
Quantification of anthracene after dermal absorption test via APCI-tandem mass spectrometry
Xinyi
Sui
 a,
Julio E.
Terán
a,
Chengcheng
Feng
a,
Killian
Wustrow
b,
Caroline J.
Smith
b and
Nelson R.
Vinueza
*a
a,
Julio E.
Terán
a,
Chengcheng
Feng
a,
Killian
Wustrow
b,
Caroline J.
Smith
b and
Nelson R.
Vinueza
*a
aDepartment of Textile Engineering, Chemistry and Science, College of Textiles, North Carolina State University, Raleigh, NC 27695, USA. E-mail: nelson_vinueza@ncsu.edu
bDepartment of Health & Exercise Science, Appalachian State University, Boone, NC 28608, USA
First published on 5th May 2020
Abstract
An analytical method for the detection and quantification of anthracene from interstitial fluid samples was developed by using Atmospheric Pressure Chemical Ionization-Tandem Mass Spectrometry (APCI-MS/MS). The anthracene samples were obtained using intradermal microdialysis to assess dermal absorption of this polycyclic aromatic hydrocarbon (PAH). The experimental considerations were evaluated based on the chemical properties of this PAH and the detection limits of the instrumentation. The addition of an isotopically labeled internal standard allows the reduction of ionization suppression due to instrumental fluctuation and run-to-run deviation. The dermal extraction samples were prepared considering the proceeding conditions for measurement enhancement. Several variables for method validation including coefficient of determination (R2 = 0.993 ± 0.003), percent error (% error = 0 ± 2%), coefficient of variation (% CV = 5 ± 1%), lowest limit of detection (LOD = 39 ± 3 ng mL−1), and lowest limit of quantification (LLOQ = 129 ± 10 ng mL−1) were obtained from the inter- and intra-day measurements for the calibration and the quality control samples. Posterior to this, actual dermal interstitial fluid samples were measured, and it was observed they fitted into the ranges defined from the method development with good accuracy and precision. It was observed that the introduction of an internal standard while performing APCI-MS/MS allows an accurate and precise measurement of the concentration of anthracene to be obtained from dermal extraction samples. This method could be further used for complex mixtures to enhance our understanding of hazardous exposure of PAH on firefighter gear.
1. Introduction
Polycyclic Aromatic Hydrocarbons (PAHs) are chemical substances that possess two or more benzene molecules bound together. These molecules do not contain any other atoms, besides carbon and hydrogen, either within their structure or as a substituent. Moreover, the physicochemical properties and behavior of these compounds depend on the arrangement of the benzene rings available. For instance, an increment in their size increases their hydrophobicity and electrochemical stability, which correlates with increased persistence and accumulation in the environment.1 Furthermore, these compounds are semi-volatile (low vapor pressure) and have high melting and boiling points, which increases the difficulty of degradation.2Considering the adverse physiological responses which PAHs generate in the body of animals, particularly humans, it is important to mention that these compounds can irritate upon contact with the skin.3 Likewise, it has been shown that PAHs increase the risk of acquiring several types of cancer upon exposure.4–7
There are several natural and anthropogenic sources of PAH generation, with one of the most common sources being the combustion of any carbon-based compound.1 Consequently, due to the nature of their jobs and training, occupational workers including firefighters, are subjected to prolonged exposure to combustion products, most of which contain PAHs. When firefighters enter a burning building, their protective equipment and tools are in constant contact with PAHs in the air, thus these compounds normally deposit onto them. However, one of the problems regarding protective clothing and PAHs is their capability of permeating and accumulating on the clothing. Consequently, dermal absorption of PAHs may occur, and continue even after the removal of these garments. This results in prolonged skin irritation5,6,8 and systemic exposure.
Several studies indicate that besides respiratory exposure, the skin is one of the main gateways for PAH absorption, with the forehead, neck, and scrotum regions more prone to affection. These anatomical sites showed significantly more permeable behavior than other parts of the body.7,8
Respiratory routes of exposure to PAHs and other carcinogens have been extensively studied, with dermal absorption receiving less attention. Dermal absorption of PAHs can be estimated via metabolite detection in blood and urine3,5 and direct dermal tests utilizing skin swabs and tape stripping methods.6,7,9,10 These methods may confirm the presence of PAHs on the skin surface, superficial layers, or systemically, but do not accurately quantify dermal absorption. One approach involves the topical application of ointments or patches coupled with interstitial fluid sampling via intradermal microdialysis to quantify absorption. The concentration of these ointments intends to replicate real exposure conditions and allow researchers to closely study this absorption phenomenon over time.8 The most commonly used analytical quantification methods reported for these dermal and urine tests include high-performance liquid chromatography (HPLC) with a fluorescence spectrophotometer11 and gas chromatography-mass spectrometry (GC-MS).12
Mass spectrometry itself is a powerful technique employed in several fields, due to its sensitivity and versatility. The possibility of detection and quantification on mass spectrometry relies heavily on the selection of the ionization method. Atmospheric pressure chemical ionization (APCI) is an ionization technique, which is effective for polar and non-polar molecules like PAHs. This ionization method has been employed in the characterization of petroleum asphaltenes,13 aromatic analytes,14 saturated and unsaturated hydrocarbons,15 and intact carbohydrates with reproducible results.16 Furthermore, reproducible and repeatable quantifications of the number of hydrocarbons can be obtained by the addition of an internal standard to the solution employed for mass spectrometry analysis. Both strategies can enhance the detection and quantification limits of the analyte of interest without generating additional modifications on the instrument.17
Mass spectrometry has been employed for PAH quantification, however, it has not been employed for evaluation on dermal based matrices.18–22 For that reason, this paper introduces an APCI-MS method in combination with a novel sampling protocol for human dermal exposure.23 The present study selected anthracene since it is a non-mutagenic, non-carcinogenic PAH and it possesses a relatively low molecular weight. These considerations made it suitable for topical application in humans and suitable for recovery via intradermal microdialysis. Furthermore, the development of this quantification method has useful applications for a better understanding of occupational exposure and dermal absorption in firefighters during fire suppression and improves our knowledge of how PAHs can interact with the human skin.
2. Methods
2.1 Reagents
2.2 Methodology
A quantity of 0.2 g of 2% Anthracene solution mixed in Aquaphor® (Beiersdorf Inc., CT. USA) was weighed and applied to the skin surface directly over the MD site. Previous pilot testing determined the solution medium appropriate for mixing and topical delivery of anthracene (unpublished). A local heating unit (Moor Instruments, UK) was attached to the skin surface over the site and clamped at 38 °C to locally warm the skin and replicate skin temperatures observed during fire suppression in firefighters.25 Dialysate sample collection was initiated 2.5 hours following anthracene application based on prior pilot work and continued intermittently until 4.5 hours following application. During this time, dialysate was collected for 15 minutes and micro perfusion pumps were intermittently turned off (no perfusion) for 30 minutes between samples to allow equilibration of anthracene between the interstitial fluid and fluid perfusing the MD fiber (dialysate). Upon completion of sample collection, the micro dialysis fiber was removed and the arm thoroughly cleaned to remove anthracene. All sites were covered for 48 hours to avoid irritation from sunlight exposure. After collection, tubes containing dialysate samples were stored in a refrigerator at 5 °C before being transferred to NC State University for APCI-MS/MS measurement. All pharmacologics and methods of delivery for this study were approved by the Food and Drug Administration (FDA) prior to experimentation (IND 141409).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 using a 10 mL volumetric flask and HPLC grade acetone.
:10 using a 10 mL volumetric flask and HPLC grade acetone.
For the internal standard solution, 1.2 mL of a 200 ppm anthracene-d10 standard solution was diluted with HPLC grade acetone into a 25 mL volumetric flask to obtain a final concentration of a 10 ppm. All resulting solutions were stored in a refrigerator at 5 °C prior use.
| Concentration [ng mL−1] | Anthracene (10 ppm) [μL] | Anthracene-d10 (10 ppm) [μL] | Acetone [μL] | Total [μL] |
|---|---|---|---|---|
| a Note: QC = quality control. | ||||
| 100 | 10 | 200 | 790 | 1000 |
| 200 | 20 | 200 | 780 | 1000 |
| 300 | 30 | 200 | 770 | 1000 |
| 500 | 50 | 200 | 750 | 1000 |
| 1000 | 100 | 200 | 700 | 1000 |
| 1500 | 150 | 200 | 650 | 1000 |
| QC-150 | 50 | 200 | 750 | 1000 |
| QC-1200 | 120 | 200 | 680 | 1000 |
After preparation, both calibrations and QC solutions were stored in a refrigerator at 5 °C before the MS analyses. The quantification experiments were done on a Velos Pro Linear Ion Trap Mass Spectrometer (LTQ-MS) from Thermo Fisher Scientific, where all calibration solutions and QC solutions were measured by APCI-MS/MS to create a calibration curve. To facilitate method validation, the preparation of calibration solutions and quantification were repeated multiple times on the same day (intra-day repeats) and on different days (inter-day repeats).
Linearity is represented by the coefficient of determination (R2) obtained from intra-day and inter-day repeats. Sensitivity is evaluated by Limit of Detection (LOD) and Lowest Limit of Quantification (LLOQ). Accuracy is evaluated by the mean percent error of calibration standard and QC standard against their nominal concentration from intra-day and inter-day repeats. Finally, precision is evaluated by the mean coefficient of variance (% CV) from the performed replications.26
Before extraction, the weight of the sample vial was measured, using an analytical balance. 300 μL of HPLC grade acetone was then added to the sample tube, which was mixed using a VWR vortex mixer for 15 seconds at 2200 rpm. The liquid in the vial was then transferred to a 2 mL HPLC vial, using a 1 mL plastic syringe.
Next, another 100 μL of HPLC grade acetone was added to the sample tube and mixed using a VWR vortex mixer for 15 seconds at 2200 rpm. This step was repeated once more. Posterior to the extraction, the container was dried and weighed again.
The solution in the HPLC vial was then mixed with 200 μL of 10 ppm anthracene-d10 solution and 300 μL of HPLC grade acetone. This HPLC vial was analyzed employing the same instrument conditions to establish the calibration curve. Abundance ratios of each sample were obtained from tandem mass spectrometry where the loss of C2H2 (C2D2 for anthracene-d10) was monitored and the ratio between the resulting non deuterated (152 m/z) and deuterated ions (160 m/z) was used to calculate anthracene concentration using the regression model established in the calibration system (Fig. 1 provides a schematic of the proposed transition).
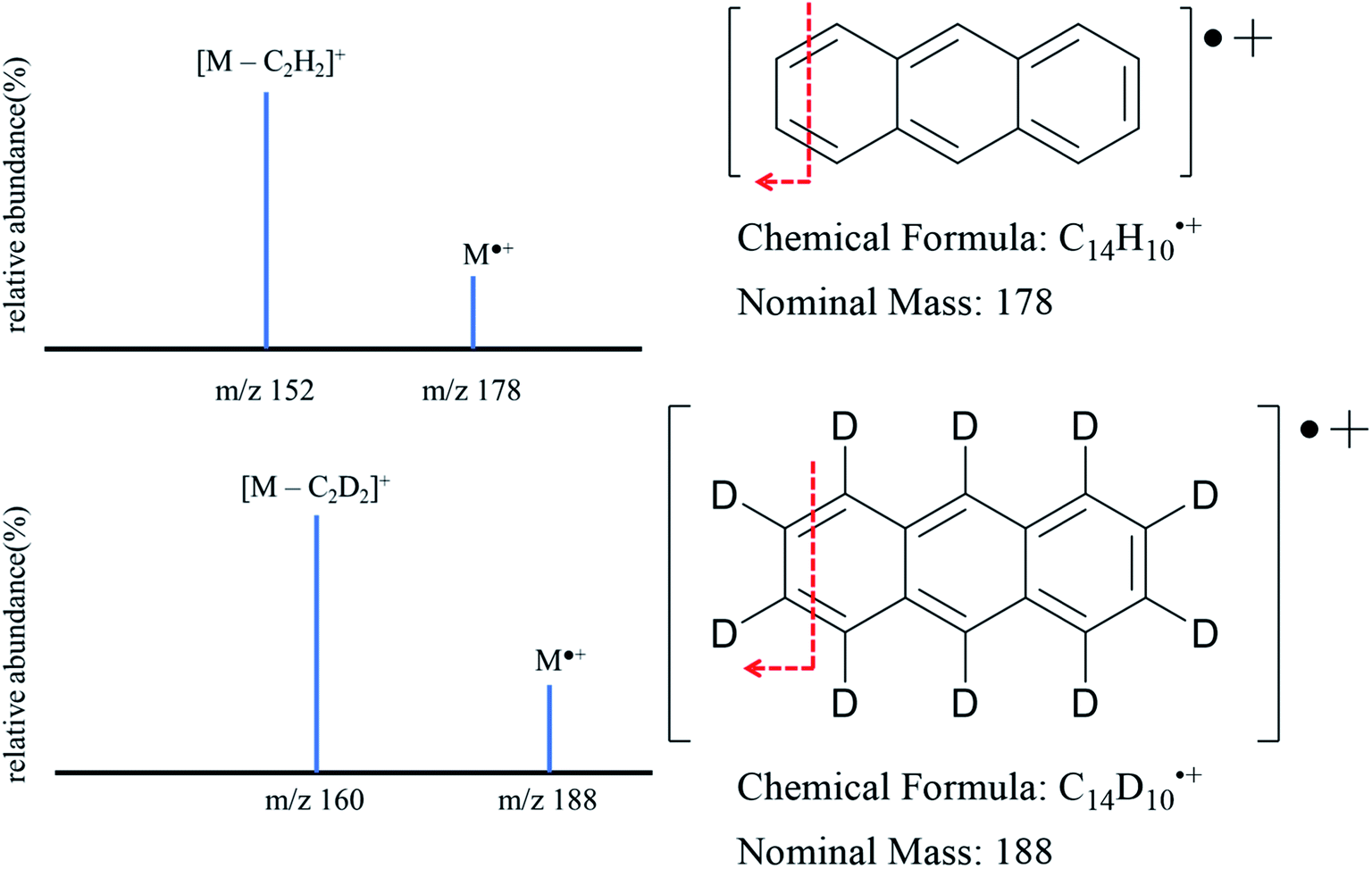 | ||
| Fig. 1 MS/MS spectra of anthracene (m/z 178 > m/z 152) and anthracene-d10 (m/z 188 > m/z 160). | ||
2.3 Instrumentation
Measurements of MS/MS abundance ratios in the calibration solution and samples from the in vivo dermal absorption study were carried out on an LTQ Velos Pro linear ion trap mass spectrometer coupled with Ultimate 3000 UHPLC system from Thermo Scientific (Fisher Scientific, PA, USA). Samples were introduced via an autosampler, with an injection volume of 30 μL. The mobile phase was made up of 95% LC-MS grade acetonitrile and 5% LC-MS grade water. The system was configured to bypass the column. The total run time for each sample was four minutes. Ionization was performed in positive mode via a commercial APCI source (Thermo Fisher Scientific™). The following source parameters are used including sheath gas (nitrogen gas): 30 arb. (arbitrary units), auxiliary gas (nitrogen gas): 5 arb., sweep gas: 0 arb., APCI discharge current 5 μA, capillary temperature: 275 °C, and S-lens RF level of 53.3%. For quantification, the best MS/MS conditions were reached by optimizing the collision energy to obtain the highest signal. The selected reaction monitoring (SRM) method recorded the following transitions for the analytes: m/z 178 to 152 for anthracene and m/z 188 to 160 for anthracene-d10. For Higher-energy Collision Dissociation (HCD) a normalized collision energy of 85 as described by the manufacturer was used (see Fig. 1). Data were collected by Tune Plus software (Thermo Scientific) and data processing was performed by using Xcalibur software (version 4.0.27.19) (Fig. 2).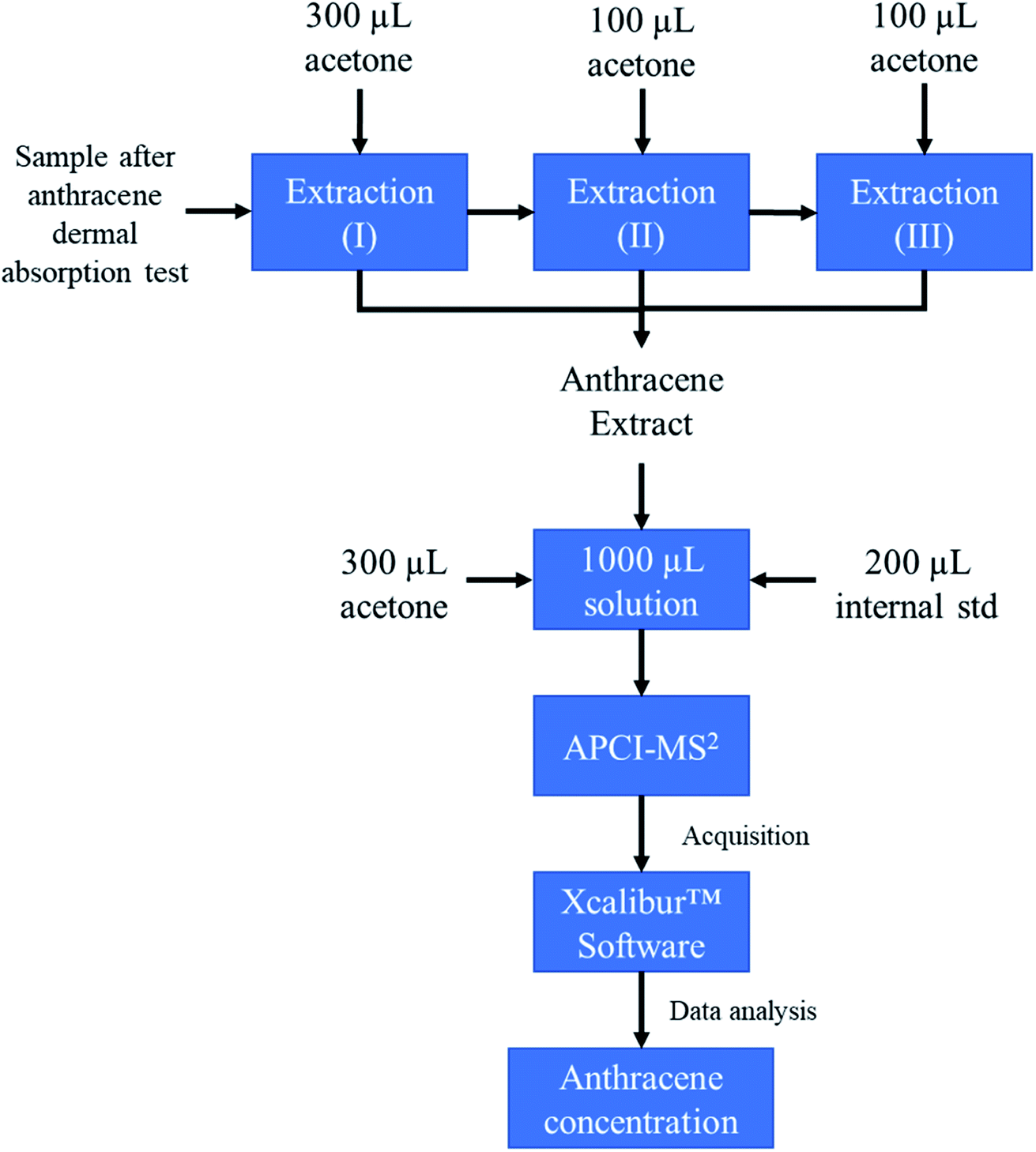 | ||
| Fig. 2 Schematic of the analytical procedure employed in this work. | ||
3. Results and discussion
3.1 Establishment of the calibration system
Table 2 summarized the linear calibration data obtained from APCI-MS/MS. Fifteen linear regression models were generated, where it was found that the mean coefficient of determination is 0.993 ± 0.010. This value suggests that, under these experimental conditions, the change of anthracene concentration in solution is highly proportional to the product ion abundance ratio between anthracene and anthracene-d10. This expected linearity between analyte concentration and abundance ratio from tandem mass spectrometry could be explained by the reduction of ionization suppression. It is important to consider that ionization will not be the same for all molecules (ionization bias) in the case of dermal samples.| R 2 | m | b | |
|---|---|---|---|
| a Note: linear regression in form y = mx + b, values are displayed to three significant figures when available. | |||
| Day 1 | 0.979 | 0.00067 | 0.107 |
| 0.996 | 0.00061 | 0.108 | |
| 0.986 | 0.00056 | 0.120 | |
| Day 2 | 0.999 | 0.00055 | 0.118 |
| 0.996 | 0.00061 | 0.100 | |
| 0.998 | 0.00059 | 0.105 | |
| Day 3 | 0.998 | 0.00061 | 0.0945 |
| 0.999 | 0.00064 | 0.0986 | |
| 0.996 | 0.00068 | 0.0843 | |
| Day 4 | 0.960 | 0.00059 | 0.117 |
| 0.997 | 0.00059 | 0.102 | |
| 0.998 | 0.00056 | 0.111 | |
| Day 5 | 0.997 | 0.00056 | 0.101 |
| 0.993 | 0.00063 | 0.0721 | |
| 0.997 | 0.00066 | 0.0623 | |
| Average | 0.993 | 0.00061 | 0.100 |
| Std error | 0.010 | 0.00001 | 0.010 |
The reasons behind the employment of APCI-MS/MS for this method development are (1) to ionize molecules which, because of their nature (non-polar molecules), will not ionize employing Electrospray ionization (ESI) and (2) to overcome the inconsistency brought by ionization suppression and instrumental fluctuation an internal standard and tandem mass spectrometry (MS/MS) are introduced. Under MS/MS conditions, the ratio between selected fragments can be established for quantification purposes. The results can be observed in the calibration curve obtained in these experiments.
3.2 Method validation
The precision and accuracy data obtained from intra-day and inter-day repeats of each concentration level are shown in Table 3. In this table, accuracy was shown as a relative error against nominal concentration (% error) in percentage and concentration measured by calibration curves.| Concentration (ng mL−1) | Intra-day repeats | Inter-day repeats | ||
|---|---|---|---|---|
| Accuracy (% error) | Precision (% CV) | Accuracy (% error) | Precision (% CV) | |
| a Note: values reported to three significant figures, CV = coefficient of variation, *quality control standard. | ||||
| 100 | 14.2 (114 ng mL−1) | 7.37 | 8.10 (108 ng mL−1) | 5.95 |
| 200 | 0.571 (201 ng mL−1) | 1.37 | −4.79 (190 ng mL−1) | 2.29 |
| 300 | 0.998 (303 ng mL−1) | 3.66 | 1.23 (304 ng mL−1) | 4.16 |
| 500 | −1.66 (492 ng mL−1) | 5.88 | −5.43 (473 ng mL−1) | 4.69 |
| 1000 | −3.32 (967 ng mL−1) | 3.15 | −0.877 (991 ng mL−1) | 7.36 |
| 1500 | 1.55 (1520 ng mL−1) | 4.99 | −0.987 (1480 ng mL−1) | 5.41 |
| 150* | 10.1 (165 ng mL−1) | 0.613 | 4.77 (157 ng mL−1) | 4.98 |
| 1200* | −0.662 (1190 ng mL−1) | 1.88 | 8.10 (1180 ng mL−1) | 2.81 |
Precision was shown as the Coefficient of Variation in percentage (% CV), alternatively called the Relative Standard Deviation in percentage (% RSD), and was calculated based on the ratio between standard deviation and mean. According to the FDA guidance, % CV from intra-day and inter-day repeats should be within ± 15%, except for concentrations around the Lowest Limit of Quantification (LLOQ), where the threshold can be widened to ± 20%. Moreover, it is noticeable that the percentages of error from all calibration levels, including QC standards, were below ± 15%, which suggests that this quantification method has sufficient accuracy, according to FDA guidelines.
Higher percentages of error are associated with the 100 ng mL−1 calibration standard and the 150 ng mL−1 QC standard. These values could be explained by considering that lower concentrations experience more influence from instrumental fluctuation and random error from sample preparation.
In addition, % CVs from all calibration standards and QC standards were below ± 10%, indicating that this quantification method developed on anthracene provides better precision than FDA requirements. In addition to precision and accuracy values at each concentration, a summary of validation parameters is provided in Table 4.
| Intra-day repeats | Inter-day repeats | |
|---|---|---|
| a Note: accuracy and precision data is reported as the average across all quantitation standards. | ||
| Linearity (R2) | 0.998 ± 0.001 | 0.993 ± 0.003 |
|
||
| Sensitivity | ||
| LLOQ | 129 ± 10 ng mL−1 | |
| LOD | 39 ± 3 ng mL−1 | |
|
||
| Accuracy (mean% error) | ||
| Quantitation standards | 2 ± 3 | 0 ± 2 |
|
||
| Quality control standards | ||
| 150 ng mL−1 | 10 ± 3 | 5 ± 9 |
| 1200 ng mL−1 | 0 ± 1 | −2 ± 1 |
| Precision (mean CV%) | 4 ± 1 | 5 ± 1 |
Overall, by performing intra-day and inter-day repeats, the anthracene quantification method based on tandem mass spectrometry and the internal standard has shown good linearity (R2 > 0.99), good accuracy (mean% error less than 15%) and good precision (mean% CV less than 15%). Another parameter which needs to be evaluated during method validation is sensitivity, which is represented by Lowest Limit of Quantification (LLOQ) and is estimated by LLOQ = 10σ/m, where σ is the standard deviation of the y-intercept of the linear regressions and m is the average slope of the linear regressions.16 Based on this equation, the LLOQ of this quantification method was determined to be 129 ± 10 ng mL−1, which is noticeably lower than the mean anthracene concentration obtained from the dermal absorption study (205 ± 5 ng mL−1). In addition, the implementation of the APCI ionization for anthracene brought specificity. Compared with other anthracene reported quantification methods, which were mainly developed for conventional matrices such as urine and blood,27 this method does not require sample preparation of the unique MD dialysate samples after dermal absorption, which are limited liquid quantities (average weight of sample extracted is 14 ± 1 mg).
4. Conclusions
A method for anthracene quantification based on APCI-MS/MS was successfully developed and validated, according to the guideline provided by the U.S. Food and Drug Administration. The combination of APCI, HCD, and deuterated internal standard ensures the linearity of calibration curves. Accuracy and precision of this method were evaluated based on intra-day and inter-day repeats of calibration curves.Finally, the concentration of anthracene in dialysate samples obtained from in vivo dermal absorption assessment via intradermal microdialysis (205 ± 5 ng mL−1) was successfully measured by this method. Further, results suggest that this quantification system has an appropriate sensitivity, as the measured anthracene concentration falls within the linear range of the calibration curve. Findings in this study could strengthen our understanding of Polycyclic Aromatic Hydrocarbon identification and provide the basis for further studies regarding firefighter carcinogen exposure and protection strategies. In terms of future steps, through a collaboration with Boone Fire Department, the measurement of anthracene concentration via APCI-MS/MS could be applied to other matrices including firefighter turonout gear and dermal absorption after simulated fire scenes, and at different body locations (e.g., neck). In addition, this method is aiming for the future use of portable MS devices28–30 to enable on-site testing of PAH.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NRV gratefully thank the NC State Chancellor’s Faculty Excellence Program.References
- A. T. Lawal, Cogent Environ. Sci., 2017, 3, 1339841 CrossRef.
- O. O. Alegbeleye, B. O. Opeolu and V. A. Jackson, Environ. Manag., 2017, 60, 758–783 CrossRef PubMed.
- J. Unwin, J. Cocker, E. Scobbie and H. Chambers, Ann. Occup. Hyg., 2006, 50, 395–403 CAS.
- A. Ben, H. Emma, U. John and F. Tony, Environ. Health Perspect., 2004, 112, 970–978 CrossRef PubMed.
- K. W. Fent, C. Toennis, D. Sammons, S. Robertson, S. Bertke, A. M. Calafat, J. D. Pleil, M. A. Geer Wallace, S. Kerber, D. L. Smith and G. P. Horn, Int. J. Hyg. Environ. Health, 2019, 222, 991–1000 CrossRef CAS PubMed.
- K. Fent, J. Eisenberg, J. Snawder, D. Sammons, J. Pleil, M. Stiegel, C. Mueller, G. Horn and J. Dalton, Ann. Occup. Hyg., 2014, 58(7), 830–845 CAS.
- A. A. Stec, K. E. Dickens, M. Salden, F. E. Hewitt, D. P. Watts, P. E. Houldsworth and F. L. Martin, Sci. Rep., 2018, 8, 2476 CrossRef PubMed.
- J. G. M. VanRooij, J. H. C. De Roos, M. M. Bodelier-Bade and F. J. Jongeneelen, J. Toxicol. Environ. Health, 1993, 38, 355–368 CrossRef CAS PubMed.
- C. S. Baxter, J. D. Hoffman, M. J. Knipp, T. Reponen and E. N. Haynes, J. Occup. Environ. Hyg., 2014, 11, D85–D91 CrossRef CAS PubMed.
- B. Strandberg, A. Julander, M. Sjöström, M. Lewné, K. A. Hatice and C. Bigert, Chemosphere, 2018, 198, 274–280 CrossRef CAS PubMed.
- F. J. Jongeneelen, R. P. Bos, R. B. M. Anzion, J. L. Theuws and P. T. Henderson, Scand. J. Work, Environ. Health, 1986, 12, 137–143 CrossRef CAS PubMed.
- B. Strandberg, A. Julander, M. Sjöström, M. Lewné, K. A. Hatice and C. Bigert, Chemosphere, 2018, 198, 274–280 CrossRef CAS PubMed.
- M. R. Hurt, D. J. Borton, H. J. Choi and H. I. Kenttämaa, Energy Fuels, 2013, 27, 3653–3658 CrossRef CAS.
- L. M. Amundson, V. A. Gallardo, N. R. Vinueza, B. C. Owen, J. N. Reece, S. C. Habicht, M. Fu, R. C. Shea, A. B. Mossman and H. I. Kenttämaa, Energy Fuels, 2012, 26, 2975–2989 CrossRef CAS.
- J. Gao, B. C. Owen, D. J. Borton, Z. Jin and H. I. Kenttämaa, J. Am. Soc. Mass Spectrom., 2012, 23, 816–822 CrossRef CAS PubMed.
- N. R. Vinueza, V. A. Gallardo, J. F. Klimek, N. C. Carpita and H. I. Kenttämaa, Fuel, 2013, 105, 235–246 CrossRef CAS.
- K. S. Boes, M. S. Roberts and N. R. Vinueza, J. Am. Soc. Mass Spectrom., 2018, 29, 535–542 CrossRef CAS PubMed.
- K. A. Anderson, M. J. Szelewski, G. Wilson, B. D. Quimby and P. D. Hoffman, J. Chromatogr. A, 2015, 1419, 89–98 CrossRef CAS PubMed.
- S. C. C. Lung and C. H. Liu, Sci. Rep., 2015, 5, 1–13 Search PubMed.
- G. Park, P. Brunswick, H. Kwok, M. Haberl, J. Yan, C. MacInnis, M. Kim, C. Helbing, G. van Aggelen and D. Shang, Anal. Methods, 2018, 10, 5559–5570 RSC.
- J. Jeffery, M. Carradus, K. Songin, M. Pettit, K. Pettit and C. Wright, Chem. Cent. J., 2018, 12, 27 CrossRef CAS PubMed.
- F. P. M. Jjunju, S. Maher, A. Li, A. K. Badu-Tawiah, S. Taylor and R. Graham Cooks, J. Am. Soc. Mass Spectrom., 2015, 26, 271–280 CrossRef CAS PubMed.
- A. E. Stanhewicz, R. S. Bruning, C. J. Smith, W. L. Kenney and L. A. Holowatz, J. Appl. Physiol., 2012, 112, 791–797 CrossRef CAS PubMed.
- L. A. Holowatz and W. L. Kenney, Am. J. Physiol.: Heart Circ. Physiol., 2007, 293, H1090–H1096 CrossRef CAS PubMed.
- G. P. Horn, R. M. Kesler, S. Kerber, K. W. Fent, T. J. Schroeder, W. S. Scott, P. C. Fehling, B. Fernhall and D. L. Smith, Ergonomics, 2018, 61, 404–419 CrossRef PubMed.
- B. Booth and L. Kux, Bioanalytical Method Validation Guidance for Industry, FDA, Washington, 2018 Search PubMed.
- Z. Li, C. D. Sandau, L. C. Romanoff, S. P. Caudill, A. Sjodin, L. L. Needham and D. G. Patterson, Environ. Res., 2008, 107, 320–331 CrossRef CAS PubMed.
- G. Huang, L. Gao, J. Duncan, J. D. Harper, N. L. Sanders, Z. Ouyang and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2010, 21, 132–135 CrossRef CAS PubMed.
- Z. E. Lawton, A. Traub, W. L. Fatigante, J. Mancias, A. E. O'Leary, S. E. Hall, J. R. Wieland, H. Oberacher, M. C. Gizzi and C. C. Mulligan, J. Am. Soc. Mass Spectrom., 2017, 28, 1048–1059 CrossRef CAS PubMed.
- P. Fedick, W. Fatigante, Z. Lawton, A. O'Leary, S. Hall, R. Bain, S. Ayrton, J. Ludwig and C. Mulligan, Instruments, 2018, 2, 5 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |