
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-residue enantioselective determination of emerging drug contaminants in seawater by solid phase extraction and liquid chromatography-tandem mass spectrometry†
Katie
McKenzie
a,
Colin F.
Moffat
ab and
Bruce
Petrie
 *a
*a
aSchool of Pharmacy and Life Sciences, Robert Gordon University, Aberdeen, AB10 7GJ, UK. E-mail: b.r.petrie@rgu.ac.uk; Tel: +44 (0)1224 262824
bOffice of the Chief Scientific Advisor Marine, Scottish Government, Marine Laboratory, 375 Victoria Road, Aberdeen, AB11 9DB, UK
First published on 15th May 2020
Abstract
This study proposes a new multi-residue enantioselective method for the determination of emerging drug contaminants in sea water by solid phase extraction (SPE) and liquid chromatography-tandem mass spectrometry (LC-MS/MS). To achieve satisfactory enantiomeric separation with a vancomycin stationary phase it was essential to limit sodium chloride in extracted samples to <1 μg per injection. This was achieved through a straightforward SPE method using a 50 mL water wash volume and analyte elution in acetonitrile. A Chiral-V enantioselective column (150 × 2.1 mm; 2.7 μm particle size) operated in polar ionic mode enabled simultaneous drug separations in 30 minutes. Analytes with enantioresolution ≥1 were the stimulants amphetamine and methamphetamine, the beta-agonist salbutamol, the beta-blockers propranolol, sotalol and acebutolol, the anti-depressants fluoxetine, venlafaxine, desmethylvenlafaxine and citalopram, and the antihistamine chlorpheniramine. Method quantitation limits were <10 ng L−1 and method trueness was 80–110% for most analytes. The method was applied to samples from the Forth and Clyde estuaries, Scotland. Chiral drugs were present at concentrations in the range 4–159 ng L−1 and several were in non-racemic form (enantiomeric fraction ≠ 0.50) demonstrating enantiomer enrichment. This emphasises the need for further enantiospecific drug exposure and effect studies in the marine environment.
1. Introduction
Emerging contaminants such as prescription, over the counter and illicit drugs are present in the environment mainly due to their incomplete removal during wastewater treatment.1 They pose a potential threat to the ecology of the receiving environment in ng L−1 to μg L−1 concentrations.2–5 Research to date has focussed on drug fate and behaviour in freshwater environments, with comparatively fewer studies in the marine environment.6 However, large cities located near coastal areas discharge wastewater effluents directly to marine waters emphasising the need for further research, especially in coastal ecosystems close to point discharges and any critical areas associated with the local hydrography. Furthermore, findings of research undertaken on drug fate and behaviour in freshwater is unlikely to reflect marine waters due to differences in the physicochemical and biological properties of fresh and marine water.6 A key consideration is the different organisms in river systems and marine systems that are exposed to these drugs.An important consideration for understanding the fate and toxicity of drugs in the environment is their stereochemistry.7 Approximately 50% of drugs are chiral and exist as two or more enantiomers.8 Most chiral drugs are manufactured and dispensed in racemic form (equimolar enantiomer distribution). However, they can be subject to enantiospecific metabolism in the human body and during wastewater treatment.9,10 This results in enrichment of one enantiomer entering the environment. The altered enantiomeric distribution cannot be measured using conventional analytical methods. Enantiomer enrichment is significant as drugs exhibit enantiospecific toxicity to organisms used as environmental toxicity indicators.11,12 For example, S(+)-fluoxetine is 30 times more toxic towards the freshwater eukaryote Tetrahymena thermophila than R(−)-fluoxetine.12 Despite this knowledge, the role of stereochemistry on chiral drug biodegradation and toxicity in the marine environment has received little attention to date.
There is a lack of enantioselective analytical methodologies reported for marine matrices. The high concentration of non-volatile salts in seawater (e.g., sodium chloride, ∼3.0–3.4%) can be detrimental to enantioselective chromatography and the high temperature ionisation source of mass spectrometers. Significant signal suppression (>50%) during electrospray ionisation has been reported for pharmaceuticals in seawater extracts analysed by achiral LC-MS/MS.13–15 However, no previous study has investigated the extent of non-volatile salts transfer through the SPE process, or the injected concentrations responsible for reduced analytical performance. Nevertheless, Coelho et al.16 reported the occurrence of a range of acidic and basic chiral drugs at the enantiomeric level in the Douro River estuary, Portugal; a single SPE protocol followed by analysis by two enantioselective LC-MS/MS methods utilising vancomycin and 1-(3,5-dinitrobenzamido)-1,2,3,4-tetrahydrophenanthrene chiral selectors. However, the salinity of studied samples was not reported, nor were whether or not there were any analytical issues (loss of chiral recognition, signal suppression etc.) experienced during method development or monitoring studies.
A wider limitation of existing multi-residue enantioselective methods for environmental analysis is chromatographic run time. Method run times of ≥55 minutes is typical for the simultaneous separation of chiral drugs from ≥5 therapeutic classes,17–19 which restricts sample throughput. A limiting factor has been stationary phase particle size. Commercially available enantioselective columns utilise particle sizes of 5 μm which limits column efficiency which could be improved if a smaller particle size became available.
A further consideration required for the establishment of new analytical protocols is the potential for analyte losses during sample collection, transport and storage. Errors associated with sampling procedures can outweigh the error of the instrumental method.20 For example, analytes can adsorb to the surface of sampling bottles,21 or degrade between sample collection and analysis.22 Analyte degradation within collected samples is particularly important for marine samples which need collected from boats, often precluding immediate processing. This can be due to the limited laboratory facilities on the vessel, or the fact that the analytical equipment will not operate on a vibrating, moving platform. Mijangos et al.23 found several drugs degraded by >20% in seawater stored at 4 °C during 31 d. However, the influence of storage conditions to the enantiomeric composition of chiral drugs in marine waters has not been investigated previously. Significant changes to the enantiomeric composition of chiral drugs during sample storage have been observed in collected freshwater samples.19
To address the lack of analytical methods reported for the enantioselective analysis of chiral drugs in marine matrices, the objectives of this work was:
(i) To develop an enantioselective LC-MS/MS instrumental method suitable for multi-residue drug separation within a short run time (≤30 minutes).
(ii) To establish a robust SPE protocol suitable for multi-residue drug extraction from seawater (30–35 practical salinity units – PSU) which limits the content of non-volatile salts (e.g., sodium chloride) in sample extracts.
(iii) To determine suitable sample storage conditions to ensure the stability of analytes and their enantiomeric composition in collected seawater samples.
In total 20 analytes were selected for method development. This included prescription, over-the-counter and illicit chiral and achiral drugs as well as other markers of municipal wastewater discharge (e.g., caffeine) (Table S1†). The developed method was assessed in terms of linearity, sensitivity, precision, recovery, trueness and signal suppression. The method was applied to samples collected from the Clyde and Forth estuaries, Scotland and can be used to further our understanding of the sources, fate and possible effects of drugs in the marine environment.
2. Materials and methods
2.1. Materials
Analytical reference standards and deuterated surrogates were purchased from Sigma Aldrich (Gillingham, UK) and Toronto Research Chemicals (North York, Canada). The reference standards were paracetamol, caffeine, carbamazepine, carbamazepine-10,11-epoxide, cotinine, R/S(±)-amphetamine, R/S(±)-methamphetamine, R/S(±)-atenolol, R/S(±)-chlorpheniramine, R/S(±)-citalopram, R/S(±)-fluoxetine, R/S(±)-3,4-methylenedioxymethamphetamine (MDMA), R/S(±)-propranolol, R/S(±)-salbutamol, R/S(±)-venlafaxine, R/S(±)-desmethylvenlafaxine, R/S(±)-bisoprolol, R/S(±)-acebutolol, R/S(±)-metoprolol and R/S(±)-sotalol. The deuterated surrogates were paracetamol-d4, caffeine-13C3, carbamazepine-d10, cotinine-d3, R/S(±)-amphetamine-d11, R/S(±)-methamphetamine-d11, R/S(±)-atenolol-d7, R/S(±)-chlorpheniramine-d6, R/S(±)-citalopram-d6, R/S(±)-fluoxetine-d6, R/S(±)-MDMA-d5, R/S(±)-propranolol-d7, R/S(±)-salbutamol-d3, R/S(±)-venlafaxine-d6, R/S(±)-bisoprolol-d5, R/S(±)-acebutolol-d5, R/S(±)-metoprolol-d7 and R/S(±)-sotalol-d6. All standards were prepared at 0.1 or 1.0 mg mL−1 in methanol and stored at −20 °C in the dark. HPLC grade methanol, acetonitrile, acetic acid and ammonium acetate, were purchased from Fischer Scientific (Loughborough, UK). Ultrapure water was 18.2 MΩ cm−1 quality. Seawater samples (10 L) used during development and validation of the analytical method were collected from the North-East coast of Scotland in high density polyethylene (HDPE) bottles.2.2. Solid phase extraction
Marine waters were filtered through GF/F filters (0.7 μm) using silanized glassware and 500 mL aliquots spiked at 100 ng L−1 with individual drug enantiomers (200 ng L−1 for achiral analytes). Oasis HLB (200 mg, 6 mL) obtained from Waters UK (Manchester, UK) were conditioned using 4 mL methanol and equilibrated using 4 mL water. Samples were loaded at 10 mL min−1 followed by a 50 mL water wash volume. The SPE sorbent was dried under vacuum at room temperature prior to elution with 6 mL acetonitrile at 1 mL min−1. The acetonitrile was then removed by drying under nitrogen at a temperature of 40 °C using a dry block. Finally, dried extracts were reconstituted in 0.25 mL methanol for enantioselective LC-MS/MS analysis (Fig. 1). During development of the SPE protocol, the water wash volume (0, 10 and 50 mL) and elution solvent (methanol and acetonitrile) were investigated to restrict the quantity of non-volatile salts present in extracted samples for analysis. Furthermore, the impact of elution solvent volume and sample pH on analyte recovery were investigated. | ||
| Fig. 1 Developed analytical workflow for the simultaneous determination of a selection of chiral and achiral drugs in seawater. Key: SPE, solid phase extraction; MRM, multiple reaction monitoring. | ||
2.3. ICP-OES
Metals were analysed by inductively coupled plasma optical emission spectrometry (ICP-OES) and used as a proxy for non-volatile salts determination in extracted samples. Samples were diluted and concentrations of sodium, potassium, calcium and magnesium determined by a Perkin Elmer Optima™ 7000 DV Spectrometer (Shelton, Connecticut, USA). The auxiliary gas flow was 0.2 L min−1, plasma gas flow 1.5 mL min−1 and nebulizer gas flow 0.8 L min−1. The radiofrequency was 1300 W and the pump flow rate was 1.5 mL min−1.2.4. Enantioselective LC-MS/MS
For chromatographic analysis an Agilent 1200 series HPLC (Cheshire, UK) was used. Analytes were separated using an InfinityLab Poroshell 120 Chiral-V column (150 × 2.1 mm; 2.7 μm particle size) fitted with a 0.2 μm pre-filter. An isocratic polar ionic mobile phase consisting of 2 mM ammonium acetate in methanol containing 0.01% acetic acid was used. A flow rate of 0.15 mL min−1 was utilised and the column temperature was 15 °C. The injection volume was 10 μL. During the development of the chromatographic method the influence of mobile phase buffer (ammonium acetate and formate), buffer concentration (0, 0.5, 1, 2, 5 mM), acid concentration (0, 0.01 and 0.05%), column temperature (15, 20 and 25 °C), flow rate (0.10, 0.15 and 0.20 mL min−1) and injection volume (10, 20 and 40 μL) on analyte separation was investigated.The HPLC was coupled to an Agilent 6420 MS/MS triple quadrupole utilising positive electrospray ionisation. The capillary voltage was 4000 V. The desolvation temperature was 350 °C with a nitrogen gas flow of 12 L min−1. The nebulizing pressure was 50 psi. Nitrogen gas was used as the nebulising, desolvation and collision gas. Two multiple reaction monitoring (MRM) transitions were monitored for each analyte for quantification and confirmation purposes (one in the case of deuterated surrogates). The optimised MS/MS transitions for each analyte are compiled (Table S2†). Other quality criteria used to ensure quality of data were pre-determined tolerances of ion ratio and retention time.24
2.5. Instrument and method performance
Linearity was established using a 13 point calibration curve ranging from 0.01 to 1000 ng mL−1. Instrument detection limits (IDLs) were determined by the lowest concentration at which the signal to noise ratio (S/N) ≥3 and instrument quantitation limit (IQL) when S/N ≥ 10. Both solvent and matrix matched calibrations were prepared. Matrix matched calibration standards were prepared directly in the dried extracts from seawater samples taken through the SPE process. The matrix matched calibration was prepared to assess analyte signal suppression during electrospray ionisation using eqn (1): | (1) |
Intra-day and inter-day instrument precision was determined by triplicate injections of 5, 50 and 250 ng mL−1 solvent standards within the same day and over three different days, respectively. Method recovery and trueness of target analytes by the developed SPE-enantioselective LC-MS/MS method was assessed by extracting 500 mL seawater samples spiked at 25, 100 and 250 ng L−1 in triplicate and determined using eqn (2) and (3):
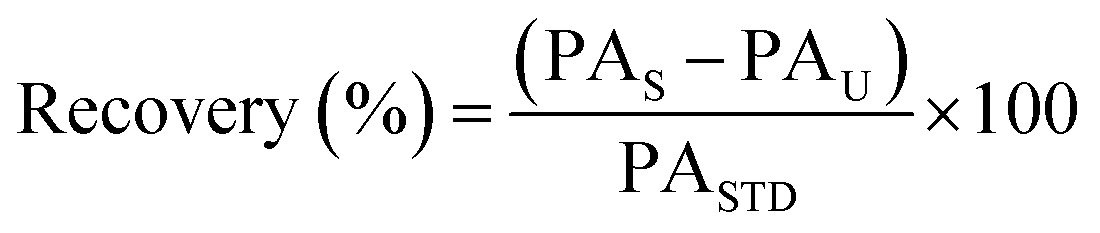 | (2) |
 | (3) |
Method detection limits (MDLs) and method quantitation limits (MQLs) of the entire SPE-enantioselective LC-MS/MS method was calculated using eqn (4) and (5):
 | (4) |
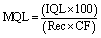 | (5) |
2.6. In-sample drug stability
The stability of analytes within collected samples was assessed under typical sample transport/storage conditions. Sacrificial 250 mL seawater samples (10 per treatment condition) were spiked with all analytes at 250 ng L−1 and stored in the dark at 18 °C, 4 °C and −20 °C. Samples were extracted in duplicate at 0, 1, 4, 7 and 14 days for samples stored at 18 °C and 4 °C. For those stored at −20 °C duplicate analysis was performed at 0, 7, 14, 21 and 28 days. The enantiomeric composition of chiral analytes was expressed as enantiomeric fraction (EF) using eqn (6) or (7):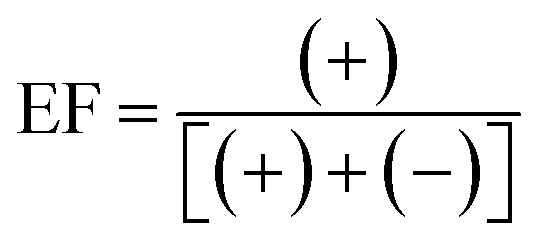 | (6) |
 | (7) |
2.7. Application to estuarine waters
The developed method was applied to samples collected from the Clyde and Forth estuaries, Scotland during June 2019. Five locations were sampled within each estuary (Table S3† and Fig. 4). Water samples were collected at a depth of 3 m from a small boat and stored in 2.5 L HDPE bottles. Collected samples were kept cool and in the dark until arrival at the laboratory (<5 h). These were then frozen at −20 °C and processed within 7 d using the SPE methodology described in Section 2.2.3. Results and discussion
3.1. Enantioselective liquid chromatography-tandem mass spectrometry method
A Poroshell 120 Chiral-V column of 2.7 μm particle size, containing the macrolide antibiotic vancomycin as a chiral selector, was used for method development. A vancomycin-based column was chosen due to its broad selectivity and multi-residue analysis capabilities.19,25,26 Vancomycin has 18 chiral centres and facilitates hydrogen bonding, dipole stacking, steric interactions, ionic binding and π–π complexation for enantioselective separation of a broad range of drugs.25During initial screening of mobile phases for drug enantioseparation it was found polar ionic mode (methanol containing ammonium acetate and acetic acid) offered broader selectivity over reversed phase and polar organic modes.19 Ammonium acetate concentration had the greatest influence on enantioselective separations,26,27 with 0.5 mM achieving best separation. However, during analysis of sample extracts a shift in retention time was noted. Increasing the ammonium acetate concentration to 2 mM overcame this issue. This enabled reproducibility in retention time whilst achieving enantiomer resolutions (RS) ≥1 for amphetamine, methamphetamine, salbutamol, propranolol, fluoxetine, venlafaxine, desmethylvenlafaxine, citalopram, sotalol, chlorpheniramine and acebutolol (Fig. 2). A minimum RS threshold of 1 is required for quantitative purposes.28 For atenolol, metoprolol and bisoprolol, where only partial separation was achieved (RS 0.6–0.8 – Fig. 2), the valley drop method was utilised for integration29 and can only be considered semi-quantitative. During method development no enantioseparation of R/S(±)-MDMA was noted. Nevertheless, these mobile phase conditions achieved suitable chromatography for simultaneous analysis of achiral analytes (Fig. 2).
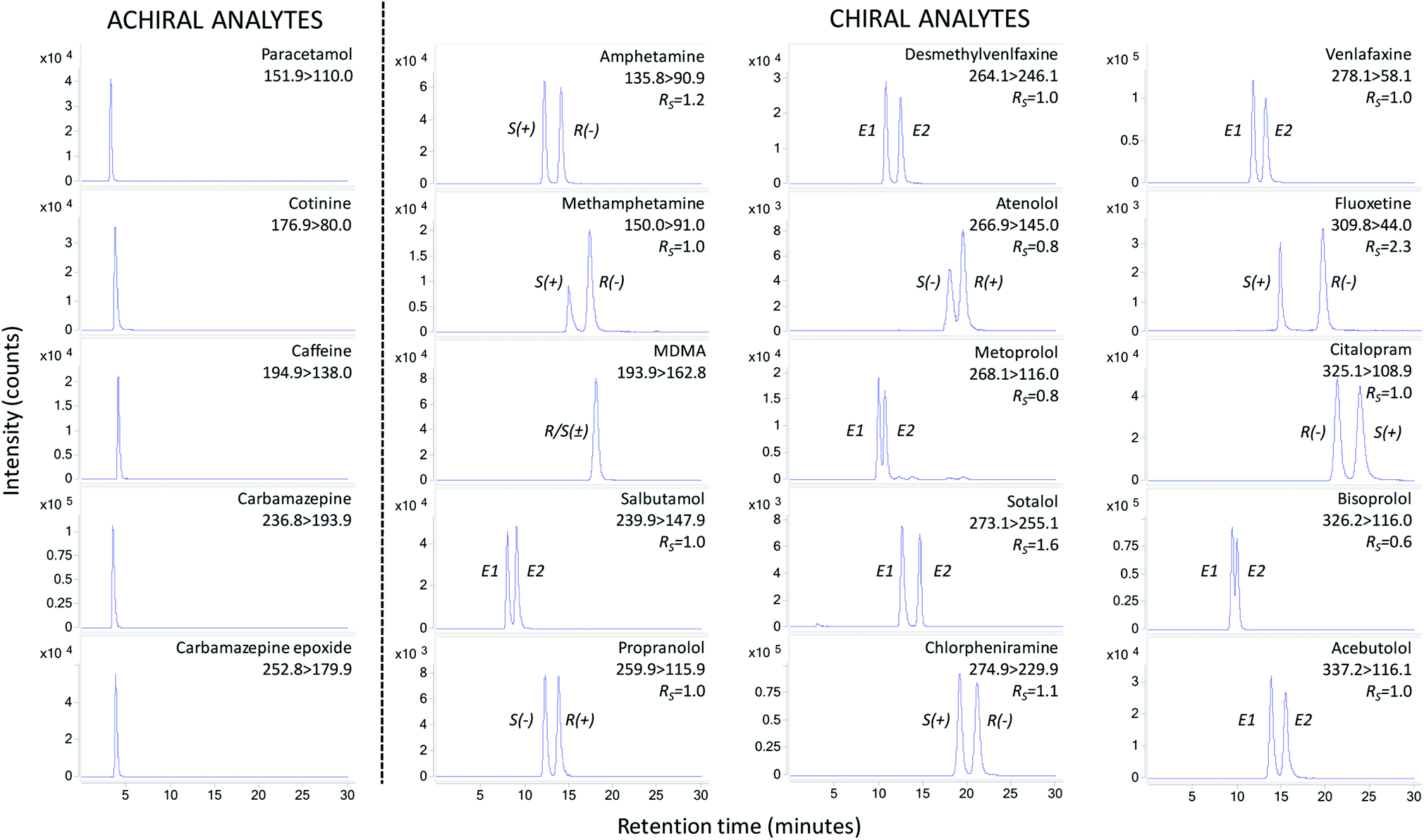 | ||
| Fig. 2 Enantioselective LC-MS/MS chromatograms of extracted seawater spiked at enantiomer concentrations of 100 ng L−1 for each enantiomer (200 ng L−1 for achiral analytes). Each chromatogram shows corresponding MRM transition and enantiomer resolution (RS). Note: cotinine is a chiral compound but was purchased as (−)-cotinine. The deuterated surrogate (±)-cotinine-d3 showed no enantioseparation. Key: MDMA, 3,4-methylenedioxymethamphetamine; E1, enantiomer 1; E2, enantiomer 2 (where order of enantiomer elution is not known). S(−), S(−)-enantiomer; R(+), R(+)-enantiomer; S(+), S(+)-enantiomer; R(−), R(−)-enantiomer (where order of enantiomer elution is known). | ||
The total chromatographic run time of 30 minutes is considerably shorter than other previous multi-residue enantioselective methodologies which report times ≥55 minutes.17–19 This is beneficial as it improves sample throughput by at least 83% over previously published methods. The use of a smaller stationary phase particle size column (2.7 μm versus 5 μm) and superficially porous particles facilitated improved efficiency and reduced run times.30 Intra-day and inter-day precision assessed at three different concentrations was <5% for the majority of analytes (Table S4†). Instrument quantitation limits were ≤10 ng mL−1 for all analytes. Performance of the developed instrumental method with respect to precision and sensitivity is comparable to previous enantioselective LC-MS/MS methods utilising vancomycin as the chiral selector.18,19,31
3.2. Solid phase extraction protocol
Oasis HLB SPE cartridges were selected for drug extraction from the seawater matrix due to the multi-residue capabilities of the mixed mode ion exchange and reversed phase co-polymer. Furthermore, they do not require sample adjustment to acidic pH conditions for drug extraction which can be detrimental to subsequent enantioselective separations.31 A further purpose of the SPE protocol here was to limit the introduction of non-volatile salts to the LC-MS/MS. Seawater with a salinity of 33.8 PSU was used to develop the SPE extraction protocol. This would ensure the method could be applicable to other marine waters of lower salinity (e.g., estuarine waters). However, the level of non-volatile salts in extracted samples that are detrimental to analysis methods is unknown. Therefore, extracted river water samples subject to long term monitoring without analytical issues using the same instrumentation were used as a target benchmark. Here, the estimated quantity of sodium chloride present in a 10 μL LC-MS/MS injection was 2.0 μg (Table S5†).Applying the same SPE extraction protocol to seawater (10 mL wash with water and methanol elution) resulted in a sodium chloride equivalent of 63 μg per injection (Table S5†). A simple method of reducing non-volatile salts in extracted SPE samples is to apply an increased volume of wash water after sample loading to remove excess salts.32 Increasing the wash water volume to 50 mL resulted in the reduction of sodium chloride to 17 μg per injection. Although this did not achieve the 2.0 μg target threshold, extracts were analysed by LC-MS/MS. However, loss of enantioseparation was noted for both acebutolol and sotalol. To further reduce the quantity of salts in extracted samples the elution solvent was changed to acetonitrile due to the poor solubility of sodium chloride in this solvent (whilst maintaining analyte solubility). The use of a 50 mL water wash volume and acetonitrile elution resulted in sodium chloride reducing to <1 μg per injection (Table S5†). In these extracts no loss of drug enantioseparation was experienced (Fig. 2), therefore this extraction protocol was selected for subsequent method performance assessments.
3.3. Method performance
The performance of the developed SPE-enantioselective LC-MS/MS method was assessed using seawater spiked at 25, 100 and 250 ng L−1. Absolute method recovery (based on peak area response) varied from 1% for S(+)-fluoxetine to 61% for desmethylvenlafaxine-E2 (Table 1). Analyte losses are mainly attributed to the extraction process and suppression during electrospray ionisation. Greater analyte recovery is achievable using a protic elution solvent such as methanol during SPE.33 However, multi-residue enantioselective analysis for such a challenging matrix requires trade-offs between analyte recovery and removal of co-extracted interferences. Using acetonitrile as the SPE elution solvent facilitated the recovery of all target analytes without loss of enantioresolution and, as highlighted above, overcame the potential issues relating to the high saline concentration of the initial samples.| Analyte class | Analyte | Method recovery ± SD (%) (n = 3) | Method trueness ± SD (%) (n = 3) | Signal suppression (%) | MDL (ng L−1) | MQL (ng L−1) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 25 ng L−1 | 100 ng L−1 | 250 ng L−1 | 25 ng L−1 | 100 ng L−1 | 250 ng L−1 | |||||
| a Key: MDL, method detection limit; MQL, method quantitation limit; MDMA, 3,4-methylenedioxymethamphetamine. b Spike concentrations were 50, 200 and 500 ng L−1. | ||||||||||
| Achiral drugs and other wastewater markers | Paracetamolb | 5 ± 1 | 4 ± 0 | 4 ± 1 | 108 ± 9 | 91 ± 4 | 88 ± 3 | 78 | 8.8 | 26.3 |
| Carbamazepineb | 18 ± 2 | 20 ± 1 | 29 ± 3 | 84 ± 6 | 96 ± 6 | 92 ± 2 | 55 | 1.0 | 3.3 | |
| Carbamazepine epoxideb | 44 ± 4 | 41 ± 3 | 41 ± 1 | 125 ± 6 | 137 ± 7 | 111 ± 9 | 22 | 0.4 | 1.3 | |
| Caffeineb | 18 ± 4 | 35 ± 2 | 28 ± 2 | 114 ± 9 | 105 ± 4 | 106 ± 5 | 30 | 2.0 | 6.6 | |
| Cotinineb | 26 ± 2 | 25 ± 2 | 32 ± 7 | 92 ± 6 | 112 ± 9 | 106 ± 3 | 36 | 0.1 | 0.2 | |
| Stimulants | S(+)-Amphetamine | 3 ± 3 | 3 ± 2 | 6 ± 4 | 106 ± 14 | 98 ± 13 | 94 ± 9 | 11 | 1.6 | 6.2 |
| R(−)-Amphetamine | 3 ± 4 | 4 ± 2 | 5 ± 3 | 99 ± 6 | 106 ± 8 | 102 ± 10 | 5 | 1.3 | 5.1 | |
| S(+)-Methamphetamine | 5 ± 3 | 7 ± 3 | 3 ± 0 | 91 ± 5 | 101 ± 6 | 91 ± 3 | 66 | 1.2 | 2.5 | |
| R(−)-Methamphetamine | 12 ± 10 | 17 ± 5 | 13 ± 5 | 92 ± 5 | 98 ± 7 | 91 ± 1 | 24 | 0.2 | 0.7 | |
| R/S(±)-MDMAb | 13 ± 10 | 17 ± 5 | 17 ± 7 | 95 ± 6 | 100 ± 3 | 100 ± 10 | 16 | 1.0 | 3.3 | |
| Anti-histamine | S(+)-Chlorpheniramine | 38 ± 5 | 38 ± 7 | 35 ± 1 | 94 ± 3 | 96 ± 9 | 95 ± 4 | 0 | 0.4 | 1.4 |
| R(−)-Chlorpheniramine | 37 ± 5 | 38 ± 6 | 35 ± 1 | 90 ± 3 | 93 ± 9 | 98 ± 4 | −2 | 0.4 | 1.4 | |
| Beta-agonist | Salbutamol-E1 | 10 ± 2 | 9 ± 4 | 12 ± 1 | 91 ± 2 | 100 ± 7 | 94 ± 2 | −25 | 0.5 | 1.5 |
| Salbutamol-E2 | 10 ± 3 | 9 ± 4 | 12 ± 1 | 85 ± 3 | 99 ± 6 | 88 ± 3 | 2 | 0.5 | 1.5 | |
| Beta-blockers | S(−)-Propranolol | 20 ± 11 | 21 ± 3 | 24 ± 4 | 95 ± 11 | 91 ± 4 | 92 ± 9 | 12 | 0.7 | 2.3 |
| R(+)-Propranolol | 23 ± 12 | 23 ± 3 | 25 ± 4 | 94 ± 11 | 92 ± 4 | 97 ± 10 | 6 | 0.6 | 2.1 | |
| S(−)-Atenolol | 41 ± 13 | 32 ± 1 | 44 ± 3 | 92 ± 4 | 97 ± 8 | 95 ± 2 | 26 | 2.0 | 6.5 | |
| R(+)-Atenolol | 33 ± 14 | 32 ± 3 | 41 ± 7 | 91 ± 5 | 94 ± 7 | 92 ± 4 | 13 | 2.2 | 7.2 | |
| Sotalol-E1 | 45 ± 2 | 47 ± 3 | 50 ± 9 | 93 ± 8 | 100 ± 9 | 101 ± 11 | −2 | 1.4 | 4.5 | |
| Sotalol-E2 | 46 ± 5 | 44 ± 3 | 47 ± 8 | 97 ± 9 | 99 ± 3 | 110 ± 6 | 21 | 1.4 | 4.6 | |
| Bisoprolol-E1 | 49 ± 16 | 45 ± 2 | 45 ± 7 | 87 ± 1 | 101 ± 9 | 90 ± 6 | 23 | 0.1 | 0.4 | |
| Bisoprolol-E2 | 27 ± 1 | 39 ± 2 | 56 ± 19 | 89 ± 12 | 103 ± 8 | 100 ± 7 | 10 | 0.1 | 0.5 | |
| Acebutolol-E1 | 53 ± 17 | 46 ± 2 | 50 ± 6 | 101 ± 8 | 104 ± 10 | 100 ± 7 | 1 | 0.1 | 0.4 | |
| Acebutolol-E2 | 37 ± 7 | 32 ± 3 | 15 ± 12 | 102 ± 12 | 107 ± 1 | 95 ± 1 | 37 | 0.2 | 0.8 | |
| Metoprolol-E1 | 35 ± 3 | 42 ± 2 | 50 ± 12 | 82 ± 3 | 97 ± 8 | 88 ± 2 | 15 | 1.0 | 3.0 | |
| Metoprolol-E2 | 36 ± 11 | 37 ± 2 | 46 ± 12 | 86 ± 5 | 103 ± 10 | 93 ± 4 | 14 | 1.0 | 3.2 | |
| Anti-depressants | S(+)-Fluoxetine | 1 ± 2 | 1 ± 1 | 1 ± 1 | 101 ± 3 | 96 ± 6 | 96 ± 11 | 44 | 21.2 | 67.7 |
| R(−)-Fluoxetine | 2 ± 2 | 2 ± 1 | 4 ± 3 | 94 ± 8 | 97 ± 8 | 97 ± 3 | 18 | 12.3 | 39.4 | |
| R(−)-Citalopram | 39 ± 6 | 41 ± 5 | 38 ± 1 | 94 ± 5 | 95 ± 10 | 92 ± 3 | 6 | 0.6 | 2.2 | |
| S(+)-Citalopram | 39 ± 6 | 44 ± 6 | 34 ± 1 | 94 ± 7 | 97 ± 7 | 93 ± 5 | 7 | 0.6 | 2.2 | |
| Venlafaxine-E1 | 55 ± 6 | 56 ± 5 | 51 ± 3 | 89 ± 6 | 97 ± 7 | 92 ± 0 | 3 | 0.1 | 0.4 | |
| Venlafaxine-E2 | 54 ± 4 | 58 ± 5 | 52 ± 3 | 87 ± 8 | 98 ± 8 | 91 ± 1 | −3 | 0.1 | 0.4 | |
| Desmethylvenlafaxine-E1 | 50 ± 5 | 48 ± 5 | 45 ± 4 | 89 ± 8 | 90 ± 7 | 105 ± 4 | −9 | 0.4 | 1.5 | |
| Desmethylvenlafaxine-E2 | 61 ± 6 | 53 ± 5 | 46 ± 2 | 98 ± 9 | 97 ± 8 | 105 ± 2 | −13 | 0.4 | 1.3 | |
Signal suppression varied from 78% for paracetamol to −25% (i.e., signal enhancement) for salbutamol-E1 (Table 1). Signal enhancement during electrospray ionisation of environmental matrices including seawater is not uncommon.15,34 In general the achiral analytes which had the least interaction with the stationary phase had the greatest signal suppression (22–78%). Significantly, several of the chiral drugs demonstrate enantiospecific signal suppression. For example, signal suppressions of 66% and 24% were determined for S(+)-methamphetamine and R(−)-methamphetamine, respectively (Table 1). This was apparent in the chromatography (Fig. 2), and demonstrated the necessity of using deuterated analogues to account for these effects at the enantiomeric level. Indeed, method trueness for the majority of target analytes was in the range 80–110% (Table 1). Method trueness for carbamazepine epoxide was 111–137% over the three spike concentration levels. This analyte did not have its own deuterated surrogate included in the method and was quantified against carbamazepine-d10, explaining the method trueness observed. All analytes demonstrated acceptable repeatability in method trueness with RSDs <20% (n = 3) at spiked concentrations of 25, 100 and 250 ng L−1.
Calculated MQLs of most analytes were suitable to monitor drugs below 10 ng L−1 (Table 1). This concentration is set as an action threshold for predicted environmental concentration of drugs in surface waters by the European Medicines Agency.35 Only MQLs for paracetamol (26.3 ng L−1), S(+)-fluoxetine (67.7 ng L−1) and R(−)-fluoxetine (39.4 ng L−1) were above this threshold. However, paracetamol is reported in seawater at concentrations >50 ng L−1 due to its high use.6,15,36,37 On the other hand fluoxetine is typically reported at concentrations below the MQLs achievable in this study.15,16 Nevertheless, the overall performance of the new methodology was similar to previous methods developed for other aqueous environmental matrices (Table S6†).16,18,19,29,38 MQLs for the majority of analytes are comparable to other SPE-enantioselective LC-MS/MS methods for surface waters.16,18,19,28,29,38 Other than being the first multi-residue enantioselective method developed for drugs in seawater, it also offers the advantage of simultaneous analysis of achiral analytes.
3.4. Drug stability in collected seawater samples
Initially, the potential loss of analytes to the surface of HDPE sample bottles was investigated. Sacrificial sample bottles filled with water were spiked with all analytes. A threshold of ±20% of the spiked concentration was considered significant as applied in previous studies.21,22,39 No significant changes to concentration of the studied analytes were observed during 14 day storage (Fig. 3 and S1–S19†). This agrees with previous studies of polar/semi-polar drugs to polypropylene sample bottles.21,40 | ||
| Fig. 3 Relative concentration and enantiomeric fraction of R/S(±)-amphetamine spiked at 500 ng L−1 (n = 2) in ultra-pure water at 18 °C (A), seawater at 18 °C (B), seawater at 4 °C (C) and seawater at −20 °C (D). Key: td, concentration at a specified number of days; t0, concentration at 0 days following spiking. | ||
To assess drug degradation during sample transport and storage, seawater was spiked with all analytes to ensure detectable concentrations. In seawater stored at 18 °C significant degradation was observed for amphetamine (Fig. 3B). Furthermore, sample degradation was found to be enantioselective. Following storage for 7 days the concentration of S(+)-amphetamine reduced from 267.1 ng L−1 to 211.9 ng L−1 (−21%). As the concentration of R(−)-amphetamine was unchanged during this time, the EF reduced from 0.49 to 0.44. Following 14 days storage, the concentration of S(+)-amphetamine and R(−)-amphetamine reduced by 62% and 44%, respectively. The corresponding EF here was 0.40. Preferential biodegradation of S(+)-amphetamine over R(−)-amphetamine has been observed in river water.41 All other drugs were considered stable over the 14 day study period (Fig. S1–S19†). Based on these results, short-term storage (≤4 days) of collected seawater at 18 °C is acceptable prior to sample processing.
The stability of R/S(±)-amphetamine in collected seawater samples was improved by storage at 4 °C. No significant degradation was observed over a 14 day period (Fig. 3). Freezing samples at −20 °C was also investigated for sample storage and no significant change to any drug was observed over 28 days (Fig. 3D and S1–S19†). This agrees with the findings of Fedorova et al.42 who showed that several of the same drugs (citalopram, atenolol, bisoprolol, carbamazepine, venlafaxine, sotalol, metoprolol) were stable for a minimum of 60 days in wastewater from a municipal WTP in the Czech Republic stored at −18 °C.
3.5. Application to estuarine water samples
The newly developed multi-residue enantioselective method was applied to water samples collected from the Clyde and Forth estuaries (n = 5 per estuary). In total 13/20 analytes and 7/20 analytes were detected at least once in samples collected from the Clyde and Forth estuaries, respectively. A maximum total analyte (ΣANALYTE) concentration of 1548 ng L−1 was found at the Kelvin confluence within the Clyde estuary, this site being the furthest ‘up-river’ of the samples collected (Fig. 4). Paracetamol (495 ng L−1) and caffeine (466 ng L−1) were present at the greatest concentrations at this site. In the Forth estuary, the highest paracetamol concentration was 1056 ng L−1 at Dunmore. Paracetamol has been previously found in the Humber estuary, North-East England at a maximum concentration of 917 ng L−1.43 At the sampling sites within the Clyde estuary other than the Kelvin confluence, caffeine was present at the greatest concentration. Caffeine concentrations were 356, 222, 102 and 37 ng L−1 at Dalmuir, Milton, Woodhall and Dunoon, respectively (see Fig. 4 for site locations). In the Forth estuary, caffeine was present in all samples ranging from 53–235 ng L−1. Caffeine was the only analyte present within all estuary samples analysed (n = 10) and is well established as an indicator of wastewater discharge.44,45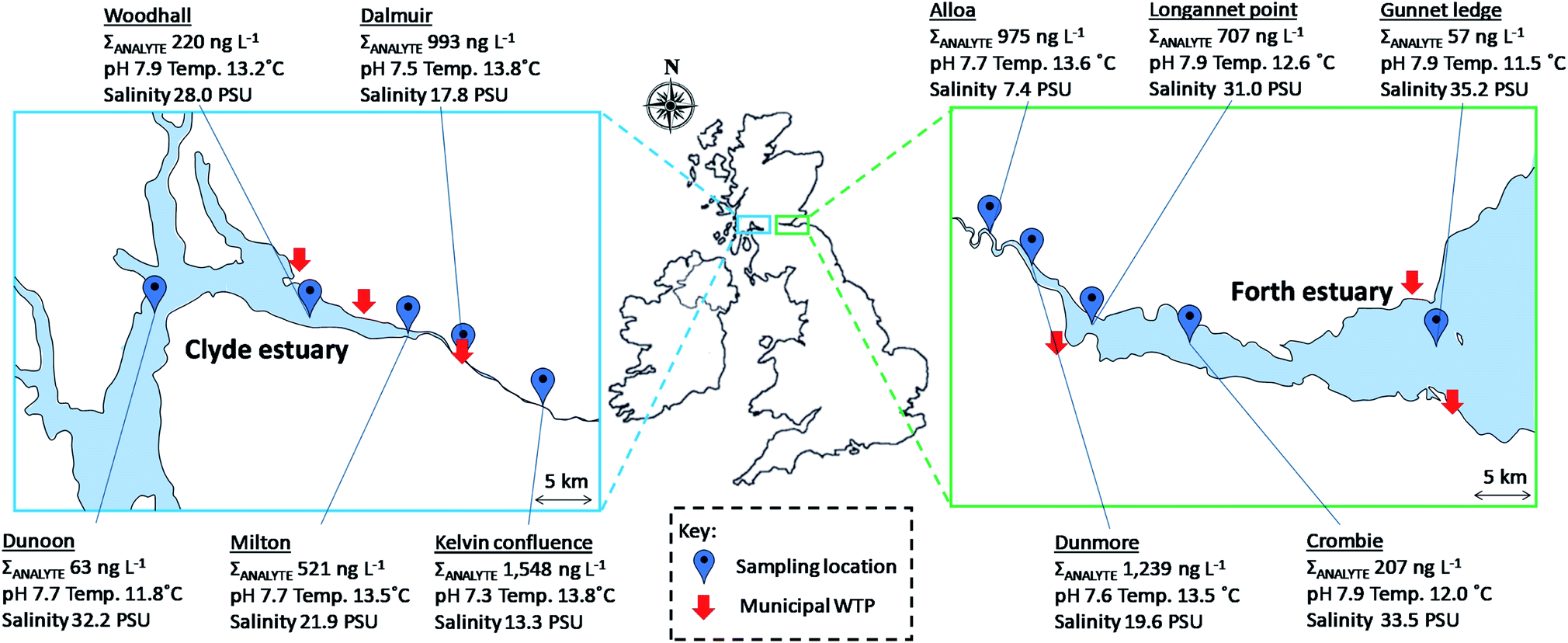 | ||
| Fig. 4 Sampling locations and total analyte concentrations in the Clyde and Forth estuaries. Key: ΣANALYTE, total analyte concentration; PSU, practical salinity units; WTP, wastewater treatment plant. | ||
Chiral drugs were more prevalent in the Clyde estuary than the Forth estuary reflecting the greater wastewater contributions to the inner Clyde estuary (Fig. 5). The beta-blockers propranolol, atenolol and bisoprolol were all detected in the Clyde estuary. Propranolol reduced in concentration from 60 ng L−1 at the Kelvin confluence to 6.5 ng L−1 at Woodhall (Fig. 5). The reduction in concentration is expected due to increased dilution as effluent discharges travel down the estuary. However, a change in the enantiomeric composition of propranolol was also apparent, whereby an EF of 0.41 ± 0.01 at the Kelvin confluence increased to 0.51 ± 0.01 at Woodhall (Fig. 5). A change in drug EF is indicative of biotic processes (e.g., biodegradation) taking place.7 However, different wastewater inputs between sampling sites and the limited number of samples collected may also contribute to the changes in EF observed. Wastewater effluents released in the environment are typically enriched with S(−)-propranolol and EFs <0.5.19,21,28,31 Differences in atenolol EF throughout the estuary were also noted. The EFs were 0.53, 0.56, 0.58 and 0.58 at the Kelvin confluence, Dalmuir, Milton and Woodhall (Fig. 5). An enrichment of R(+)-atenolol could be significant for marine risk assessments as this enantiomer is twice as toxic to the indicator species Daphnia magna as S(−)-atenolol.46 Bisoprolol was quantifiable at the Kelvin confluence, Dalmuir and Milton with EFs >0.5 (Fig. 5). The other beta-blockers acebutolol, sotalol, metoprolol, and the beta-agonist salbutamol were not quantifiable in any of the samples analysed.
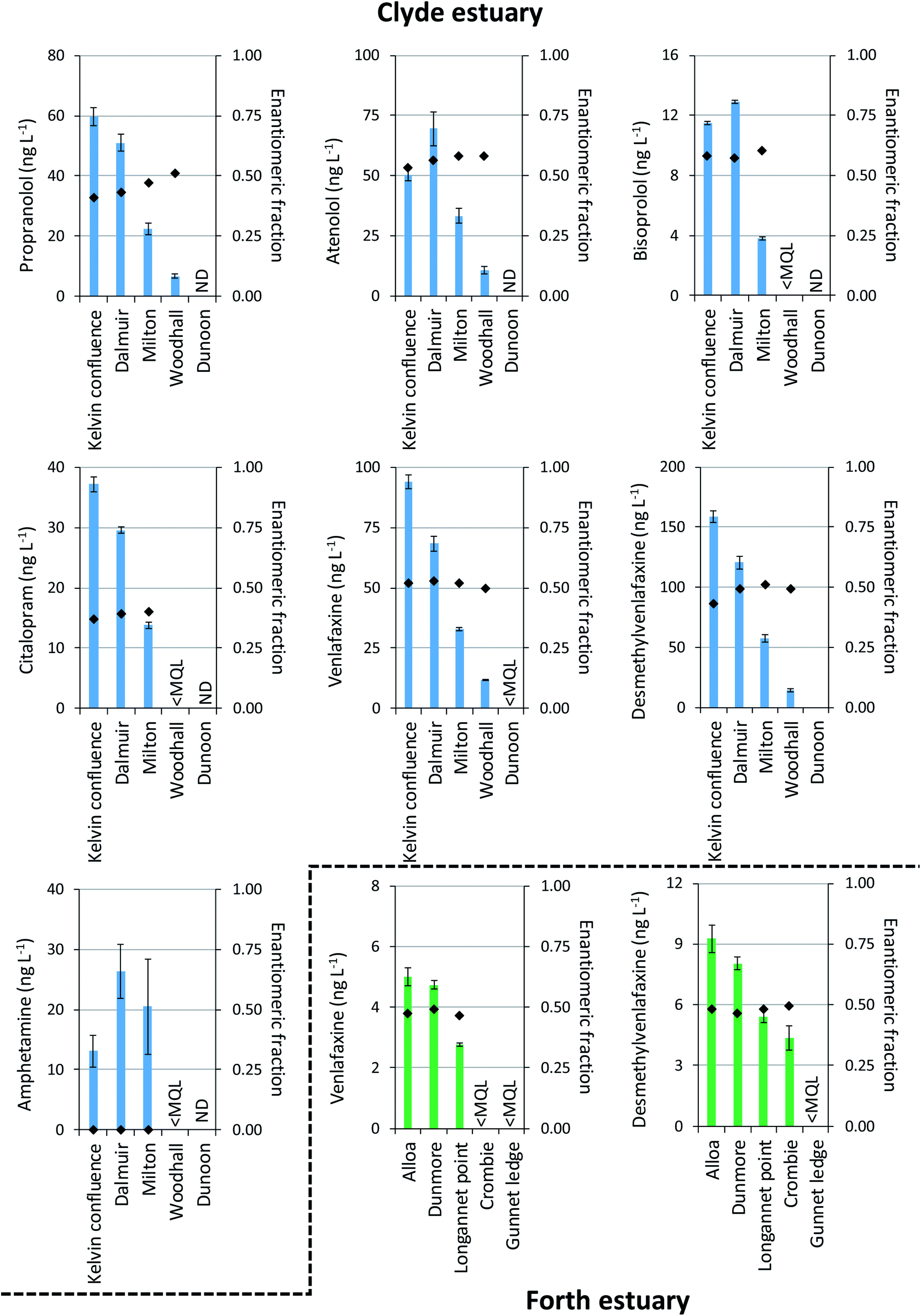 | ||
| Fig. 5 Total concentrations (sum of both enantiomers) and enantiomeric fractions of chiral drugs determined in the Clyde and Forth estuaries. Bars represent concentration (primary axis) and markers represent the enantiomeric fraction (secondary axis). Note: error bars represent the standard deviation of n = 3 replicates; sampling locations (X-axis, sample location name) correspond to those in Fig. 4. <MQL = less than method quantitation limit; ND = not detected. | ||
The anti-depressant citalopram was in the range 14–37 ng L−1 at the Kelvin confluence, Dalmuir and Milton within the Clyde estuary (Fig. 5). Significantly, the EFs were 0.37–0.40 representing an enrichment of the R(−)-enantiomer which is the pharmacologically less active enantiomer.47 This is in agreement with observations in wastewater effluent.19,21 Assuming pharmacological activity reflects toxicity in the environment, this observation would result in an overestimation of environmental risk if enantioselective analysis is not undertaken. Citalopram has previously been reported in UK estuaries at concentrations <50 ng L−1,43 albeit not at the enantiomeric level. Venlafaxine and desmethylvenlafaxine were the chiral drugs found at the greatest concentration. Maximum concentrations were 94 ng L−1 and 159 ng L−1 in the Clyde estuary (Fig. 5). Although venlafaxine and desmethylvenlafaxine were detected in all Forth estuary samples, concentrations were <10 ng L−1. In both estuaries EFs were ∼0.50 showing little enantiomeric enrichment which is in agreement with that found in UK freshwaters48 and wastewater effluent.31
On the other hand, the stimulant amphetamine was detected exclusively as the R(−)-enantiomer at concentrations ≤26 ng L−1 in the Clyde estuary (Fig. 5). An enrichment of R(−)-amphetamine (EFs <0.2) has been reported previously in UK river water.49 In this case R(−)-amphetamine has half the stimulant activity of S(+)-amphetamine,7 but no data exists on the enantiospecific toxicity of amphetamine to environmental organisms.
Although only a limited number of samples were collected in this study to demonstrate the method's application, several drugs were present >10 ng L−1 and in non-racemic form (Fig. 5). More robust monitoring is now required to better assess the distribution and composition of chiral drugs in marine waters. This information can then be used to inform toxicity driven assessments to better understand the risk posed to the marine environment. Currently, no data exists on the enantiospecific toxicity of chiral drugs to marine organisms.
4. Conclusions
A new multi-residue SPE-enantioselective LC-MS/MS method was successfully developed for the determination of emerging contaminants in seawater. Limiting the sodium chloride concentration in SPE extracts was vital for attaining suitable enantioselective chromatography. This was achieved using a 50 mL water wash volume following sample loading and acetonitrile as the elution solvent. The sensitivity of the method was demonstrated in seawater with MQLs <10 ng L−1 for the majority of studied analytes. Application of the method found several chiral drugs in the Clyde estuary to be present in non-racemic form. This demonstrates that further investigation is needed on the enantiospecific behaviour of chiral drugs in the marine environment, which can be supported using this newly developed method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Royal Society (RGS\R1\191250). The authors wish to thank Marine Scotland Science and the Scottish Environment Protection Agency (SEPA) for providing the samples used in this study. In particular Matt Blackburn and Jack Bloodworth (both SEPA) are acknowledged for their assistance. Contains SEPA data© Scottish Environment Protection Agency and database right 2019. All rights reserved.References
- S. R. Hughes, P. Kay and L. E. Brown, Global synthesis and critical evaluation of pharmaceutical data sets collected from river systems, Environ. Sci. Technol., 2013, 47(2), 661–677, DOI:10.1021/es3030148.
- K. Fent, A. A. Weston and D. Caminada, Ecotoxicology of human pharmaceuticals, Aquat. Toxicol., 2006, 76(2), 122–159, DOI:10.1016/j.aquatox.2005.09.009.
- K. A. Kidd, P. J. Blanchfield, K. H. Mills, V. P. Palace, R. E. Evans, J. M. Lazorchak and R. W. Flick, Collapse of a fish population after exposure to a synthetic estrogen, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(21), 8897–8901, DOI:10.1073/pnas.0609568104.
- L. H. M. L. M. Santos, A. N. Araújo, A. Fachini, A. Pena, C. Delerue-Matos and M. C. B. S. M. Montenegro, Ecotoxicological aspects related to the presence of pharmaceuticals in the aquatic environment, J. Hazard. Mater., 2010, 175(1–3), 45–95, DOI:10.1016/j.jhazmat.2009.10.100.
- H. Watanabe, I. Tamura, R. Abe, H. Takanobu, A. Nakamura, T. Suzuki, A. Hirose, T. Nishimura and N. Tatarazako, Chronic toxicity of an environmentally relevant mixture of pharmaceuticals to three aquatic organisms (alga, daphnid, and fish), Environ. Toxicol. Chem., 2016, 35(4), 996–1006, DOI:10.1002/etc.3285.
- S. Gaw, K. V. Thomas and T. H. Hutchinson, Sources, impacts and trends of pharmaceuticals in the marine and coastal environment, Philos. Trans. R. Soc., B, 2014, 369(1656), 20130572, DOI:10.1098/rstb.2013.0572.
- B. Kasprzyk-Hordern, Pharmacologically active compounds in the environment and their chirality, Chem. Soc. Rev., 2010, 39(11), 4466–4503, 10.1039/c000408c.
- E. Sanganyado, Z. Lu, Q. Fu, D. Schlenk and J. Gan, Chiral pharmaceuticals: a review on their environmental occurrence and fate processes, Water Res., 2017, 124, 527–542, DOI:10.1016/j.watres.2017.08.003.
- N. H. Hashim, L. D. Nghiem, R. M. Stuetz and S. J. Khan, Enantiospecific fate of ibuprofen, ketoprofen and naproxen in a laboratory-scale membrane bioreactor, Water Res., 2011, 45(18), 6249–6258, DOI:10.1016/j.watres.2011.09.020.
- D. Camacho-Muñoz, B. Petrie, L. Lopardo, K. Proctor, J. Rice, J. Youdan, R. Barden and B. Kasprzyk-Hordern, Stereoisomeric profiling of chiral pharmaceutically active compounds in wastewaters and the receiving environment – A catchment-scale and a laboratory study, Environ. Int., 2019, 127, 558–572, DOI:10.1016/j.envint.2019.03.050.
- J. K. Stanley, A. J. Ramirez, C. K. Chambliss and B. W. Brooks, Enantiospecific sublethal effects of the antidepressant fluoxetine to a model aquatic vertebrate and invertebrate, Chemosphere, 2007, 69(1), 9–16, DOI:10.1016/j.chemosphere.2007.04.080.
- M. J. Andrés-Costa, K. Proctor, M. T. Sabatini, A. P. Gee, S. E. Lewis, Y. Pico and B. Kasprzyk-Hordern, Enantioselective transformation of fluoxetine in water and its ecotoxicological relevance, Sci. Rep., 2017, 7(1), 15777, DOI:10.1038/s41598-017-15585-1.
- J. Wu, X. Qian, Z. Yang and L. Zhang, Study on the matrix effect in the determination of selected pharmaceutical residues in seawater by solid-phase extraction and ultra-high-performance liquid chromatography-electrospray ionization low-energy collision-induced dissociation tandem mass spectrometry, J. Chromatogr. A, 2010, 1217(9), 1471–1475, DOI:10.1016/j.chroma.2009.12.074.
- M. Borecka, A. Białk-Bielińska, G. Siedlewicz, K. Kornowska, J. Kumirska, P. Stepnowski and K. Pazdro, A new approach for the estimation of expanded uncertainty of results of an analytical method developed for determining antibiotics in seawater using solid-phase extraction disks and liquid chromatography coupled with tandem mass spectrometry technique, J. Chromatogr. A, 2013, 1304, 138–146, DOI:10.1016/j.chroma.2013.07.018.
- P. Paíga, L. H. M. L. M. Santos and C. Delerue-Matos, Development of a multi-residue method for the determination of human and veterinary pharmaceuticals and some of their metabolites in aqueous environmental matrices by SPE-UHPLC–MS/MS, J. Pharm. Biomed. Anal., 2017, 135, 75–86, DOI:10.1016/j.jpba.2016.12.013.
- M. M. Coelho, A. R. Lado Ribeiro, J. C. G. Sousa, C. Ribeiro, C. Fernandes, A. M. T. Silva and M. E. Tiritan, Dual enantioselective LC–MS/MS method to analyse chiral drugs in surface water: monitoring in Douro River estuary, J. Pharm. Biomed. Anal., 2019, 170, 89–101, DOI:10.1016/j.jpba.2019.03.032.
- B. Kasprzyk-Hordern, V. V. R. Kondakal and D. R. Baker, Enantiomeric analysis of drugs of abuse in wastewater by chiral liquid chromatography coupled with tandem mass spectrometry, J. Chromatogr. A, 2010, 1217(27), 4575–4586, DOI:10.1016/j.chroma.2010.04.073.
- R. López-Serna, B. Kasprzyk-Hordern, M. Petrović and D. Barceló, Multi-residue enantiomeric analysis of pharmaceuticals and their active metabolites in the Guadalquivir River basin (South Spain) by chiral liquid chromatography coupled with tandem mass spectrometry, Anal. Bioanal. Chem., 2013, 405(18), 5859–5873, DOI:10.1007/s00216-013-6900-7.
- S. Ramage, D. Camacho-Muñoz and B. Petrie, Enantioselective LC-MS/MS for anthropogenic markers of septic tank discharge, Chemosphere, 2019, 219, 191–201, DOI:10.1016/j.chemosphere.2018.12.007.
- C. Ort, M. G. Lawrence, J. Reungoat and J. F. Mueller, Sampling for PPCPs in wastewater systems: comparison of different sampling modes and optimization strategies, Environ. Sci. Technol., 2010, 44(16), 6289–6296, DOI:10.1021/es100778d.
- B. Petrie, K. Proctor, J. Youdan, R. Barden and B. Kasprzyk-Hordern, Critical evaluation of monitoring strategy for the multi-residue determination of 90 chiral and achiral micropollutants in effluent wastewater, Sci. Total Environ., 2017, 579, 569–578, DOI:10.1016/j.scitotenv.2016.11.059.
- O. Hillebrand, S. Musallam, L. Scherer, K. Nödler and T. Licha, The challenge of sample-stabilisation in the era of multi-residue analytical methods: a practical guideline for the stabilisation of 46 organic micropollutants in aqueous samples, Sci. Total Environ., 2013, 454–455, 289–298, DOI:10.1016/j.scitotenv.2013.03.028.
- L. Mijangos, O. Urain, L. Ruiz-Rubio, H. Ziarrusta, M. Olivares, O. Zuloaga, A. Prieto and N. Etxebarria, Short-term stability assessment for the analysis of emerging contaminants in seawater, Environ. Sci. Pollut. Res., 2019, 26(23), 23861–23872, DOI:10.1007/s11356-019-05172-4.
- Official Journal of the European Communities, Commission decision (2002/657/EC): implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results, 2002, available from: http://faolex.fao.org/docs/pdf/eur49615.pdf Search PubMed.
- S. E. Evans and B. Kasprzyk-Hordern, Applications of chiral chromatography coupled with mass spectrometry in the analysis of chiral pharmaceuticals in the environment, Trends Environ. Anal. Chem., 2014, 1, e34–e51, DOI:10.1016/j.teac.2013.11.005.
- B. Petrie, J. Mrazova, B. Kasprzyk-Hordern and K. Yates, Multi-residue analysis of chiral and achiral trace organic contaminants in soil by accelerated solvent extraction and enantioselective liquid chromatography tandem–mass spectrometry, J. Chromatogr. A, 2018, 1572, 62–71, DOI:10.1016/j.chroma.2018.08.034.
- E. Sanganyado, Z. Lu and J. Gan, Mechanistic insights on chaotropic interactions of liophilic ions with basic pharmaceuticals in polar ionic mode liquid chromatography, J. Chromatogr. A, 2014, 1368, 82–88, DOI:10.1016/j.chroma.2014.09.054.
- J. P. Bagnall, S. E. Evans, M. T. Wort, A. T. Lubben and B. Kasprzyk-Hordern, Using chiral liquid chromatography quadrupole time-of-flight mass spectrometry for the analysis of pharmaceuticals and illicit drugs in surface and wastewater at the enantiomeric level, J. Chromatogr. A, 2012, 1249, 115–129, DOI:10.1016/j.chroma.2012.06.012.
- D. Camacho-Muñoz and B. Kasprzyk-Hordern, Simultaneous enantiomeric analysis of pharmacologically active compounds in environmental samples by chiral LC–MS/MS with a macrocyclic antibiotic stationary phase, J. Mass Spectrom., 2017, 52(2), 94–108, DOI:10.1002/jms.3904.
- P. Yang, T. McCabe and M. Pursch, Practical comparison of LC columns packed with different superficially porous particles for the separation of small molecules and medium size natural products, J. Sep. Sci., 2011, 34(21), 2975–2982, DOI:10.1002/jssc.201100530.
- S. E. Evans, P. Davies, A. Lubben and B. Kasprzyk-Hordern, Determination of chiral pharmaceuticals and illicit drugs in wastewater and sludge using microwave assisted extraction, solid-phase extraction and chiral liquid chromatography coupled with tandem mass spectrometry, Anal. Chim. Acta, 2015, 882, 112–126, DOI:10.1016/j.aca.2015.03.039.
- M. D. Gil-García, D. Barranco-Martínez, M. Martínez-Galera and P. Parrilla-Vázquez, Simple, rapid solid-phase extraction procedure for the determination of ultra-trace levels of pyrethroids in ground and sea water by liquid chromatography/electrospray ionization mass spectroscopy, Rapid Commun. Mass Spectrom., 2006, 20(16), 2395–2403, DOI:10.1002/rcm.2600.
- L. Maldaner and I. C. S. F. Jardim, Determination of some organic contaminants in water samples by solid-phase extraction and liquid chromatography-tandem mass spectrometry, Talanta, 2012, 100, 38–44, DOI:10.1016/j.talanta.2012.08.006.
- K. Wille, H. Noppe, K. Verheyden, J. Vanden Bussche, E. De Wulf, P. Van Caeter, C. R. Janssen, H. F. De Brabander and L. Vanhaecke, Validation and application of an LC-MS/MS method for the simultaneous quantification of 13 pharmaceuticals in seawater, Anal. Bioanal. Chem., 2010, 397(5), 1797–1808, DOI:10.1007/s00216-010-3702-z.
- EMEA, Guideline on the Environmental Risk Assessment of Medicinal Products for Human Use, EMEA/CHMP/SWP/4447/00 2006 Search PubMed.
- A. Lolić, P. Paíga, L. H. M. L. Santos, S. Ramos, M. Correia and C. Delerue-Matos, Assessment of non-steroidal anti-inflammatory and analgesic pharmaceuticals in seawaters of North of Portugal: occurrence and environmental risk, Sci. Total Environ., 2015, 508, 240–250, DOI:10.1016/j.scitotenv.2014.11.097.
- P. Paíga, A. Lolić, F. Hellebuyck, L. H. M. L. Santos, M. Correia and C. Delerue-Matos, Development of a SPE-UHPLC-MS/MS methodology for the determination of non-steroidal anti-inflammatory and analgesic pharmaceuticals in seawater, J. Pharm. Biomed. Anal., 2015, 106, 61–70, DOI:10.1016/j.jpba.2014.06.017.
- D. Camacho-Muñoz and B. Kasprzyk-Hordern, Multi-residue enantiomeric analysis of human and veterinary pharmaceuticals and their metabolites in environmental samples by chiral liquid chromatography coupled with tandem mass spectrometry detection, Anal. Bioanal. Chem., 2015, 407(30), 9085–9104, DOI:10.1007/s00216-015-9075-6.
- M. Llorca, M. Gros, S. Rodríguez-Mozaz and D. Barceló, Sample preservation for the analysis of antibiotics in water, J. Chromatogr. A, 2014, 1369, 43–51, DOI:10.1016/j.chroma.2014.09.089.
- J. J. Palmgrén, J. Mönkkönen, T. Korjamo, A. Hassinen and S. Auriola, Drug adsorption to plastic containers and retention of drugs in cultured cells under in vitro conditions, Eur. J. Pharm. Biopharm., 2006, 64(3), 369–378, DOI:10.1016/j.ejpb.2006.06.005.
- J. Bagnall, L. Malia, A. Lubben and B. Kasprzyk-Hordern, Stereoselective biodegradation of amphetamine and methamphetamine in river microcosms, Water Res., 2013, 47(15), 5708–5718, DOI:10.1016/j.watres.2013.06.057.
- G. Fedorova, O. Golovko, T. Randak and R. Grabic, Storage effect on the analysis of pharmaceuticals and personal care products in wastewater, Chemosphere, 2014, 111, 55–60, DOI:10.1016/j.chemosphere.2014.02.067.
- S. Letsinger, P. Kay, S. Rodríguez-Mozaz, M. Villagrassa, D. Barceló and J. M. Rotchell, Spatial and temporal occurrence of pharmaceuticals in UK estuaries, Sci. Total Environ., 2019, 678, 74–84, DOI:10.1016/j.scitotenv.2019.04.182.
- I. J. Buerge, T. Poiger, M. D. Müller and H.-R. Buser, Caffeine, an anthropogenic marker for wastewater contamination of surface waters, Environ. Sci. Technol., 2003, 37(4), 691–700, DOI:10.1021/es020125z.
- C. Potera, Caffeine in wastewater is a tracer for human fecal contamination, Environ. Health Perspect., 2012, 120(3), A108–A109 CrossRef PubMed.
- F. de Andrés, G. Castañeda and Á. Ríos, Use of toxicity assays for enantiomeric discrimination of pharmaceutical substances, Chirality, 2009, 21(8), 751–759, DOI:10.1002/chir.20675.
- C. Jacquot, D. J. David, A. M. Gardier and C. Sánchez, Escitalopram and citalopram: the unexpected role of the R-enantiomer, Encephale, 2007, 33(2), 179–187, DOI:10.1016/S0013-7006(07)91548-1.
- S. Evans, J. Bagnall and B. Kasprzyk-Hordern, Enantiomeric profiling of a chemically diverse mixture of chiral pharmaceuticals in urban water, Environ. Pollut., 2017, 230, 368–377, DOI:10.1016/j.envpol.2017.06.070.
- B. Kasprzyk-Hordern and D. R. Baker, Enantiomeric profiling of chiral drugs in wastewater and receiving waters, Environ. Sci. Technol., 2012, 46(3), 1681–1691, DOI:10.1021/es203113y.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ay00801j |
| This journal is © The Royal Society of Chemistry 2020 |