively charged sulfate and carboxyl groups of heparin participate in electrostatic interactions with the positively charged amine groups of aminated PLCL. This surface modification was considered as a control, whereas, during the reaction with crosslinking agents (CL), heparin was first activated in the presence of EDC and NHS and then crosslinked to the amine groups of aminated PLCL via covalent amide bonds. In both cases, first-hand visual confirmation of successful amination was obtained by toluidine blue staining. The aminated PLCL control samples remained white, whereas the heparinized PLCL test samples showed a distinct purple colour (Fig. 4C), in good agreement with the earlier reports.38 A high intensity of the purple colour in the chemically crosslinked heparinized PLCL sample demonstrated the effectiveness of the covalent heparin grafting in comparison with the physical adsorption method.The chemical surface composition of the physically adsorbed (PA-Hep-PLCL) and chemically crosslinked (CL-Hep-PLCL) heparinized PLCL surfaces was analysed by ATR-FTIR and XPS. The ATR-FTIR spectra (Fig. 3B) of pure heparin showed heparin's characteristic peaks for asymmetrical stretching of SO in CH2-SO3− at 1010 cm−1, symmetrical stretching of SO in NH-SO3− at 1220 cm−1, N–H bending at 1420 cm−1, CO stretching at 1610 cm−1 and OH stretching at 3400 cm−1.46 Most of these peaks were found in both the PA-H-PLCL and CL-H-PLCL samples; however, in contrast to PA-H-PLCL, the chemically crosslinked CL-H-PLCL exhibited a new peak at 1640 cm−1 that was characteristic of the amide bond created as a result of the covalent crosslinking between heparin and aminated PLCL. Our data are consistent with an earlier report by Singh et al., who observed a similar peak after chemical crosslinking of heparin and PCL.38
Further evidence supporting the successful heparinization of the PLCL capsules was obtained from XPS analysis. The physical adsorption of heparin on the Am-PLCL surfaces induced only minor changes in the C 1s spectrum. At the same time, the covalent binding of heparin caused an increase in the amide contribution at 287.6 ± 0.2 eV. Notably, both PA-Hep-PLCL and CL-Hep-PLCL (Fig. 3C) samples exhibited a new contribution in the high-resolution S 2p spectrum at 168.4 ± 0.2 eV (with a separation between the 2p3/2–2p1/2 spin-split doublet of approximately 0.6 eV) that corresponds to the –SO4H and –NH–SO3H moieties of heparin. Such a peak was also observed by Kolar et al. in heparin-functionalized PET tubes47 and by Sharkawi et al. in the heparin-functionalized PDLLA films.42
The successful heparinization of the PLCL capsules led to changes in their surface wettability. The heparinization resulted in surface hydrophilization. As shown in Fig. 3D, the contact angles of PA-Hep-PLCL and CL-Hep-PLCL were 81 ± 3 and 53 ± 5°, respectively; this corresponds to reduction by 20 and 47%, respectively, compared to the aminolysed Am-PLCL (101°), and by 44 and 63%, respectively, compared to pristine PLCL (145°). Similar changes in surface wettability were reported by Khandwekar et al., who found a 43% reduction in the advancing contact angles of heparinized PCL surfaces compared to those of pristine PCL surfaces.48 The observed profound difference in the contact angles of the PA-Hep-PLCL and CL-Hep-PLCL surfaces also indirectly showed that the chemical crosslinking approach made it possible to immobilize a higher (and/or more stable) amount of heparin on the polymer surface than was accomplished via the physical adsorption method. The heparin content of the PLCL capsules quantified by the toluidine blue assay was 0.33, 1.08, 2.10 and 2.39 μg per mg of the scaffold in the 15 hPA-Hep-PLCL, 1 hCL-Hep-PLCL, 6 hCL-Hep-PLCL and 15 hCL-Hep-PLCL samples, respectively (Fig. 4C). Consistent with the qualitative results, the efficiency of heparin immobilization by physical adsorption was lower than that achieved by chemical crosslinking. Furthermore, the heparin content increased with crosslinking time. Our observation agrees with the findings of Wei et al., who reported a similar difference in the adsorbed vs. crosslinked heparin content of chitosan/graphene oxide hydrogels,49 and with those of Singh et al., who reported 1.5 and 7.5 μg heparin per PCL scaffold (discs of 6 × 2 mm) after a crosslinking time of 1 and 15 h, respectively.38 However, although the heparin content and crosslinking time appeared to be directly correlated, as shown in Fig. 5C, the relationship was not linear. The data suggest that the rate of heparin immobilization may be linear during the first few hours but that it may then reach a plateau due to a lack of sufficient binding sites. Since a detailed kinetic study was beyond the scope of the current investigation, based on our observations we used a 6 h crosslinking time as the optimum reaction time for all following experiments.
The changes in the surface topography of the scaffolds before and after heparinization were investigated by AFM using model solvent-cast films. Fig. 5 shows typical topographies and distinct differences between physically and covalently immobilized heparin layers. The topography of the physically adsorbed Hep surface closely follows the topography of the original aminolysed surface (Fig. 5C) reflecting the low amount of Hep adsorbed as evaluated by the toluidine blue assay (Fig. 4C). On the other hand, we observed spherically shaped objects (hills) in the samples that had reacted for 6 h to produce covalently immobilized Hep layers (Fig. 5D); in these samples, an amount of heparin greater than one order of magnitude was deposited. There was also a significant increase in the mean square roughness, Rq. The Rq of Am-PLCL was 2.9 nm, whereas Rq increased to 4.4 nm in the case of 15 hPA-Hep-PLCL and to 41.4 nm in the case of 6 hCL-Hep-PLCL.
On SEM analysis, the aminolysed capsule surface appeared smooth, whereas globular structures were clearly seen in the Hep-PLCL capsules crosslinked for 6 and 15 h (Fig. 6); in the case of CL-Hep-PLCL capsules, we can consider that a gradual increase in the granule size and granule surface density occurred with increasing crosslinking time. Elahi et al. recently reported similar morphological changes in silk fibroin fabric after deposition of polyelectrolyte multilayers (PEMs) followed by EDC/NHS-mediated heparin immobilization; the authors speculated that these changes reflect the non-uniform deposition and distribution of PEMs and heparin on the fabric surface.50,51 Based on our findings and on their similarity to the findings of Elahi et al., we conclude that the observed morphological changes were the result of EDC/NHS-mediated heparin immobilization.
 | ||
| Fig. 6 Morphological changes after heparinization: SEM images suggested that the deposition of heparin was achieved by both cross-linking (CL) and physical adsorption (PA) methods. However, the heparin content was higher in the CL approach with a direct correlation with the CL reaction time. The changes in the surface morphology were clearly visible at a higher magnification, yet the gross morphology of the samples remained largely unaltered. The sample details: (a) the pristine PLCL sample, (b–d) the PLCL samples heparinized via CL for 1, 6 and 15 h respectively, and (e) the sample heparinized via PA for 15 h. | ||
3.4 VEGF immobilization
VEGF is a potent proangiogenic molecule that has been shown to promote infiltration by and proliferation of endothelial cells leading to efficient vascularization of scaffolds.52 Taking advantage of the strong bioaffinity between VEGF and heparin, we immobilized VEGF on the heparinized surfaces of PLCL capsules without using any crosslinkers, i.e., by physical adsorption. To study the capability of modification, VEGF was loaded at two model concentrations, i.e., 0.08 μg mg−1 of scaffold (i.e., in total 2 μg per capsular scaffold; heparin/VEGF_2) and 0.4 μg mg−1 of scaffold (i.e., in total 10 μg per capsular scaffold; heparin/VEGF_10). The success of the VEGF loading was qualitatively determined by IHC staining using an antibody against VEGF. As presented in Fig. 7A, staining was negative in the control sample that lacked VEGF and was positive in the samples loaded with VEGF. The staining intensity was lower in the heparin/VEGF_2 sample than in the heparin/VEGF_10 sample, confirming the difference in VEGF loaded in these two test samples. The amount of VEGF loaded onto the sample evaluated by ELISA was approximately 970 ng in the case of heparin/VEGF_2 and approximately 4200 ng in the case of heparin/VEGF_10; therefore, the loading yield was 97% and 87%, respectively. The loading efficiency appeared to be inversely proportional to the concentration of VEGF. An inverse relationship was also observed by Singh et al. who obtained loadings of approximately 60% and 45% at initial concentrations of 1 and 10 μg per sample, respectively.38 This effect may be due to well-known ligand–receptor binding-related issues such as saturation, competitive binding, and other factors. Nevertheless, the loading obtained in the current study was relatively high compared to the values reported by Singh et al.,38 suggesting that the conditions explored in the current study are optimal for loading.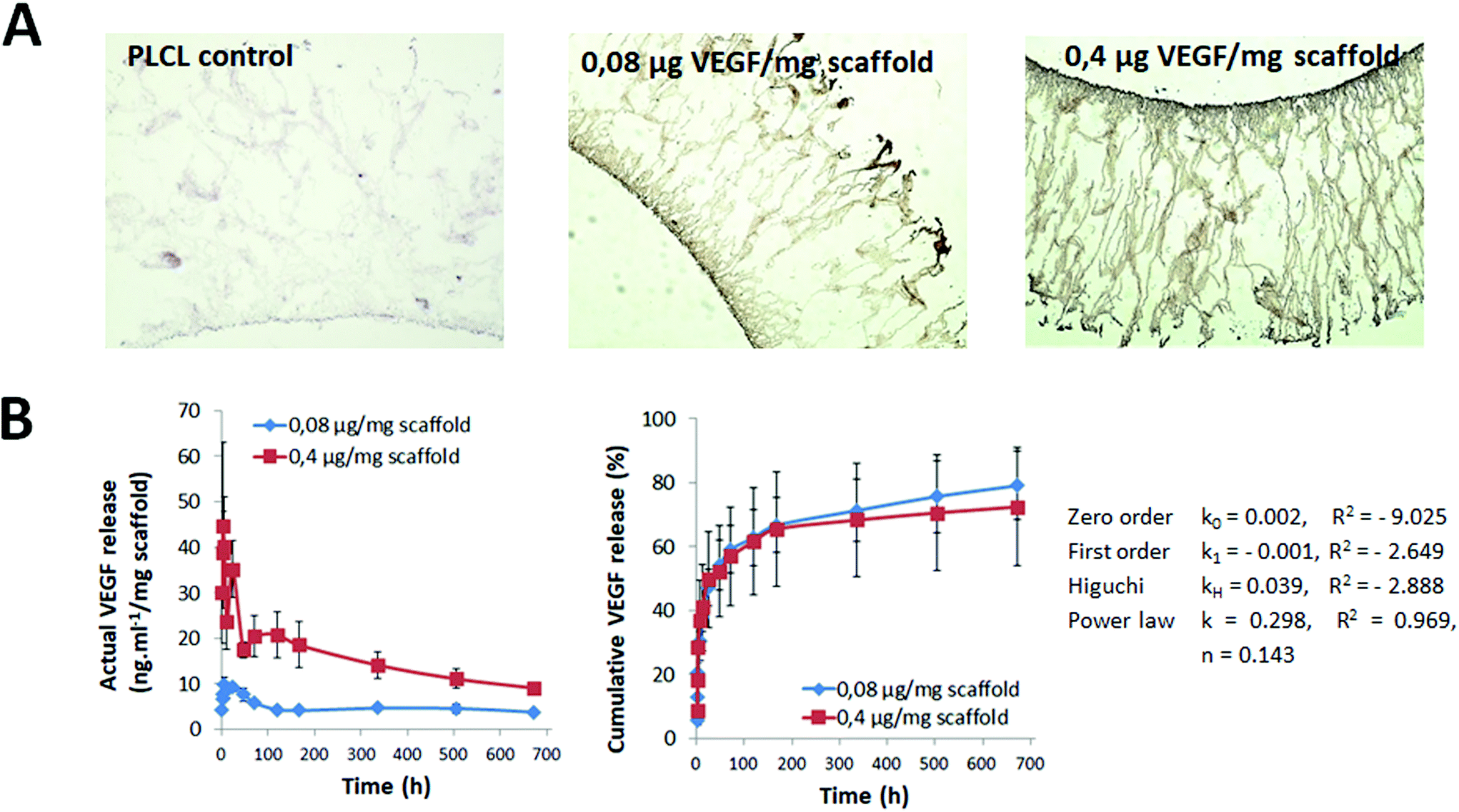 | ||
| Fig. 7 Qualitative and quantitative data on VEGF loading and in vitro release: (A) Anti-VEGF antibody staining was negative in the control PLCL, but positive in heparin/VEGF_2 (0.08 μg mg−1 of scaffold) and heparin/VEGF_10 (0.4 μg mg−1 of scaffold) samples; a difference in the signal intensity was related to the VEGF dose, magnification: 10×; (B) VEGF release kinetics (ELISA); the power law model was found to fit with drug release kinetics data (a value of the R2 parameter is positive and close to 1) and therefore suggested diffusion as the predominant mechanism of the VEGF release. | ||
The in vitro release profile of VEGF under physiological conditions was investigated. For both heparin/VEGF_2 and heparin/VEGF_10 samples, a typical two-phase release profile with a relatively short burst release phase followed by a slow and sustained release phase was observed (Fig. 7B). The actual (absolute) amount of VEGF released varied depending on the amount that was initially loaded; however, the release profile was comparable in both cases. The cumulative release was ∼25% at 3 h. A high initial burst of VEGF release was also reported by Murphy et al. for VEGF-loaded PLGA scaffolds53 and by Singh et al. for VEGF-loaded PCL scaffolds38 and can be explained by the rapid release of weakly bound VEGF molecules especially from the top of the heparin layer. Then the release decreased resulting in the cumulative release of ∼60% after 120 h. Finally, VEGF continued to be released slowly and steadily until the end of the study, i.e., at four weeks, during which ∼25% VEGF retention was observed. The obtained profile also suggests that additional continuous release of a low amount of VEGF may occur for more than 28 days with the cumulative release of approximately 80%. This can be of potential benefit for efficient support of capsule vascularization when the capsule is implanted in vivo. Furthermore, the drug release kinetics data were found to fit the power law model (Fig. 7B). This indicates that VEGF bound to heparinized-PLCL by physical adsorption was released by diffusion.
Based on the results obtained from all optimization studies, the scaffolds for in vivo experiments were first aminolysed at 22 °C for 30 min, then a heparin layer was covalently immobilized via EDC-NHS chemistry for 6 h and finally VEGF was loaded overnight at 4 °C.
3.5 In vivo response
An unlimited supply of oxygen is crucial for beta cell function as a glycaemic sensor and for adequate insulin release. It is also beneficial for islet engraftment. Most studies in which an artificially created pouch is described emphasize the detrimental effect of inflammation caused by the surgery itself and by reaction to the foreign body. Therefore, the maturation of the cavity was optimized according to fully finished healing after scaffold implantation.11,29 Some papers reported promising support of islets with oxygen releasing substances,54 but, despite this enhanced oxygen supply, a large graft to correct glycaemia had to be used.In previous studies, we have tested the in vivo behaviour of pristine PLCL capsules and found that the capsules were infiltrated mostly by fibroblasts;20 preliminary experiments also showed no islet survival and functioning when transplanted in such a cavity (ESI Fig. 3†). Then we also tested the PLCL capsules supplemented with VEGF that should additionally support the formation of a capillary network on the internal surface of the artificial cavity. When the VEGF-loaded scaffolds were implanted subcutaneously in rats, fibrous tissue growth was insufficient and slow, which resulted in the deficiency of tissue to be stimulated by VEGF present within the scaffold. Therefore, the engraftment and long-term survival of transplanted islets was not improved in that model (data not presented). However, the high specific surface of the PLCL scaffolds21 in combination with the attached VEGF holds promise for achieving much better tissue stimulation than that achieved in our previous experiments. Thus, in this study, another site of implantation that is predominantly more compliant to incorporate the scaffold, i.e., in the greater omentum, was tested. As shown previously,20 the greater omentum provides a fast-growing tissue. In addition to that, the drainage of blood coming from the graft to the portal vein is much more physiological; it provides a natural drainage of the graft blood towards a portal vein allowing the first pass effect for insulin. This feature is important especially due to the first pass effect of the liver removing approximately 50% of insulin from blood flowing through.
Scaffold implantation was followed by prompt recovery and did not cause any side effects in experimental animals either immediately after the surgery or during the 28-day post-implantation period. The scaffolds maintained their physical integrity and were effectively engrafted to the host tissues throughout the duration of the experiment (ESI Fig. 4a and b†). A typical sound of a vacuum release generated while removing the Teflon bar from the scaffold lumen indicated the airtightness of the cavity within the scaffold and showed that it had been fully penetrated by connective tissue.
Generally, after 4-week implantation, haematoxylin and eosin (H&E) staining, Masson's trichrome (TRI) staining and special immunohistochemical techniques showed that the fully matured scaffolds were surrounded by a very thin layer of fibrous tissue. Beneath the fibrous capsule, a foreign body-type granulomatous reaction in the presence of mononuclear and multinucleate macrophages was discernible. There were no signs of adhesion or other complications. The connective tissue penetrating the walls of the scaffolds was composed of cellular components and the extracellular matrix. The cellular components consisted predominantly of fibroblasts and myofibroblasts, sporadic inflammatory cells and vascular endothelial cells (ESI Fig. 4c, d and e†). The neovessels were detectable as either thin non-luminized strands of endothelial cells or fully mature branching vascular structures with patent lumina containing red blood cells, lined by CD31 positive endothelial cells and a thin layer of smooth muscle cells. A mild inflammatory reaction involving predominantly small lymphocytes and histiocytes was slightly accentuated along the outer surface of the scaffolds. Importantly, no accumulation of neutrophils and/or eosinophilic leukocytes indicating purulent or hypersensitivity reaction was observed in any animal. The non-cellular component of the connective tissue was composed of small amounts of the ground substance and strands of collagen fibres oriented predominantly radially around the internal cavity with the highest fibre density along the outer surface of each scaffold, reflecting the orientation and size of the pores. Interestingly, no elastin fibres were discernible in the walls of the scaffolds.
 | ||
| Fig. 8 Dose-dependent experiment (evaluation of the VEGF quantity for sufficient stimulation of cell ingrowth and formation of new vessels two weeks after implantation into the greater omentum): histochemical and immuno-histochemical analyses of heparin-PLCL (a–c), heparin/VEGF_10 (d–f; 0.4 μg VEGF per mg scaffold), heparin/VEGF_50 (g–i, 2 μg VEGF per mg scaffold) and heparin/VEGF_100 ((j–l, 4 μg VEGF per mg scaffold) capsules. H&E, TRI and anti-CD31 (black arrows indicate formation of vessels in the scaffold wall) staining; cross-sections, * indicates the internal cavity of the capsule, original magnification: 200×. | ||
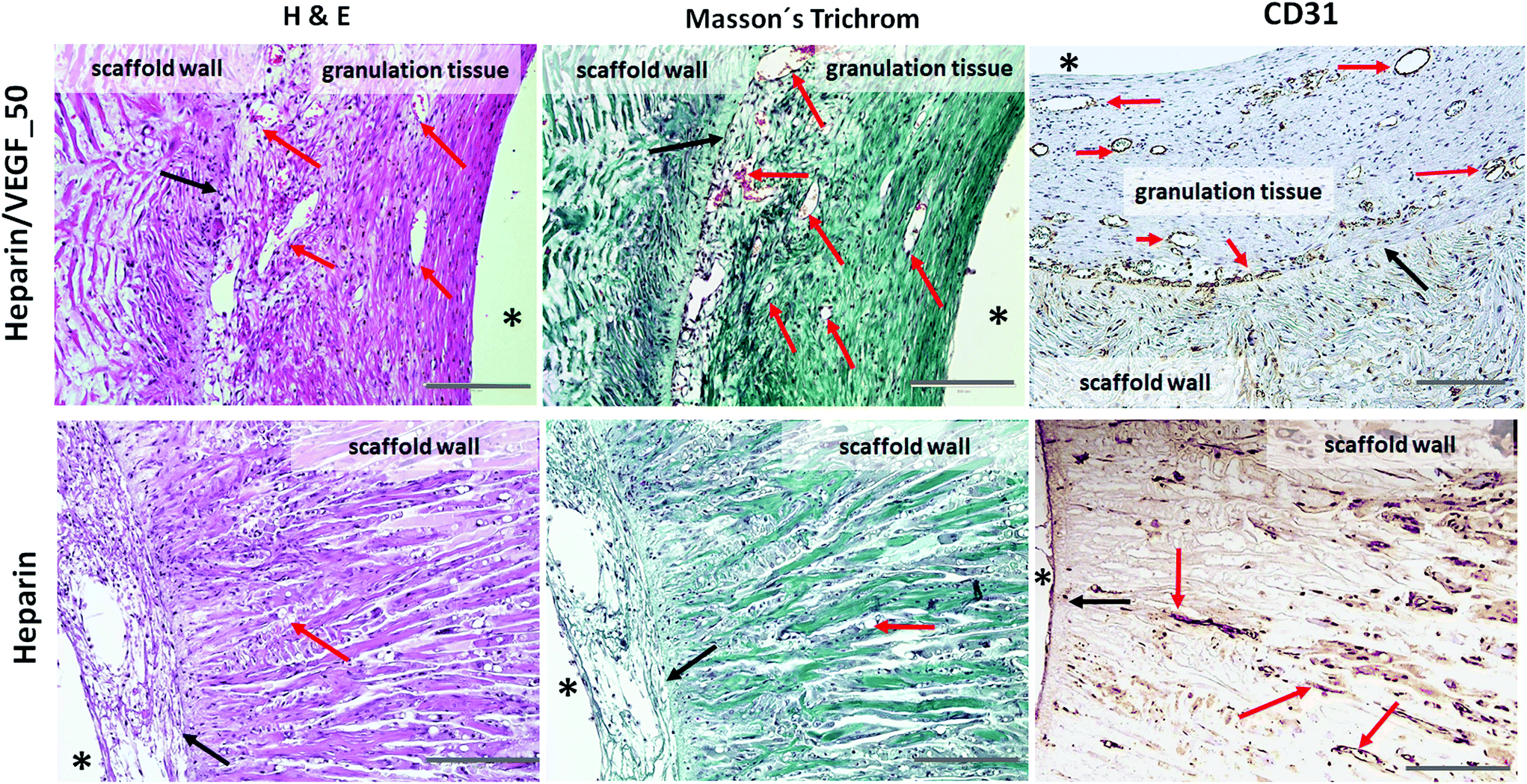 | ||
| Fig. 9 Time-dependent experiment: heparin and heparin/VEGF_50 loaded PLCL capsules four weeks after implantation. The heparin/VEGF_50 coating stimulated the ingrowth of tissue rich in vessels; there is a thin layer of granulation tissue under the internal surface of the scaffold, i.e. inside the cavity intended for islet transplantation. H&E, Masson's Trichrome and anti-CD31 staining; red arrows – neovessels, black arrows – the inner surface of the capsule, * – indicates the internal cavity of the capsule (original magnification ×100, bar 200 μm). | ||
To summarize, both dose- and time-optimization studies showed that the pre-vascularized pouch can be obtained after a 4-week integration of the heparin/VEGF_50 scaffolds with the host tissue.
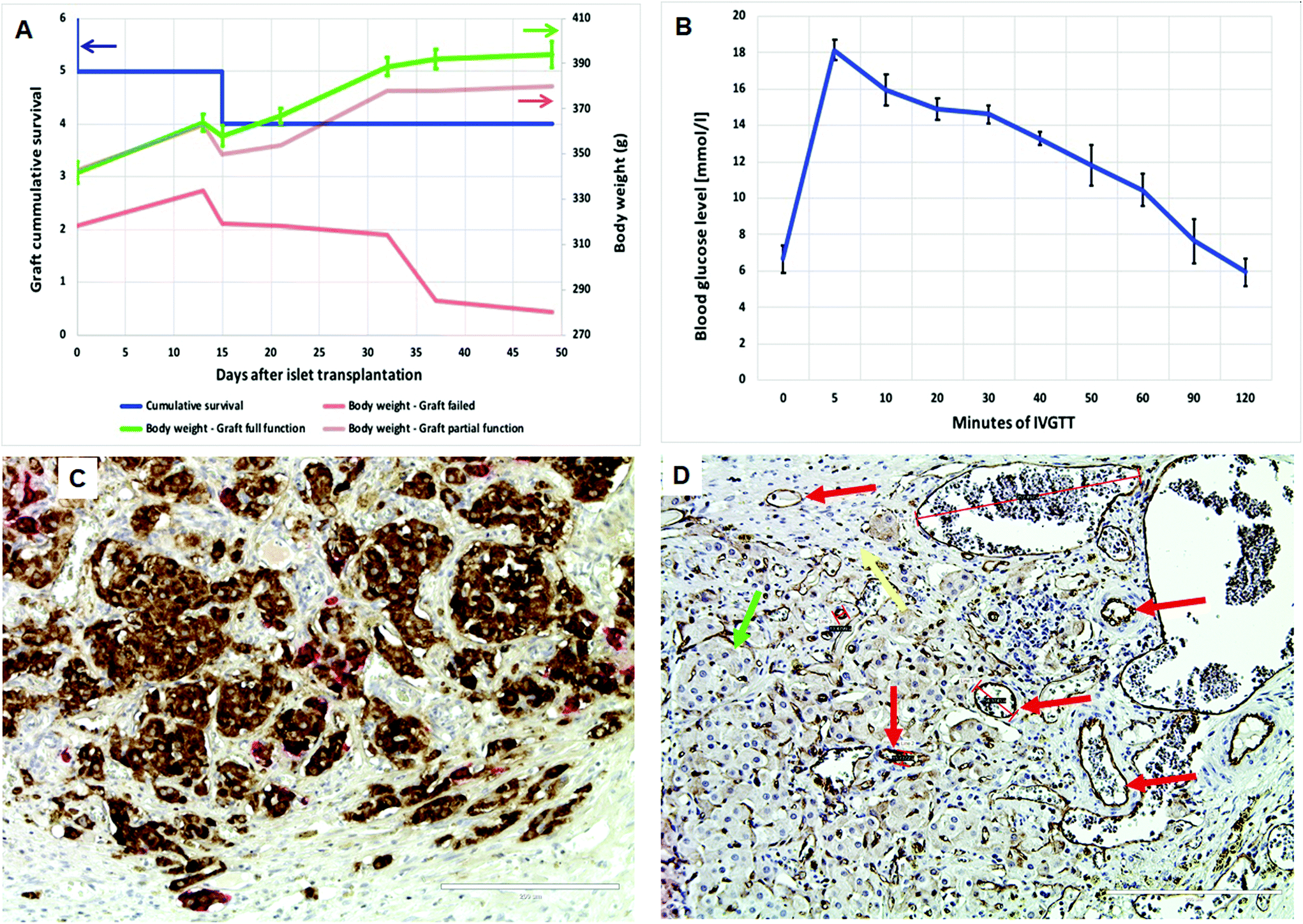 | ||
| Fig. 10 Evaluation of islet survival in a pre-vascularized pouch prepared using the VEGF-loaded PLCL scaffold (heparin/VEGF-50, 4-week pre-implantation): (A) Cumulative survival of islet grafts (the left y-axis, n = 6, blue line). Two grafts failed to function within the first two weeks after transplantation; the body weight of animals (the right y-axis) with a full graft function (green line) and a partial graft function (brown line) increased with time. The body weight in the animal with the non-functional graft (red line) dropped down in 50 days after transplantation. (B) Intravenous glucose tolerance test revealed normal glucose assimilation in animals with a full function of the graft. (C) Immunohistology of the graft seven weeks after transplantation – double anti-insulin (brown color) and anti-glucagon (red color) staining proved the presence of beta-cells and alpha-cells within engrafted islets. (D) Immunohistology of the graft seven weeks after transplantation – anti-CD31 staining proved the presence of endothelial cells in the graft and surrounding tissues. Fibrotic tissue is indicated by yellow arrows, endothelial cells by red arrows and endocrine cells of islets by green arrows. Bar – 200 μm. The negative controls for both immunohistological staining are presented in ESI Fig. 11.† | ||
A pancreatic islet graft normalized the blood glucose levels (under 10 mmol l−1) in five of six diabetic recipients, and in four of them the full function of islets was maintained until the end of the study, i.e., 50 days after islet transplantation (Fig. 10a, the left y-axis and ESI Fig. 9†). The graft did not start to function in one animal, and the function failure appeared in the second animal three weeks after transplantation. Importantly, the body weight of recipients increased in time continuously in the animals with the functioning graft (Fig. 10a, the right y-axis). This observation confirms an adequate insulin secretion by the graft, which prevents the massive loss of glucose (a key nutrient) and stimulates a growth of tissues (as insulin acts also as a powerful growth factor). Furthermore, the intravenous glucose challenge test performed 40 days after islet transplantation confirmed a full function of the graft in four animals (Fig. 10b). This means that the basal insulin secretion can be adequately stimulated by the i.v. administration of glucose (0.5 g kg−1).
Using anti-insulin and anti-glucagon double-staining, large pancreatic islet conglomerates (Fig. 10c and ESI Fig. 10† at lower magnification) rich in vascular networks were detected in all animals. The observed frequency and localization of alpha-cells resemble their distribution in normal islets. This finding indicates that a cellular dedifferentiation of beta-cells resulting from the transplantation procedure likely did not occur. The good function of the grafts indicated that an even distribution within a hosting tissue (as is typical in portal vein transplantation) is not necessary for adequate insulin secretion and graft survival until the end of the study. However, the good function of the graft is strongly related to its efficient reconnection to the recipient bloodstream (capillaries close to the internal surface of the pouch) early after its transplantation. Indeed, the presence of vessels within the graft was proved by anti-CD31 staining of the samples; Fig. 10d shows the luminized blood vessels together in the islet grafts and surrounding tissues as well.
In the present work, we made a strong emphasis on the characterization of all performed surface modifications to verify that the modification processes described for 2D model surfaces can be successfully adapted for real 3D scaffolds of particular size and morphology. We proved that the pro-angiogenic factor VEGF, when used in an appropriate dose, can adequately stimulate microvascular network infiltration through the micro-channelled porous scaffold following the fibroblast ingrowth under conditions of optimal timing. In vivo studies indeed confirm the successful creation of a realistic pre-vascularized bed for cellular transplantation.14,57,58 Using a rodent model, we have shown that the created pre-vascularized pouch can successfully host transplanted islets; the newly formed vascular network inside the cavity led to the graft survival and functioning for at least 50 days. In addition, the chosen rat animal model used in this study makes possible testing of the islet/mass (i.e., islet per kg of body weight) ratio similar to the ratio required for clinical applications. In mice, the device must be very small, and the surface, which can be provided to islets for engraftment, is inadequately small. In clinical practice, the proposed concept would be a promising approach particularly for islet auto-transplantations, which can be precisely scheduled; therefore, islet transplantation can be performed during the optimal phase of healing. We believe that this can be achieved using our approach providing an adequate blood supply to islets at the time of transplantation.
Nevertheless, we expect that further optimization of the coatings with respect to faster cell in-growth through the scaffold wall and application of the “cocktail” of factors supporting vascularization would even more increase the efficiency of islet transplantation and a long-term graft function.
4. Conclusions
We studied the feasibility of adapting modification processes described for planar model surfaces to complex three-dimensional porous scaffolds. We described the 3-step surface functionalization of the PLCL capsules prepared by the Dip-TIPS method and its promising application in bioengineering a realistic pre-vascularized pouch for potential cell transplantation applications.Importantly, all modification steps preserved the anisotropic channelled pore structure of the capsules as confirmed by microscopic analysis. The success of amination and heparinization of the PLCL surface was proven by ATR-FTIR and XPS spectroscopy analysis. Although the decrease in the Mw of the PLCL polymer induced by aminolysis was time- and temperature-dependent and could reach 50%, the chosen conditions resulted in only a 10% Mw decrease and had no effect on the gross capsule morphology. The covalent coupling approach enabled us to vary the amount of heparin immobilized on the capsule surface from 1.08 to 2.39 μg per mg of scaffold with increasing reaction time. Performing the EDC/NHS crosslinking for 6 h led to sufficiently high heparin content while the open character of inner pores in the capsule lumen was maintained. Heparinization led to a significant decrease in wettability from 144° for the pristine capsule to 53°, a more convenient value for cell ingrowth in general. VEGF release studies showed that sustained release that followed the power law-based diffusion kinetics occurred for at least four weeks.
The results of in vivo studies in Lewis rats revealed that the scaffolds coated with heparin or heparin/VEGF implanted in the greater omentum are fully biocompatible. Although heparin itself slightly decreased the rate of ingrowth of fibrous tissue and endothelial cells, with stimulation by VEGF the infiltration of the formed microvascular network was deeper than that previously found in native scaffolds. When an appropriate dose of VEGF was used, a suitable pre-vascularized pouch was obtained after a 4-week implantation. The prepared pouch provided for the islet graft survival and functioning for 50 days after islet transplantation in rats. The proposed concept of creation of the pre-vascularized pouch represents a promising approach for clinical cell transplantation.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The study was supported by Czech Health Research Council, Project No. 16-28254A, the Ministry of Education, Youth and Sports within the National Sustainability Program II (Project BIOCEV-FAR LQ1604) and by the BIOCEV project (CZ.1.05/1.1.00/02.0109), the Ministry of Health Czech Republic – DRO (Institute for Clinical and Experimental Medicine IKEM, IN00023001) and by the Charles University, project GA UK No. 100217.References
- O. Iorio, V. Petrozza, A. De Gori, M. Bononi, N. Porta, G. De Toma and G. Cavallaro, J. Invest. Surg., 2018, 1–8, DOI:10.1080/08941939.2018.1441344.
- T. F. Davies, Thyroid, 2013, 23, 139–141 CrossRef.
- A. Dworzynska, K. Paduszynska and L. Pomorski, Pol. Przegl. Chir., 2018, 90, 22–28 Search PubMed.
- Y. Fan, A. Tajima, S. K. Goh, X. Geng, G. Gualtierotti, M. Grupillo, A. Coppola, S. Bertera, W. A. Rudert, I. Banerjee, R. Bottino and M. Trucco, Mol. Ther., 2015, 23, 1262–1277 CrossRef CAS.
- T. Anazawa, H. Okajima, T. Masui and S. Uemoto, Ann. Gastroenterol. Surg., 2019, 3, 34–42 CrossRef.
- K. Gao, P. Kumar, E. Cortez-Toledo, D. Hao, L. Reynaga, M. Rose, C. Wang, D. Farmer, J. Nolta, J. Zhou, P. Zhou and A. Wang, Stem Cell Res. Ther., 2019, 10, 34 CrossRef CAS.
- S. K. Vishwakarma, A. Bardia, C. Lakkireddy, N. Raju, S. A. B. Paspala, M. A. Habeeb and A. A. Khan, Mater. Sci. Eng., C, 2019, 98, 861–873 CrossRef CAS.
- T. Kunath, A. Natalwala, C. Chan, Y. Chen, B. Stecher, M. Taylor, S. Khan and M. M. K. Muqit, Eur. J. Neurosci., 2019, 49, 453–462 CrossRef.
- P. Assinck, G. J. Duncan, B. J. Hilton, J. R. Plemel and W. Tetzlaff, Nat. Neurosci., 2017, 20, 637–647 CrossRef CAS.
- M. Omami, J. J. McGarrigle, M. Reedy, D. Isa, S. Ghani, E. Marchese, M. A. Bochenek, M. Longi, Y. Xing, I. Joshi, Y. Wang and J. Oberholzer, Curr. Diabetes Rep., 2017, 17, 47 CrossRef.
- A. Pileggi, R. D. Molano, C. Ricordi, E. Zahr, J. Collins, R. Valdes and L. Inverardi, Transplantation, 2006, 81, 1318–1324 CrossRef.
- A. R. Pepper, B. Gala-Lopez, R. Pawlick, S. Merani, T. Kin and A. M. J. Shapiro, Nat. Biotechnol., 2015, 33, 518–523 CrossRef CAS.
- J. Kriz, D. Jirak, T. Koblas, G. Vilk, P. Girman, M. Hajek and F. Saudek, Transplant. Proc., 2011, 43, 3226–3230 CrossRef CAS.
- N. A. Forster, A. J. Penington, A. A. Hardikar, J. A. Palmer, A. Hussey, J. Tai, W. A. Morrison and S. J. Feeney, Islets, 2011, 3, 271–283 CrossRef.
- A. J. Hussey, M. Winardi, X. L. Han, G. P. L. Thomas, A. J. Penington, W. A. Morrison, K. R. Knight and S. J. Feeney, Tissue Eng., Part A, 2009, 15, 3823–3833 CrossRef CAS.
- A. M. Smink, S. R. Li, D. T. Hertsig, B. J. de Haan, L. Schwab, A. A. van Apeldoorn, E. de Koning, M. M. Faas, J. R. T. Lakey and P. de Vos, Transplantation, 2017, 101, E112–E119 CrossRef CAS.
- B. L. Gala-Lopez, A. R. Pepper, P. Dinyari, A. J. Malcolm, T. Kin, L. R. Pawlick, P. A. Senior and A. M. J. Shapiro, CellR4, 2016, 4, e2132 Search PubMed.
- A. R. Pepper, A. Bruni, R. L. Pawlick, B. Gala-Lopez, Y. Rafiei, J. Wink, T. Kin and A. M. Shapiro, Islets, 2016, 8, 186–194 CrossRef CAS.
- M. Najjar, V. Manzoli, M. Abreu, C. Villa, M. M. Martino, R. D. Molano, Y. Torrente, A. Pileggi, L. Inverardi, C. Ricordi, J. A. Hubbell and A. A. Tomei, Biotechnol. Bioeng., 2015, 112, 1916–1926 CrossRef CAS.
- N. Kasoju, D. Kubies, E. Fábryová, J. KříŽ, M. M. Kumorek, E. Sticová and F. Rypáček, Physiol. Res., 2015, 64, S75–S84 CAS.
- N. Kasoju, D. Kubies, M. M. Kumorek, J. Kriz, E. Fabryova, L. Machova, J. Kovarova and F. Rypacek, PLoS One, 2014, 9, e108792 CrossRef.
- F. A. Auger, L. Gibot and D. Lacroix, Annu. Rev. Biomed. Eng., 2013, 15, 177–200 CrossRef CAS.
- N. Kasoju, D. Kubies, T. Sedlacik, O. Janouskova, J. Koubkova, M. M. Kumorek and F. Rypacek, Biomed. Mater., 2016, 11, 015002 CrossRef.
- D. Kubies, F. Rypacek, J. Kovarova and F. Lednicky, Biomaterials, 2000, 21, 529–536 CrossRef CAS.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139–157 CrossRef CAS.
- T. Higuchi, J. Pharm. Sci., 1963, 52, 1145–1149 CrossRef CAS.
- N. Kasoju and U. Bora, J. Biomed. Mater. Res., Part B, 2012, 100, 1854–1866 CrossRef.
- J. Kriz, G. Vilk, D. M. Mazzuca, P. M. Toleikis, P. J. Foster and D. J. G. White, Am. J. Surg., 2012, 203, 793–797 CrossRef.
- G. R. Mitchell and A. Tojeira, Procedia Eng., 2013, 59, 117–125 CrossRef.
- F. E. Kohn, J. G. Van Ommen and J. Feijen, Eur. Polym. J., 1983, 19, 1081–1088 CrossRef CAS.
- S. Noel, B. Liberelle, L. Robitaille and G. De Crescenzo, Bioconjugate Chem., 2011, 22, 1690–1699 CrossRef CAS.
- T. I. Croll, A. J. O'Connor, G. W. Stevens and J. J. Cooper-White, Biomacromolecules, 2004, 5, 463–473 CrossRef CAS.
- Y. Zhu, C. Gao, X. Liu and J. Shen, Biomacromolecules, 2002, 3, 1312–1319 CrossRef CAS.
- Y. Zhu, Z. Mao, H. Shi and C. Gao, Sci. China: Chem., 2012, 55, 2419–2427 CrossRef CAS.
- E. L. de Mulder, G. Hannink, M. J. Koens, D. W. Lowik, N. Verdonschot and P. Buma, J. Biomed. Mater. Res., Part A, 2013, 101, 919–922 CrossRef.
- Y. B. Zhu, C. Y. Gao, T. He, X. Y. Liu and J. C. Shen, Biomacromolecules, 2003, 4, 446–452 CrossRef CAS.
- S. Singh, B. M. Wu and J. C. Dunn, Biomaterials, 2011, 32, 2059–2069 CrossRef CAS.
- A. Akkouch, Z. Zhang and M. Rouabhia, J. Biomed. Mater. Res., Part A, 2011, 96, 693–704 CrossRef.
- B. Yuan, Y. Jin, Y. Sun, D. Wang, J. Sun, Z. Wang, W. Zhang and X. Jiang, Adv. Mater., 2012, 24, 890–896 CrossRef CAS PubMed.
- S. J. Yuan, G. Xiong, X. Y. Wang, S. Zhang and C. Choong, J. Mater. Chem., 2012, 22, 13039–13049 RSC.
- T. Sharkawi, V. Darcos and M. Vert, J. Biomed. Mater. Res., Part A, 2011, 98, 80–87 CrossRef.
- Y. Hong, C. Y. Gao, Y. Xie, Y. H. Gong and J. C. Shen, Biomaterials, 2005, 26, 6305–6313 CrossRef CAS.
- M. J. Collins, S. H. Zeronian and M. L. Marshall, J. Macromol. Sci., Part A, 1991, 28, 775–792 CrossRef.
- M. Drobota, Z. Persin, L. F. Zemljic, T. Mohan, K. Stana-Kleinschek, A. Doliska, M. Bracic, V. Ribitsch, V. Harabagiu and S. Coseri, Cent. Eur. J. Chem., 2013, 11, 1786–1798 CAS.
- Q. H. Zuo, R. Guo, Q. Liu, A. Hong, Y. F. Shi, Q. Kong, Y. X. Huang, L. M. He and W. Xue, Biomed. Mater., 2015, 10, 035008 CrossRef.
- M. Kolar, M. Mozetic, K. Stana-Kleinschek, M. Frohlich, B. Turk and A. Vesel, Materials, 2015, 8, 1526–1544 CrossRef CAS.
- A. P. Khandwekar, D. P. Patil, Y. Shouche and M. Doble, J. Biomater. Appl., 2011, 26, 227–252 CrossRef CAS.
- H. L. Wei, L. L. Han, Y. C. Tang, J. Ren, Z. B. Zhao and L. Y. Jia, J. Mater. Chem. B, 2015, 3, 1646–1654 RSC.
- M. F. Elahi, G. P. Guan, L. Wang and M. W. King, Materials, 2014, 7, 2956–2977 CrossRef CAS.
- M. F. Elahi, G. P. Guan, L. Wang, X. Z. Zhao, F. J. Wang and M. W. King, Langmuir, 2015, 31, 2517–2526 CrossRef CAS.
- Y. H. Shen, M. S. Shoichet and M. Radisic, Acta Biomater., 2008, 4, 477–489 CrossRef CAS.
- W. L. Murphy, M. C. Peters, D. H. Kohn and D. J. Mooney, Biomaterials, 2000, 21, 2521–2527 CrossRef CAS.
- M. M. Coronel, R. Geusz and C. L. Stabler, Biomaterials, 2017, 129, 139–151 CrossRef CAS PubMed.
- G. Marchioli, A. D. Luca, E. de Koning, M. Engelse, C. A. Van Blitterswijk, M. Karperien, A. A. Van Apeldoorn and L. Moroni, Adv. Healthcare Mater., 2016, 5, 1606–1616 CrossRef CAS.
- D. Mao, M. Zhu, X. Zhang, R. Ma, X. Yang, T. Ke, L. Wang, Z. Li, D. Kong and C. Li, Acta Biomater., 2017, 59, 210–220 CrossRef CAS.
- P. Stiegler, V. Matzi, E. Pierer, O. Hauser, S. Schaffellner, H. Renner, J. Greilberger, R. Aigner, A. Maier, C. Lackner, F. Iberer, F. M. Smolle-Juttner, K. Tscheliessnigg and V. Stadlbauer, Xenotransplantation, 2010, 17, 379–390 CrossRef.
- K. Ogawa, K. Asonuma, Y. Inomata, I. Kim, Y. Ikada, Y. Tabata and K. Tanaka, Cell Transplant., 2001, 10, 723–729 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01280j |
| ‡ Current address: Division of Tissue Culture, BMT Wing Sree Chitra Tirunal Institute for Medical Sciences & Technology, Trivandrum, India. |
| This journal is © The Royal Society of Chemistry 2020 |