
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycerylphytate as an ionic crosslinker for 3D printing of multi-layered scaffolds with improved shape fidelity and biological features†
Ana
Mora-Boza
 ab,
Małgorzata K.
Włodarczyk-Biegun
c,
Aránzazu
del Campo
cd,
Blanca
Vázquez-Lasa
*ab and
Julio San
Román
ab
ab,
Małgorzata K.
Włodarczyk-Biegun
c,
Aránzazu
del Campo
cd,
Blanca
Vázquez-Lasa
*ab and
Julio San
Román
ab
aInstitute of Polymer Science and Technology, ICTP-CSIC, Juan de la Cierva 3, 28006 Madrid, Spain
bCIBER-BBN. Health Institute Carlos III, C/Monforte de Lemos 3-5, Pabellón 11, 28029 Madrid, Spain
cINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany
dChemistry Department, Saarland University, 66123 Saarbrücken, Germany
First published on 5th November 2019
Abstract
The fabrication of intricate and long-term stable 3D polymeric scaffolds by a 3D printing technique is still a challenge. In the biomedical field, hydrogel materials are very frequently used because of their excellent biocompatibility and biodegradability, however the improvement of their processability and mechanical properties is still required. This paper reports the fabrication of dual crosslinked 3D scaffolds using a low concentrated (<10 wt%) ink of gelatin methacryloyl (GelMA)/chitosan and a novel crosslinking agent, glycerylphytate (G1Phy) to overcome the current limitations in the 3D printing field using hydrogels. The applied methodology consisted of a first ultraviolet light (UV) photopolymerization followed by a post-printing ionic crosslinking treatment with G1Phy. This crosslinker provides a robust framework and avoids the necessity of neutralization with strong bases. The blend ink showed shear-thinning behavior and excellent printability in the form of a straight and homogeneous filament. UV curing was undertaken simultaneously to 3D deposition, which enhanced precision and shape fidelity (resolution ≈150 μm), and prevented the collapse of the subsequent printed layers (up to 28 layers). In the second step, the novel G1Phy ionic crosslinker agent provided swelling and long term stability properties to the 3D scaffolds. The multi-layered printed scaffolds were mechanically stable under physiological conditions for at least one month. Preliminary in vitro assays using L929 fibroblasts showed very promising results in terms of adhesion, spreading, and proliferation in comparison to other phosphate-based traditional crosslinkers (i.e. TPP). We envision that the proposed combination of the blend ink and 3D printing approach can have widespread applications in the regeneration of soft tissues.
Introduction
Native organs and tissues are complex and highly organized structures. The fabrication of three-dimensional (3D) scaffolds which reproduce these intricate geometries is of great interest for tissue engineering and regenerative medicine. Traditional fabrication techniques, such as freeze drying or solution casting, render random macro- and microstructures with poor control over the final architecture.1 Porosity is a key parameter in the development of tissue-like structures: the pore size and distribution determine the colonization by cells, and their distribution within the scaffold.2,3 3D printing has arisen as a promising tool for the development of scaffolds with complex and well defined geometries, as it allows layer-by-layer fabrication of 3D constructs with flexible selection of customized geometries, sizes and materials.1,4,5 Hydrogels are preferred materials for 3D printing because of their excellent biocompatibility and biodegradability. However, their high water content leads to poor processability in 3D printing methodologies.6 Moreover, their intrinsic softness is insufficient for self-supporting of the printed structures.1,4,7–10 A current trend to overcome these limitations is to combine different materials that can together fulfil the essential requirements for good printability.11–14 Such properties are: (i) shear-thinning behaviour while printing, (ii) mechanical stability for maintaining shape fidelity after printing, (iii) good structural integrity under physiological conditions, and (iv) cytocompatibility.15 Extensive efforts have been recently made in the field to develop crosslinking processes that can stabilize the scaffold immediately after printing, such as photocuring of methacrylated polymers.4,10,16Gelatin hydrogels have been widely used for 3D printing in medical applications. Gelatin is a denatured form of collagen that has several advantages. Gelatin shows less antigenicity compared to collagen, but it maintains in the backbone the RGD peptide sequences for cell attachment, and the matrix metalloproteinase-sensitive degradation domains, typical of collagen. Gelatin is commonly used in the tissue engineering and regenerative medicine fields due to its low cost and easy processability.16 However, its gelation kinetics is too slow to be efficient for the printing process. Therefore, GelMA has been extensively used in the last few years.6,11,13,15–20 Methacrylation allows fast covalent crosslinking in the presence of a photoinitiator and light exposure.16,21 Methacrylation does not affect the RGD domains and allows the synthesis of materials with tunable mechanical properties.21,22 Chitosan is a natural polysaccharide that can promote tissue regeneration through the activation of inflammatory and fibroblast cells.23–26 Chitosan supports cell proliferation and differentiation better than alginate, the quintessential printable material.23 However, its use in 3D printing has been limited due to its weak mechanical properties.27,28 In the last few years, a few studies have reported the use of chitosan for 3D printing.5,7,23,29–31 Wu et al. studied different chitosan-based inks by dissolving the chitosan in a mixture of different acids. Gelation of printed scaffolds was achieved by post-printing immersion in NaOH solution, which neutralized the amine groups of chitosan and reduced its solubility. However, the authors did not assess the biological response of the printed structures.7 Demirtas et al. developed a bioprintable form of chitosan by adding β-glycerolphosphate to the ink, which provided thermosensitiveness to the system. In this case, the scaffold demonstrated favourable biological features, but the 3D printed structures showed poor shape fidelity.23 Therefore, the development of successful strategies to overcome current limitations in the 3D printing field using hydrogels is demanded. The present work proposes the use of the crosslinker glycerylphytate (G1Phy) developed by our group32 in the fabrication of dual crosslinked 3D scaffolds using a low concentrated (<10 wt%) ink of GelMA/chitosan. Although other studies have been focused on the combination of gelatin with chitosan because of their ability to form together polyelectrolyte complexes,15,24,25,33 the blend ink composed of GelMA/chitosan has not be reported so far.
G1Phy plays a key role since it provides robust networks and avoids the necessity of neutralization and washing steps.32 The as-obtained 3D printed structures exhibit good printability, adequate mechanical properties and long-term stability. Thus, our approach involves a two-step crosslinking process that combined UV photopolymerization of GelMA followed by post-printing ionic crosslinking with G1Phy. Two-step 3D printing approaches, which usually consist of the combination of GelMA photopolymerization and ionic crosslinking processes, have been widely applied in the 3D printing field.11,34–36 In these studies, ionic crosslinking is commonly first applied followed by photocuring of GelMA. In the present work, simultaneous deposition and photopolymerization of the 3D structures has been performed. This approach improves resolution and shape fidelity without the necessity of using sacrificial polymers or template agents, which are common techniques applied to water-based ink solutions.11,37,38 This approach and the incorporation of a novel crosslinker such as G1Phy for subsequent ionic gelation not only provides appropriate processability properties to the scaffolds but also bioactive properties32 in comparison to traditional alginate–Ca2+ ionic crosslinking systems frequently applied in dual-step 3D printing technology.
In this work, 3D scaffolds printed with a pneumatic-based 3D printer show excellent shape fidelity (resolution ≈150 μm). Ionic post-treatment mediated by G1Phy, a hybrid derivative of phytic acid of reduced toxicity, provides a fast and homogeneous ionic crosslinking between phosphate groups present in G1Phy and amine groups of chitosan and GelMA which is crucial for long-term stability properties of the crosslinked polymeric networks. Since 3D printing technology aims to mimic intricate structures and geometries with high resolution, control over stability and swelling properties are essential for cell culture and tissue regeneration in the field of hydrogel 3D printing. Finally, preliminary in vitro results of the 3D printed scaffolds crosslinked with G1Phy using L929 fibroblasts display favourable biological performance in terms of biocompatibility, cell proliferation, and cytocompatibility.
Experimental
Materials
Chitosan powder (with a degree of deacetylation of 90% and Mw = 300 kDa) was purchased from Altakitin (São Julião do Tojal, Portugal) and used as received. Gelatin from porcine skin (type A, ∼300 bloom), methacrylic anhydride (MA), poly(ethylene glycol)dimethacrylate (PEGDMA, Mn 20 kDa), Irgacure2959, sodium tripolyphosphate (TPP), Triton, and Dulbecco's phosphate-buffered saline (DPBS) were purchased from Sigma-Aldrich (St Louis, MO, USA). Bovine serum albumin (BSA) was purchased from PAN-Biotech and paraformaldehyde (PFA) from Electron Microscopy Sciences (Hatfield, PA, USA). The G1Phy crosslinker was synthesized in our lab as published elsewhere.32 The dispensing tips were purchased from VIEWEG GmbH Dosier- und Mischtechnik (Kranzberg, Germany) and dispensing Optimum® cartridges from Nordson (Erkrath, Germany).Ink design and preparation
GelMA was synthesized by adapting a previously reported method.39 5 g of gelatin were dissolved in 50 mL of DPBS at 50 °C and stirred for 30 min until completely dissolved. 8 mL of MA were added gradually to the solution and the reaction was allowed to proceed for 3 h at 50 °C. The reaction was stopped by adding 150 mL of DPBS. The final solution was dialyzed against distilled water (MWCO 3.5 kDa) at 40 °C for 7 days. The resulting product was freeze-dried and stored at 4 °C in a dark container. The degree of methacrylation (70%) was calculated by 1H-nuclear magnetic resonance (NMR) in D2O at 37 °C (ref. 40) (Fig. S1†).For the preparation of the polymeric ink, GelMA was dissolved at different concentrations (2 to 5 wt%) in distilled water at 1% (v/v) of acetic acid and 1 wt% of PEGDMA at 40 °C. Chitosan powder was added to the solution to obtain different concentrations (1 to 4 wt%) in the final ink volume. Irgacure 2959 was used as a photoinitiator and was added to the ink solutions at a final concentration of 0.5% (w/v). The ink solutions were stirred at 40 °C for 3 h under dark conditions to obtain a homogeneous solution and transferred to 10 mL volume cartridges. Finally, the cartridges were centrifuged for 5 min at 800 rpm to remove air bubbles.
The viscosity of the ink solutions was measured with a rotational rheometer (DHR3, TA Instruments, USA) in oscillatory mode by increasing the shear rate from 1 to 1000 s−1 at 22 °C using a stainless-steel parallel Peltier plate geometry (12 mm diameter) with a solvent trap. The photocrosslinking reaction was followed on the same rheometer during in situ illumination using a parallel plate geometry (20 mm) at room temperature (22 °C), and a UV light source OmniCure S2000 (Excelitas Technologies, Ontario, Canada). The UV light source was previously calibrated with a UV meter. The UV intensity was 50 mW cm−2 and a 365 nm filter was used.
3D printing methodology
The 3D printing approach is summarized in Fig. 1. A pneumatic extrusion 3D printer (BioScaffolder 3.2, GeSiM, Germany) was used and the scaffolds were printed in 6 well-plates. Metal straight needles with an inner diameter of 150 μm and 200 μm were used. Printing speed and air pressure were adjusted to 5 mm s−1 and 500 kPa, respectively, for a 150 μm tip, and 8 mm s−1 and 400 kPa for a 200 μm tip.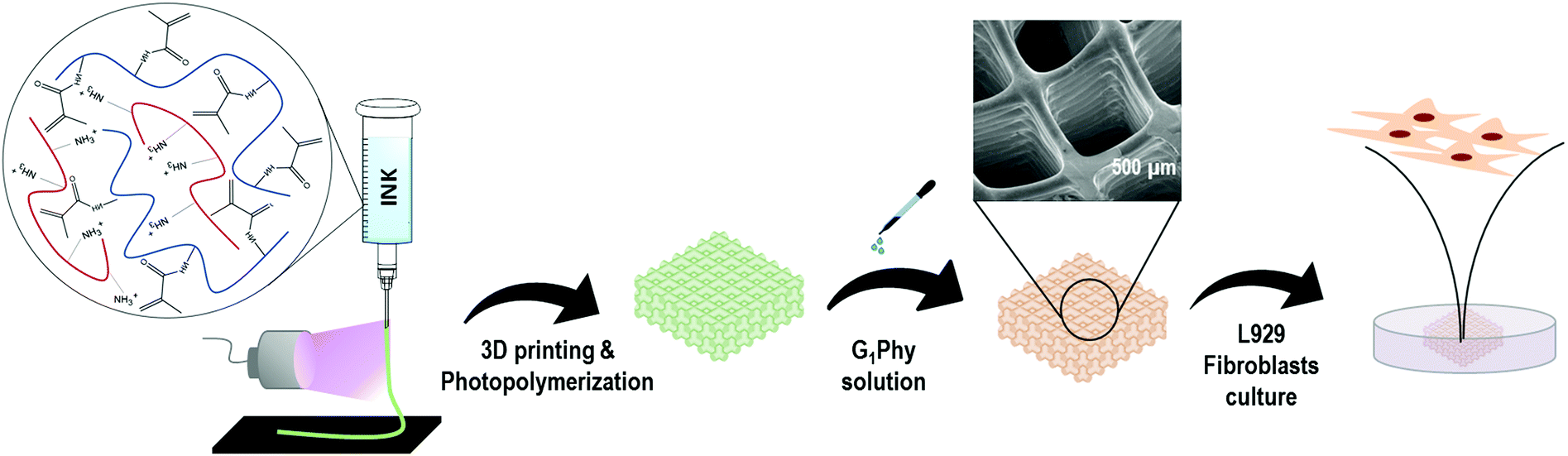 | ||
| Fig. 1 3D printing approach applied for GelMA/chitosan 3D polymeric scaffolds. | ||
A dual-step crosslinking methodology was used. In the first step, simultaneously with material deposition, UV light (OmniCure S1500, Excelitas Technologies, Ontario, Canada; filter: 320–500 nm, 50 mW cm−2) was applied to initiate photopolymerization of GelMA in each layer of the scaffold. The light was focused on the needle outlet. After printing, the 3D scaffold was immersed for 5 min in a G1Phy crosslinker solution (15 mg mL−1, 16.3 mM) for ionic crosslinking of the amine groups present in chitosan and GelMA. The scaffolds were washed once with distilled water to remove the excess crosslinker prior to further experiments. During printing, the temperatures in the cartridge and in the printer stage were maintained at 40 °C and 15 °C, respectively. Printing parameters (air pressure, printing speed and layer height) were adjusted depending on the needle used. For comparison, printed scaffolds were also crosslinked with TPP crosslinker solution (15 mg mL−1, 40.7 mM).
3D scaffold characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. The quantification of P was performed with an ICP-OES Ultima 2 (HORIBA, Kyoto, Japan). A conical nebulizer at 2.15 bar and 0.81 l min−1 flow was used, and the analytical wavelength for P was 214.914 nm. The measurements were conducted in triplicate for each sample and the data obtained were expressed as mean values ± standard deviations (SD).
:10. The quantification of P was performed with an ICP-OES Ultima 2 (HORIBA, Kyoto, Japan). A conical nebulizer at 2.15 bar and 0.81 l min−1 flow was used, and the analytical wavelength for P was 214.914 nm. The measurements were conducted in triplicate for each sample and the data obtained were expressed as mean values ± standard deviations (SD).
Energy Dispersive X-ray (EDX) analysis of P, C, N and O contents was performed with a Bruker XFlash model with a 5030 detector. P maps were recorded for all fabricated samples.
Light microscopy characterization of the samples was done using an Olympus (Hamburg, Germany) SZX16 stereomicroscope under opaque illumination.
For cryo-scanning electron microscopy (SEM) evaluation, the samples were cut in 1 cm × 2 cm pieces and dried carefully with a Kimtech Science (Kimberly-Clark, Koblenz, Germany) tissue before plunging in liquid ethane at −165 °C using a Gatan (Pleasanton, CA, USA) CP3 cryo plunger. The samples were transferred under liquid nitrogen to a self-made cryo-SEM holder and placed onto the stage of a FEI (Hillsboro, OR, USA) Quanta 400 FEG SEM. Cryo-SEM observation was performed under high vacuum conditions using accelerating voltage between 1.5 and 5 keV. Depending on the amount of ice generated at the sample surface during the preparation process, the samples were freeze-dried carefully inside the SEM before taking secondary electron images using an Everhart–Thornley detector (ETD).
The degradation rate was calculated gravimetrically by drying and weighing the scaffolds after incubation in PBS at 37 °C for different time points (2, 4, 7, 10, 20 and 30 days). The measurements were conducted in triplicate for each sample and the data obtained were expressed as mean values ± standard deviations (SD).
Crosslinker release was followed by measuring the content of P in the supernatant after incubating the scaffolds in medium at increasing times. G1Phy and TPP samples were incubated in Tris-HCl buffer at pH 7.4 and 37 °C in order to avoid interference between P in the sample and in PBS. Aliquots were taken at 2, 4, 7, 10 and 14 days of incubation and the P content was determined by ICP-OES. The measurements were conducted in triplicate for each sample and the data obtained were expressed as mean values ± standard deviations (SD).
Biological behaviour
:1000 vinculin rabbit antibody (Thermo Fisher) solution for cytoskeleton labelling and 1:200 Alexa fluor-546 phalloidin (Thermo fisher) solution for focal adhesion staining in red color for ∼1 h. Then, they were rinsed twice with PBS and incubated with secondary antibody (Alexa flour-488 goat antirabbit, Thermo Fisher) 1:500 solution to visualize the cytoskeleton in green. Finally, the samples were stained with 1:1000 DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride, Thermo Fisher) solution for nuclei visualization in blue color and washed with PBS (Sigma). Imaging was performed using a Nikon Ti-Eclipse (Nikon Instruments Europe B.V., Germany) with a Sola SE 365 II (Lumencor Inc., Beaverton, USA) solid state illumination device and an Andor Clara CCD camera for detection.
Results
2D printability and rheological evaluation of GelMA/chitosan polymeric inks
The printability, i.e. the extrusion of a continuous thread through the printer needle, of GelMA/chitosan mixtures with different compositions was evaluated first (Fig. 2a). Monocomponent GelMA solutions at concentrations below 5 wt% were low viscous and formed droplet-like structures when printed (Fig. 2a). Note that GelMA solutions at concentration ≥12.5 wt% are printable.6 The addition of chitosan at concentration <2 wt% into GelMA solutions increased the viscosity of the solution and allowed the printing of mixed droplet and filament structures. Smooth filaments were deposited when chitosan concentration in the mixture was increased from 2 wt% to 4 wt%, at total polymer concentrations between 6 wt% and 8 wt%, respectively, and GelMA/chitosan ratios ≥1 (Fig. 2a). These observations indicated that the addition of chitosan to GelMA improved the printability of GelMA at polymer concentrations >4 wt%. The addition of higher concentrated solutions of chitosan (>5 wt%) was not further investigated as the resulting inks had too high viscosity to be extruded. The composition 4 wt% chitosan and 4 wt% GelMA was selected for further studies and fabrication of 3D scaffolds because continuous, smooth and uniform filaments could be easily extruded (Fig. 2a). Better shape fidelity was also obtained in comparison to other printable formulations (Fig. 2b). The high content of amino groups due to the high chitosan content (4 wt%) was positively taken into consideration because they can contribute to the ionic crosslinks for stabilization of the final scaffold. Fig. 2c shows the viscosity of the blend ink compared to the viscosity of the monocomponent solutions of gel and chitosan at 4 wt%. A clearly shear-thinning behavior was observed for the mixture at the studied shear rates. The zero-shear viscosity of the GelMA/chitosan 4/4 w/w% ink was higher than the zero-viscosity of the tested individual components, which is expected because of the higher polymer content.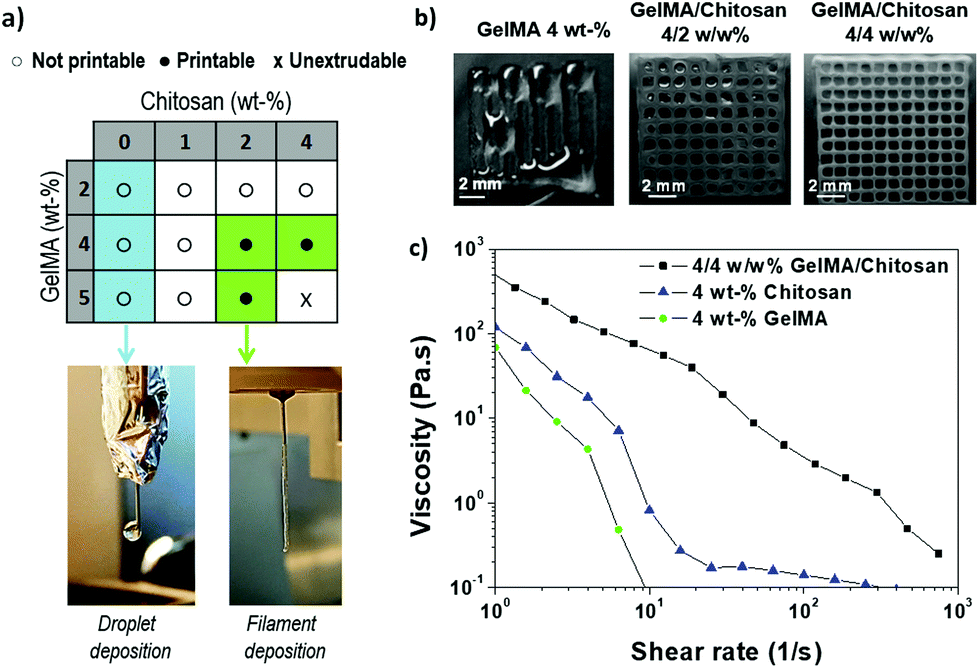 | ||
| Fig. 2 (a) Printability of GelMA/chitosan inks with different polymer concentrations. Blue boxes indicate droplet-like deposition and green boxes indicate filament deposition; the photographs show the two types of depositions. (b) Light microscopy pictures of 3D scaffolds printed with GelMA 4 wt%, GelMA/chitosan 4/2 w/w% and GelMA/chitosan w/w% mixtures; (c) viscosity measurements for 4 wt% GelMA (green circles), 4 wt% chitosan (blue triangles), and mixture (black squares). | ||
Optimization of printing parameters for improved shape fidelity and 3D printing
A dual-step crosslinking approach (Fig. 1) was used for printing the 4/4 w/w% GelMA/chitosan mixtures. The first crosslinking involves the radical polymerization of the acrylate groups and it occurs during ink deposition upon exposure to UV light. The light dose is a relevant parameter that determines the kinetics of the gelation process and the stability of the filament after extrusion. Using a power density of 50 mW cm−2, a stable filament that retained its shape was obtained, and no obstruction of the tip occurred during printing. Lower exposure doses led to widespread strands and collapse between consecutive printed layers. A parallel rheological study of the UV-crosslinked solution indicated that the shear modulus of the UV-crosslinked mixture was G′ = 20 kPa after full exposure (Fig. S2†). Adjustment of the printing parameters was necessary for each tip diameter. 150 and 200 μm metallic needles required printing speed and air pressure adjusted to 5 mm s−1 and 500 kPa, and 8 mm s−1 and 400 kPa, respectively. Under these conditions, good shape fidelity and resolution were achieved during printing and the fabrication of scaffolds with several superposed layers was possible. It is worth pointing out that adherence between layers can be compromised when consecutive printed layers are fully photopolymerized. The optimization of printing height allowed better contact between consecutively printed layers and delamination was avoided. Layer heights of 80 and 100 μm were used when printing with 150 and 200 μm needles, respectively. Scaffolds of up to 28 layers were obtained without collapse or delamination.The second crosslinking step involves ionic interactions and is performed after printing. The 3D scaffolds were immersed in a 15 mg mL−1 solution of G1Phy crosslinker. The phosphate groups of G1Phy are expected to form ionic crosslinks with the amine groups of chitosan and GelMA. This ionic crosslink forms instantaneously and permits fast gel formation and consolidation of structural integrity. In addition, this step provides long-term stability and allows the tuning of the degradation kinetics of the 3D hydrogel structures. Fig. 3 shows two 3D printed scaffolds: with and without ionic treatment. The dual crosslinked scaffold shows higher shape fidelity and controlled swelling, which were lost when the ionic post-treatment was skipped. This result indicates that both crosslinking processes, photopolymerization and ionic crosslinking, contribute to print 3D scaffolds with structural integrity and long-term stability. This methodology allowed fast printing without using supporting templates21,36,41–45 or neutralization and washing steps.5,7,24,28,30,46 The as-printed scaffolds were ready to be used for biological tests.
 | ||
| Fig. 3 Visual examination and structural integrity changes that a photochemically 3D printed scaffold (four layers) immediately experienced after immersion in distilled water containing or not the G1Phy or TPP. Scale bar is 0.5 cm. | ||
The resolution of the 3D printed scaffolds fabricated with GelMA/chitosan 4/4 w/w% composition was characterized by microscopy. The grid structures showed regular edges and corners (Fig. 4a–d), threads with uniform width (∼150 μm), very close to the inner diameter of the used needle (= 150 μm) (Fig. 4b). It is important to note that only a few studies in the last few years have reported strand diameters smaller than 200 μm for 3D printed hydrogel-based materials.14 Cryo-SEM evaluation confirmed the structural integrity of the printed scaffolds and the interconnectivity between pores without internal collapse of the 3D structures (Fig. 4e–g). A maximum of 28 consecutive layer scaffolds was printed with self-standing properties and good adherence between layers (Fig. 4f). Finally, the 3D polymeric scaffolds showed inherent porosity in their microstructure (Fig. 4h).
 | ||
| Fig. 4 Light microscopy pictures of a 4-layer GelMA/chitosan scaffold with quadrangular pore morphology (a and b) and angular geometries (c and d) at low (a and c) and higher (b and d) resolution. In (b) the local width of the printed strand at three different positions is shown (150 μm needle). Images (e–h) show cryo-SEM micrographs of 3D printed scaffolds of 28 layers with quadrangular geometry (e and f), and 4 layers with angular geometry (g). In (h) the microstructure of the printed hydrogel is shown. | ||
3D scaffold characterization
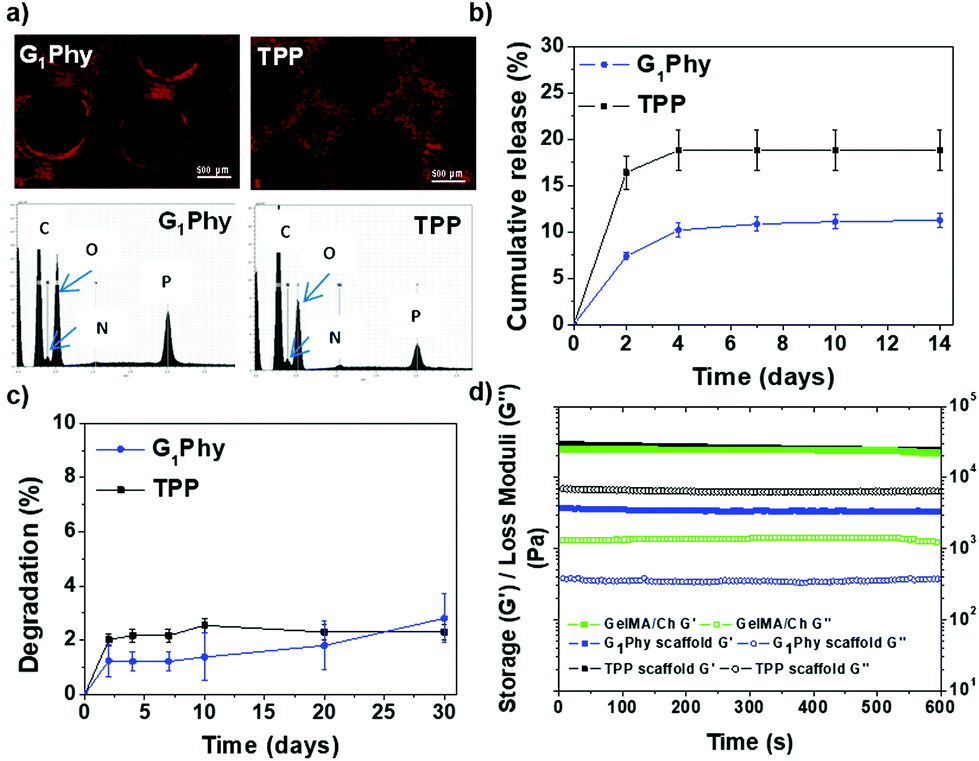 | ||
| Fig. 5 (a) EDAX map distribution of P in G1Phy and TPP scaffolds, accompanied by EDAX spectra; (b) cumulative release crosslinker profiles for G1Phy and TPP scaffolds incubated in Tris-HCl buffer at pH 7.4 and 37 °C until 14 days; (c) degradation (%) for crosslinked scaffolds incubated in PBS at pH 7.4 and 37 °C until 30 days; (d) storage and loss moduli for UV-crosslinked GelMA/Ch gel and G1Phy and TPP scaffolds. | ||
The crosslinker release of the scaffold under physiological conditions (pH 7.4, 37 °C) was studied (Fig. 5b). G1Phy showed a fast release (7.3%, which corresponded to 10.32 μM) during the first 2 days of incubation, reaching a plateau after 4 days (11.2%, which corresponded to 16.03 μM), which was maintained until 30 days of incubation. TPP showed a burst release (16.4%, which corresponded to 14.74 μM) at 2 days. A plateau was achieved at 4 days (18.8%, which corresponded to 16.31 μM), which was sustained until the end of the incubation period. This different performance of the release crosslinkers can be associated with the differences in the distribution of the crosslinkers in the scaffold, and different ionic interactions of the crosslinkers with the polymeric backbones.
Swelling behaviour of the scaffolds was examined by measuring the strand widths by light microscopy (Fig. S3†). No swelling was observed for printed G1Phy nor TPP crosslinked scaffolds after incubation in PBS under physiological conditions for 10 days. This indicated that the structures achieved their maximum water absorption after ionic crosslinking. The stability of the scaffolds was followed gravimetrically by measuring the weight loss after different incubation times (Fig. 5c). G1Phy crosslinked scaffolds showed a weight loss of 1.2 ± 0.6% during the first 2 days. The scaffolds remained stable for the following 10 days and degraded slowly up to 2.8 ± 0.9% during the next 20 days. At short time points (2 days), TPP crosslinked scaffolds showed a relatively higher degradation rate (2.0 ± 0.2%) than G1Phy scaffolds and degradation progressively increased until 10 days (2.5 ± 0.3%), to remain stable up to the end of the experiment (30 days). At the final time no significant (p < 0.5) differences between G1Phy and TPP samples were observed.
Rheological characterization of UV cured GelMA/chitosan discs after crosslinking with G1Phy or TPP was performed to compare the viscoelastic properties of the 3D printed structures as a function of the type of ionic crosslinker. Fig. 5d shows the shear and loss moduli for crosslinked discs. G1Phy crosslinked discs exhibited lower storage modulus than TPP crosslinked discs and GelMA/Ch photocrosslinked gel. The storage modulus values of TPP crosslinked scaffolds approached those of GelMA/Ch photocrosslinked gel but with higher loss modulus than this last one.
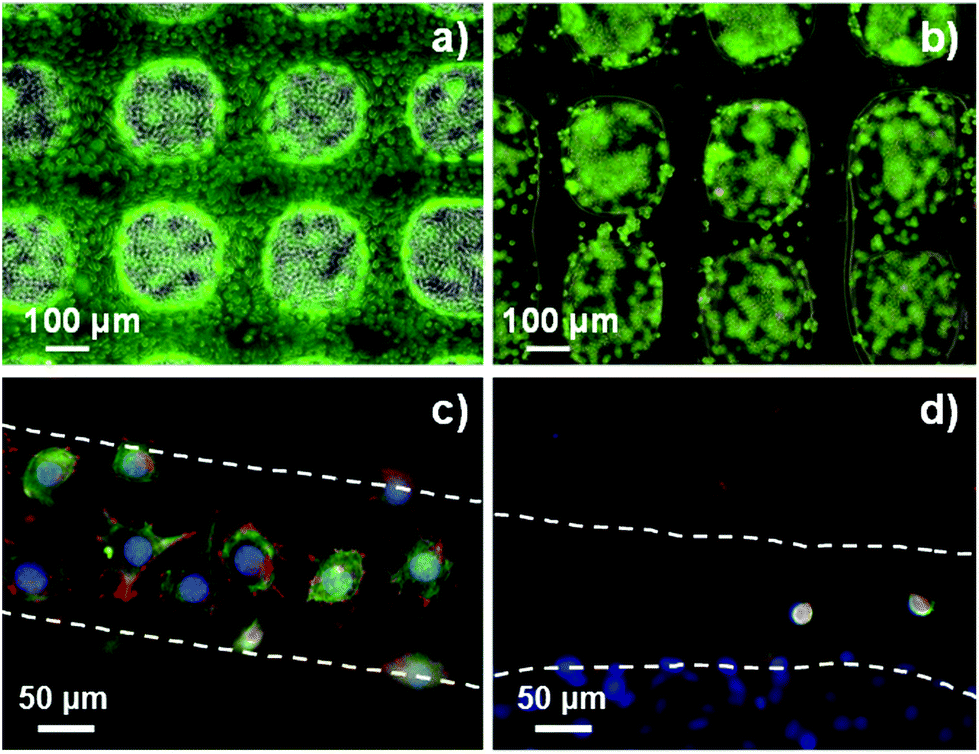 | ||
| Fig. 6 Live/dead assay on G1Phy (a) and TPP (b) scaffolds after 24 h of incubation. Pictures were taken of scaffold strands; confocal immunostaining assay performed on G1Phy (c) and TPP (d) scaffolds after 24 h of incubation. | ||
The proliferation of L929 fibroblasts on the scaffolds was quantified using the Alamar Blue assay at days 2, 4, 7 and 10 of incubation. L929 proliferation on G1Phy scaffolds increased over time and the proliferation level was at any time higher than that on TPP scaffolds (Fig. 7a). Finally, the possible cytotoxic effects caused by the release of crosslinkers or low molecular weight residues from the 3D printed scaffolds was examined by incubating L929 fibroblasts with the supernatants from the scaffolds after 2, 4, 7 and 10 days of soaking in cell culture medium. No cytotoxic effects were observed from any of the 3D printed scaffolds (Fig. 7b).
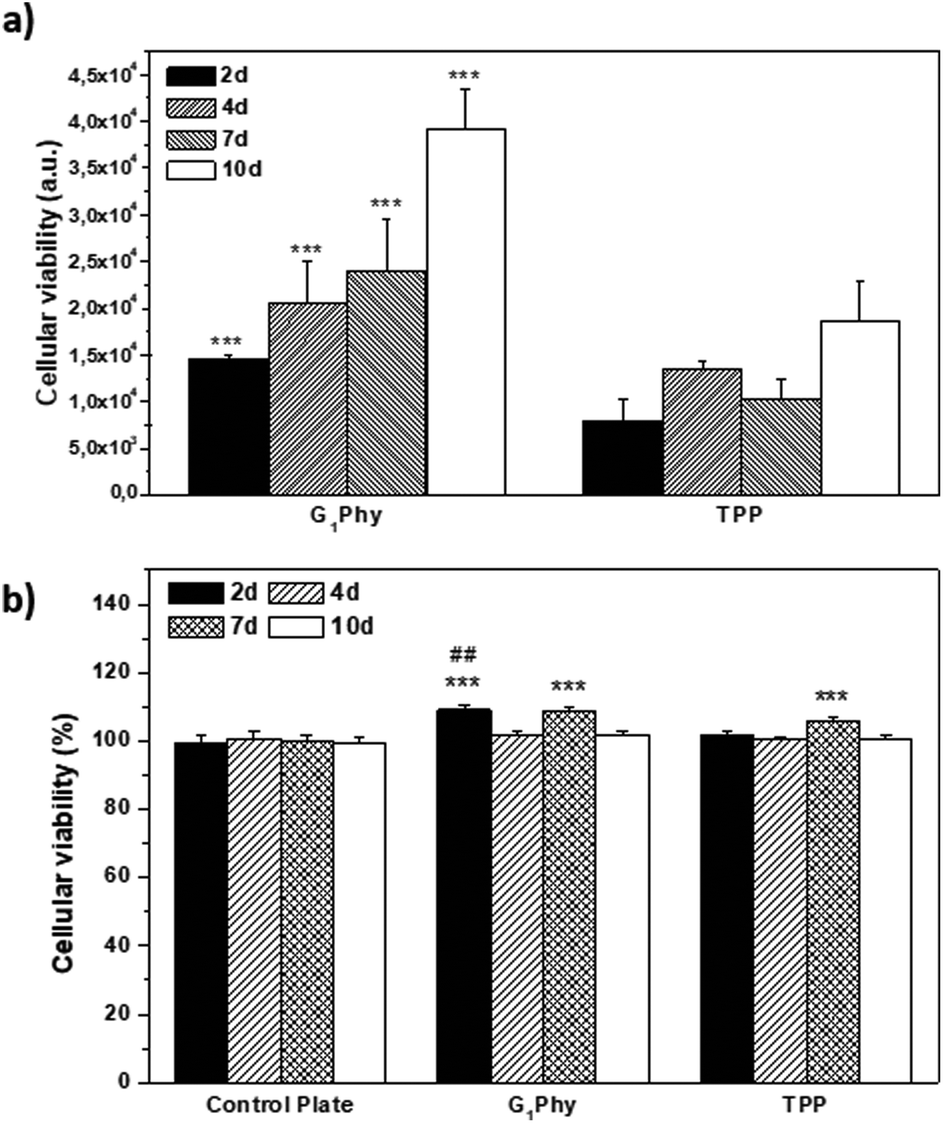 | ||
| Fig. 7 (a) Alamar Blue assay performed on G1Phy and TPP scaffolds at 2, 4, 7 and 10 days of incubation. Analysis of variance (ANOVA) of the results for G1Phy samples was performed with respect to TPP samples at each time at a significance level of ***p < 0.001; (b) MTT assay performed with extracts of G1Phy and TPP scaffolds at 2, 4, 7 and 10 days. Control plate refers to cells incubated in DMEM. Analysis of variance (ANOVA) of the results for G1Phy and TPP samples was performed with respect to the control plate at each time at a significance level of ***p < 0.001, and for G1Phy samples with respect to TPP samples at each time at a significance level of ##p < 0.01. | ||
Discussion
Among all requirements that an ink must fulfil to allow successful printing homogeneity and deposition as a uniform filament, shear-thinning behaviour, and appropriate viscosity are key features.23 In addition, the 3D printed structures should be biocompatible and maintain structural integrity under physiological conditions, avoiding delamination during and after printing.15,47 Blend inks that combine different types of polymers are commonly used to fulfil all the mechanical and biological requirements.48 We decided to work with a mixture of polysaccharide (chitosan) and protein (GelMA) hydrogel. We expected that the GelMA/chitosan combination provides a printing ink with the biological advantages of both polymers, and this mixture overcomes the weak mechanical properties of chitosan.15 Previous studies have reported the use of gel/chitosan blend inks.2,15,24,25,33,49 However, the mixture of chitosan with GelMA, which would result in superior biological properties and mechanical stability, has not been explored before.Parameter optimization was performed to identify suitable printability windows for the GelMA/chitosan ink. Wu et al. reported that 3D printing of chitosan using polymer concentrations below 10 wt% was difficult because of the low viscosity of the polymer solution.7 On the other hand, other studies reported that it is possible to print scaffolds using a low polymeric content, even with good cytocompatibility.50,51 For example, Yin et al. reduced the total polymer concentration of the ink to 8/5 w/w% GelMA/gel mixture, and claimed that the obtained shape fidelity was similar to those found with 30 wt% GelMA solutions.6 In this work, we have developed a chitosan-based ink with low polymer concentration to favour cell growth, but with appropriate viscosity for free-standing filament dispensing. Among the tested formulations (green box Fig. 2a), the GelMA/chitosan 4/4 wt% mixture showed the optimal property combination of low polymer content and easy printability as a uniform straight filament. The 8 wt% polymer content falls within the range of published studies using gel/chitosan blends,25,49 but since GelMA is used in our work, full comparison cannot be performed. Finally, scaffolds fabricated with this composition could be printed with high shape fidelity and maintain structural integrity after printing (Fig. 4). Briefly, the selected composition of chitosan and GelMA polymers had a synergistic effect, leading to better printability in comparison to each of the monocomponent polymers and made possible the deposition of a printed ink hydrogel with superior shape fidelity.
The optimization of printing parameters was done hand-in-hand with the formulation of the ink and the adjustment of the crosslinking parameters. A dual crosslinking process was applied (Fig. 1): a first step based on covalent UV crosslinking while printing and a second step based on ionic crosslinking after material deposition. The dual-step approach is commonly used for processing inks at low polymer concentrations.6 The GelMA/chitosan 4/4 w/w% ink showed shear-thinning behaviour, which is a key property for successful deposition using extrusion-based 3D printing.15,23 In addition, the photopolymerization kinetics of the ink was suitable for the fabrication of 3D structures at a speed of 5–8 mm s−1 depending on the used tip, with high printing fidelity and without the collapse of consecutive layers. Rheological tests of the crosslinked material demonstrated the viscoelastic properties and hydrogel formation, because storage modulus was constant and higher than loss modulus (Fig. 5d).
The simultaneous photocrosslinking and 3D deposition allowed the direct photocrosslinking of individual layers, avoiding common complications associated with post-printing photocrosslinking as a consequence of low light penetration into the 3D structure.6 It should be noted that simultaneous crosslinking might lead to a gradient in exposure dose across the layers. However, the post-printing characterization did not show differences in dimensions or swelling between individual layers. To date, a high number of 3D printing reports use post-printing photopolymerization,11,15–18,21,22,27,45,47 while only a few studies have used simultaneous UV curing during printing.6,39,52,53 Yin et al., who performed a detailed study of GelMA/gel ink printability, also used the simultaneous photopolymerization and 3D deposition approach applying the same UV light intensity (50 mW cm−2) which is applied in this work. Although comparisons are difficult since 3D printing approaches and ink composition differed somewhat, in our work a lower total polymer content (8 vs. 13 wt%) can be printed with better filament resolution (150 vs. 260 μm). Nevertheless, it is important to consider that a rigorous comparison of these results depends deeply on 3D printing parameters and used polymers.
The second crosslinking step took place after the immersion of the 3D printed scaffolds in G1Phy as the ionic crosslinker. G1Phy crosslinked scaffolds exhibited better biocompatibility than commonly used ionic crosslinking agents (i.e. TPP).54 In addition, the organic segment incorporated into the phytic acid structure favours interaction with cells.32 This second crosslinking step contributed to maintained shape fidelity and positively influenced swelling and long-term stability of the scaffolds (Fig. 4b). We speculate that the crosslinking of the amine groups of the chitosan and GelMA chains with the anionic crosslinker is essential to control the water uptake of the printed structures. This stabilization step did not require neutralization or washing steps, which is the common approach used for 3D printing of chitosan-based inks. For example, Wu et al. printed 10 wt% chitosan solutions obtaining 3D structures with high resolution (∼30 μm) and intricate shapes, but neutralization with 1 M NaOH solution for 4 h was required.7 Elviri et al. were able to print lower chitosan solutions (6 wt%) in a cryogenic chamber followed by the subsequent coagulation in a KOH (8 w/v%) bath.30 Moreover, the described manufacturing protocols could potentially lead to shrinking and shape deformation of the scaffolds,5,7,24,28,30,55 a post-printing phenomenon that it was not observed in our scaffolds after ionic crosslinking (Fig. S4†). Our polymer concentration is among the range of published ones (±2 wt%), while it avoids such sophisticated post-processing techniques, being more similar to the traditional alginate–CaCl2 system widely applied in 3D printing methodologies.29
The dual step approach followed in our work also avoided the use of sacrificial or template materials, which is a widely used strategy to improve printing resolution with hydrogels.11,37,38 G1Phy crosslinking led to improved mechanical properties of the scaffolds compared to TPP crosslinking. We expect G1Phy to form less compact and softer networks because of the two main reasons: (i) the organic content of this crosslinker provides some viscoelasticity to the network, and (ii) its higher molecular weight with respect to TPP provides less dense frameworks. All these features together contributed to obtaining softer and viscoelastic gels with long-term stability and controlled swelling, which are essential characteristics for tissue engineering applications.1,10,56 To the best of our knowledge, crosslinkers derived from phytic acid have not been used for the development of 3D printed scaffolds so far.
The 3D structures demonstrated excellent shape fidelity. The ink did not accumulate in the edges or in the corners of scaffolds, where printing speeds and direction change abruptly. The scaffolds showed smooth surfaces and constant widths. Moreover, printing of multiple consecutive layers without collapsing was possible, leading to the structures of up to 28 layers height. The best reported line width for hydrogel printing was approximately 100–200 μm, which is among the best resolution degree that can be currently achieved with hydrogel inks.14 This resolution is highly dependent on the diameter of the needle, and could be potentially improved using narrower tips. Porous and interconnected structures were printed, which are interesting geometries for tissue engineering applications.14,57 Finally, the inherent microstructure (Fig. 4h) of the polymeric scaffolds is also a decisive property for the correct distribution and diffusion of oxygen and nutrients of the ingrowth tissues.58,59 Summarizing, the developed GelMA/chitosan hydrogel with a dual crosslinking mechanism allows the printing of 3D structures with complex designs at high resolution.
Cytocompatibility studies indicated that G1Phy scaffolds supported better cell attachment and proliferation than TPP crosslinked scaffolds, which could be due to the chemical composition as well as morphological properties of these scaffolds. Nevertheless, since the initial composition (GelMA/chitosan) is the same for both crosslinked scaffolds and assuming that the chitosan surface exposure is rather similar in both types of scaffolds according to P distribution (Fig. 5a), for short time periods, the different biological responses should only be due to the incorporation of G1Phy or TPP into their structures. G1Phy has demonstrated to be a highly biocompatible crosslinker which exhibits an organic composition that can enhance cellular interaction.32 In addition, the different molecular weights of G1Phy and TPP could play a role in the final mechanical properties of the ionically crosslinked networks. In fact, the rheological behaviour showed that the G1Phy crosslinked polymer network was softer than the TPP crosslinked one. All this together with the sustained release of G1Phy and the slower degradation of this scaffold can favourably contribute to the higher cell proliferation of the samples in long-term periods.
Conclusions
In summary, this work shows the implementation and optimization of a 3D printing methodology using the novel G1Phy crosslinker. The methodology consisted of a dual-step crosslinking that allows the 3D printing of low concentrated GelMA/chitosan based-ink (total polymer concentration <10 wt%). This approach permitted the fabrication of 3D hydrogel scaffolds with excellent shape fidelity and resolution. The 3D printed scaffolds displayed long-term stability and excellent properties regarding swelling behaviour, and mechanical and biological properties. In particular, the use of the G1Phy crosslinker enhanced cell adhesion and proliferation on the 3D scaffolds in comparison to TPP, widely used as a traditional ionic crosslinking agent. These results open a door for the extrusion of hydrogel-based inks employing phytic acid derived crosslinkers for the fabrication of complex structures with excellent biological properties that can be used in soft tissue engineering applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank financial support from Ministry of Science, Innovation and Universities (Spain) (MAT2017-2017-84277-R), “La Caixa” Foundation (ID 100010434, scholarship of Ana Mora-Boza, code LCF/BQ/ES16/11570018) and DAAD Research Grants-Short-term grants 2017. Support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI) is also acknowledged.The authors are indebted to Dr Marcus Koch (Leibniz-INM) for excellent technical assistance with SEM and light microscopy experiments, and to Dr Claudia Fink-Straube (Leibniz-INM) for ICP-OES experiment performance.
References
- A. V. Do, B. Khorsand, S. M. Geary and A. K. Salem, Adv. Healthcare Mater., 2015, 4, 1742–1762 CrossRef CAS PubMed.
- J. Huang, H. Fu, Z. Wang, Q. Meng, S. Liu, H. Wang, X. Zheng, J. Dai and Z. Zhang, RSC Adv., 2016, 6, 108423–108430 RSC.
- Q. L. Loh and C. Choong, Tissue Eng., Part B, 2013, 19, 485–502 CrossRef CAS.
- M. K. Wlodarczyk-Biegun and A. Del Campo, Biomaterials, 2017, 134, 180–201 CrossRef CAS PubMed.
- C. Intini, L. Elviri, J. Cabral, S. Mros, C. Bergonzi, A. Bianchera, L. Flammini, P. Govoni, E. Barocelli, R. Bettini and M. McConnell, Carbohydr. Polym., 2018, 199, 593–602 CrossRef CAS PubMed.
- J. Yin, M. Yan, Y. Wang, J. Fu and H. Suo, ACS Appl. Mater. Interfaces, 2018, 10, 6849–6857 CrossRef CAS PubMed.
- Q. Wu, D. Therriault and M.-C. Heuzey, ACS Biomater. Sci. Eng., 2018, 4, 2643–2652 CrossRef CAS.
- R. Levato, W. R. Webb, I. A. Otto, A. Mensinga, Y. Zhang, M. van Rijen, R. van Weeren, I. M. Khan and J. Malda, Acta Biomater., 2017, 61, 41–53 CrossRef CAS PubMed.
- S. Stratton, N. B. Shelke, K. Hoshino, S. Rudraiah and S. G. Kumbar, Bioact. Mater., 2016, 1, 93–108 CrossRef PubMed.
- S. Derakhshanfar, R. Mbeleck, K. Xu, X. Zhang, W. Zhong and M. Xing, Bioact. Mater., 2018, 3, 144–156 CrossRef PubMed.
- W. Jia, P. S. Gungor-Ozkerim, Y. S. Zhang, K. Yue, K. Zhu, W. Liu, Q. Pi, B. Byambaa, M. R. Dokmeci, S. R. Shin and A. Khademhosseini, Biomaterials, 2016, 106, 58–68 CrossRef CAS PubMed.
- K. Schutz, A. M. Placht, B. Paul, S. Bruggemeier, M. Gelinsky and A. Lode, J. Tissue Eng. Regener. Med., 2017, 11, 1574–1587 CrossRef PubMed.
- H. Stratesteffen, M. Kopf, F. Kreimendahl, A. Blaeser, S. Jockenhoevel and H. Fischer, Biofabrication, 2017, 9, 045002 CrossRef PubMed.
- X. Wang, C. Wei, B. Cao, L. Jiang, Y. Hou and J. Chang, ACS Appl. Mater. Interfaces, 2018, 10, 18338–18350 CrossRef CAS PubMed.
- H. Li, Y. J. Tan, S. Liu and L. Li, ACS Appl. Mater. Interfaces, 2018, 10, 11164–11174 CrossRef CAS PubMed.
- B. J. Klotz, D. Gawlitta, A. Rosenberg, J. Malda and F. P. W. Melchels, Trends Biotechnol., 2016, 34, 394–407 CrossRef CAS PubMed.
- N. Celikkin, S. Mastrogiacomo, J. Jaroszewicz, X. F. Walboomers and W. Swieszkowski, J. Biomed. Mater. Res., Part A, 2018, 106, 201–209 CrossRef CAS PubMed.
- W. Liu, M. A. Heinrich, Y. Zhou, A. Akpek, N. Hu, X. Liu, X. Guan, Z. Zhong, X. Jin, A. Khademhosseini and Y. S. Zhang, Adv. Healthcare Mater., 2017, 6, 1601451 CrossRef PubMed.
- N. A. Chartrain, C. B. Williams and A. R. Whittington, Acta Biomater., 2018, 74, 90–111 CrossRef CAS PubMed.
- M. Layani, X. Wang and S. Magdassi, Adv. Mater., 2018, 30, e1706344 CrossRef PubMed.
- K. Yue, G. Trujillo-de Santiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini, Biomaterials, 2015, 73, 254–271 CrossRef CAS PubMed.
- Y. L. Cheng and F. Chen, Mater. Sci. Eng., C, 2017, 81, 66–73 CrossRef CAS PubMed.
- T. T. Demirtas, G. Irmak and M. Gumusderelioglu, Biofabrication, 2017, 9, 035003 CrossRef PubMed.
- W. L. Ng, W. Y. Yeong and M. W. Naing, Int. J. Bioprint., 2016, 2(1), 53–62 CAS.
- W. L. Ng, W. Y. Yeong and M. W. Naing, Procedia CIRP, 2016, 49, 105–112 CrossRef.
- I. C. Carvalho and H. S. Mansur, Mater. Sci. Eng., C, 2017, 78, 690–705 CrossRef CAS PubMed.
- J. Radhakrishnan, A. Subramanian, U. M. Krishnan and S. Sethuraman, Biomacromolecules, 2017, 18, 1–26 CrossRef CAS PubMed.
- I. H. Liu, S. H. Chang and H. Y. Lin, Biomed. Mater., 2015, 10, 035004 CrossRef PubMed.
- A. R. Akkineni, T. Ahlfeld, A. Lode and M. Gelinsky, Biofabrication, 2016, 8, 045001 CrossRef PubMed.
- L. Elviri, R. Foresti, C. Bergonzi, F. Zimetti, C. Marchi, A. Bianchera, F. Bernini, M. Silvestri and R. Bettini, Biomed. Mater., 2017, 12, 045009 CrossRef PubMed.
- D. Lee, J. P. Park, M. Y. Koh, P. Kim, J. Lee, M. Shin and H. Lee, Biomater. Sci., 2018, 6, 1040–1047 RSC.
- A. Mora-Boza, M. L. López-Donaire, L. Saldaña, N. Vilaboa, B. Vazquez-Lasa and J. S. Román, Sci. Rep., 2019, 9, 11491 CrossRef PubMed.
- A. Zolfagharian, A. Kaynak, S. Y. Khoo and A. Z. Kouzani, 3D Print. Addit. Manuf., 2018, 5, 138–150 CrossRef.
- M. M. Fares, E. Shirzaei Sani, R. Portillo Lara, R. B. Oliveira, A. Khademhosseini and N. Annabi, Biomater. Sci., 2018, 6, 2938–2950 RSC.
- J. Visser, B. Peters, T. J. Burger, J. Boomstra, W. J. A. Dhert, F. P. W. Melchels and J. Malda, Biofabrication, 2013, 5, 035007 CrossRef PubMed.
- A. Tamayol, A. H. Najafabadi, B. Aliakbarian, E. Arab-Tehrany, M. Akbari, N. Annabi, D. Juncker and A. Khademhosseini, Adv. Healthcare Mater., 2015, 4, 2146–2153 CrossRef CAS PubMed.
- P. Datta, B. Ayan and I. T. Ozbolat, Acta Biomater., 2017, 51, 1–20 CrossRef CAS PubMed.
- M. Muller, J. Becher, M. Schnabelrauch and M. Zenobi-Wong, J. Visualized Exp., 2013, e50632, DOI:10.3791/50632.
- C. D. O'Connell, C. Di Bella, F. Thompson, C. Augustine, S. Beirne, R. Cornock, C. J. Richards, J. Chung, S. Gambhir, Z. Yue, J. Bourke, B. Zhang, A. Taylor, A. Quigley, R. Kapsa, P. Choong and G. G. Wallace, Biofabrication, 2016, 8, 015019 CrossRef PubMed.
- M. Bartnikowski, R. Wellard, M. Woodruff and T. Klein, Polymers, 2015, 7, 1539 CrossRef.
- Y. Zhang, Y. Yu and I. T. Ozbolat, J. Nanotechnol. Eng. Med., 2013, 4(2), 0210011–0210017 Search PubMed.
- Y. S. Zhang, K. Yue, J. Aleman, K. M. Moghaddam, S. M. Bakht, J. Yang, W. Jia, V. Dell'Erba, P. Assawes, S. R. Shin, M. R. Dokmeci, R. Oklu and A. Khademhosseini, Ann. Biomed. Eng., 2017, 45, 148–163 CrossRef PubMed.
- I. S. Kinstlinger and J. S. Miller, Lab Chip, 2016, 16, 2025–2043 RSC.
- J. S. Lee, J. M. Hong, J. W. Jung, J. H. Shim, J. H. Oh and D. W. Cho, Biofabrication, 2014, 6, 024103 CrossRef PubMed.
- M. Costantini, J. Idaszek, K. Szöke, J. Jaroszewicz, M. Dentini, A. Barbetta, J. E. Brinchmann and W. Święszkowski, Biofabrication, 2016, 8(3), 035002 CrossRef PubMed.
- J. Zhang, B. J. Allardyce, R. Rajkhowa, Y. Zhao, R. J. Dilley, S. L. Redmond, X. Wang and X. Liu, ACS Biomater. Sci. Eng., 2018, 4, 3036–3046 CrossRef CAS.
- S. Sayyar, S. Gambhir, J. Chung, D. L. Officer and G. G. Wallace, Nanoscale, 2017, 9, 2038–2050 RSC.
- K. W. M. Boere, M. M. Blokzijl, J. Visser, J. E. A. Linssen, J. Malda, W. E. Hennink and T. Vermonden, J. Mater. Chem. B, 2015, 3, 9067–9078 RSC.
- K. D. Roehm and S. V. Madihally, Biofabrication, 2017, 10, 015002 CrossRef PubMed.
- J. Malda, J. Visser, F. P. Melchels, T. Jungst, W. E. Hennink, W. J. Dhert, J. Groll and D. W. Hutmacher, Adv. Mater., 2013, 25, 5011–5028 CrossRef CAS PubMed.
- I. Pepelanova, K. Kruppa, T. Scheper and A. Lavrentieva, Bioengineering, 2018, 5(3), 55 CrossRef CAS PubMed.
- X. Cui, G. Gao, T. Yonezawa and G. Dai, J. Visualized Exp., 2014,(88), e51294 Search PubMed.
- G. Gao, A. F. Schilling, K. Hubbell, T. Yonezawa, D. Truong, Y. Hong, G. Dai and X. Cui, Biotechnol. Lett., 2015, 37, 2349–2355 CrossRef CAS PubMed.
- HERA, 2003.
- A. Vitale and J. T. Cabral, Materials, 2016, 9(9), 760 CrossRef PubMed.
- K. N. Bardakova, T. S. Demina, E. A. Grebenik, N. V. Minaev, T. A. Akopova, V. N. Bagratashvili and P. S. Timashev, IOP Conference Series: Materials Science and Engineering, 2018347 Search PubMed, conference 1.
- J. Wang, Z. Nor Hidayah, S. I. A. Razak, M. R. A. Kadir, N. H. M. Nayan, Y. Li and K. A. M. Amin, Compos. Interfaces, 2018, 1–14, DOI:10.1080/09276440.2018.1508266.
- P. Thangavel, B. Ramachandran and V. Muthuvijayan, J. Biomed. Mater. Res., Part B, 2016, 104, 750–760 CrossRef CAS PubMed.
- K. Zhang, Y. Fan, N. Dunne and X. Li, Regener. Biomater., 2018, 5, 115–124 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01271k |
| This journal is © The Royal Society of Chemistry 2020 |