DOI:
10.1039/C9AY01976F
(Paper)
Anal. Methods, 2020,
12, 97-103
Pre-lab proteolysis for dried serum spots – a paper-based sampling concept targeting low abundant biomarkers†
Received
11th September 2019
, Accepted 26th November 2019
First published on 29th November 2019
Abstract
Paper-based sampling of biological matrices in combination with mass spectrometry has proven to be a promising technique for bottom-up analysis of proteins. However, these techniques have been challenging for determining low abundance proteins (pg mL−1 to low ng mL−1) due to low sampling volumes and adsorption of proteins to the sampling material. Here we are introducing a paper-based sampling concept in chip format with in-device proteolysis combined with selective proteotypic epitope peptide extraction. The sampling concept is demonstrated with the low abundant biomarker proGRP and showed promising performance from 500 pg mL−1 to 1000 ng mL−1 (R2 = 0.99) of proGRP spiked to human serum samples (all RSDs within 26%) in a five point concentration curve (n = 3). LOD and LOQ were determined to 150 pg mL−1 and 500 pg mL−1, respectively. The combination of paper-based sampling with in-device proteolysis and subsequent proteotypic epitope peptide immunocapture showed promising performance for other clinical applications.
1 Introduction
During the recent years new paper-based sampling techniques such as in-device immunocapture1 and in-device proteolysis2,3 have been introduced to simplify sampling and analysis of proteins in biological matrices sampled on paper. In addition to the paper-based devices, other sampling means with proteolysis,4,5 immunocapture6–8 or plasma separation membranes9 have been demonstrated. These devices allow easy and integrated sample preparation and have shown to significantly simplify and shorten the laboratory labor and time associated with the paper-based sampling methodology in protein analysis. Other efforts on improving sample preparation with respect to cleaner sample extracts after sample preparation for protein analyses (buffered solution or venipuncture as sampling) have also been introduced: by specifically capturing and enriching a proteotypic peptide (post proteolysis) rather than capturing intact protein before it is digested has shown to yield clean sample extracts. This has been described using both peptide capture by anti-protein antibodies,10–12 and molecular imprinted polymers (MIPs; also called plastic antibodies),13 as well as for peptide capture using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA).14 Recently we introduced a paper-based sampling concept in a chip format15 that holds the potential to be immobilized with trypsin3 to digest proteins in-device during the sampling stage. This device simplifies the workflow related to these analysis by integrating proteolysis within the sampling device and has also shown to be feasible with proteins needing reduction and alkylation.2,3 Combining the wax printed device introduced in chip format to hold liquid static in the immobilized sampling material (with immobilized trypsin) carry the potential to simplify and shorten conventional laboratory time for protein analysis. The latter combined with proteotypic peptide enrichment could make for a promising workflow providing highly clean sample extracts for mass spectrometric determinations. Therefore, the aim of this study was to explore paper-based smart sampling with integrated proteolysis combined with (immobilized with monoclonal antibodies (mAb)) for a fast and high performing targeted sampling method for low abundant proteins in serum. For the demonstration, a serum marker for small-cell lung cancer pro-gastrin-releasing peptide (ProGRP) was used as model analyte, however, the concept could readily be adapted to other proteins as well.
2 Experimental
2.1 Chemicals and reagents
Acetonitrile (99.9%, (ACN)), sodium phosphate dodecahydrate (99–102%), sodium phosphate monobasic (99–102%) were purchased from Merck (Darmstadt, Germany). Ethanolamine (≥99%) and ammonium bicarbonate (BioUltra, ≥99.5%, (ABC)) were purchased from Fluka (Milwaukee, WI, USA). Formic acid (≥98%, (FA)), benzamidine (≥95%), N,N-dimethylformamide anhydrous (DMF), 3-(trimethoxysilyl)propyl methacrylate (γ-MAPS, 98%), 1-heptanol, 2,2-diphenyl-1-picrylhydrazyl hydrate (DPPH), 2-hydroxyethyl methacrylate (HEMA, 97%, containing 200–220 ppm monomethyl ether hydroquinone), initiator 2,2′-azobis(2-methylpropionitrile) (98%, (AIBN)) and trypsin from bovine pancreas TPCK treated (≥10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 BAEE units per mg protein) were purchased from Sigma-Aldrich. 2-Vinyl-4,4-dimethylazlactone (VDM) was purchased from Polysciences Inc. (Warrington, PA, USA). Water used in this experiment was filtrated through a Merck Millipore Milli-Q integral 3 water dispenser (resistivity: 18.2 MΩ cm−1). ProGRP isoform 1 (cloned) and anti-ProGRP (monoclonal antibody (mAb) M18) were provided by the Central Laboratory, Norwegian Radium Hospital, Oslo University Hospital (Oslo, Norway). Tosyl-activated M280 Dynabeads was purchased from Life Technologies (Oslo, Norway). The peptide internal standard [NH2-ALGNQQPSWDSEDSSNF(K*)-COOH] (purity: >95%), labeled 13C/15N at K*, was bought from Innovagen (Lund, Sweden).
000 BAEE units per mg protein) were purchased from Sigma-Aldrich. 2-Vinyl-4,4-dimethylazlactone (VDM) was purchased from Polysciences Inc. (Warrington, PA, USA). Water used in this experiment was filtrated through a Merck Millipore Milli-Q integral 3 water dispenser (resistivity: 18.2 MΩ cm−1). ProGRP isoform 1 (cloned) and anti-ProGRP (monoclonal antibody (mAb) M18) were provided by the Central Laboratory, Norwegian Radium Hospital, Oslo University Hospital (Oslo, Norway). Tosyl-activated M280 Dynabeads was purchased from Life Technologies (Oslo, Norway). The peptide internal standard [NH2-ALGNQQPSWDSEDSSNF(K*)-COOH] (purity: >95%), labeled 13C/15N at K*, was bought from Innovagen (Lund, Sweden).
2.2 Preparation of sampling chips
Pre-cut DMPK-C circles (6.0 mm in diameter) were silanized and polymerized as previously described by Skjærvø et al.3 with minor modifications. The polymerized circles were further immobilized with trypsin and installed in wax printed sampling cartridges. The polymerization, immobilization and fabrication of the wax printed sampling cartridges is presented in S1.
2.3 Preparation of antibody immobilized magnetic beads
Immobilization of the monoclonal antibody M18 to magnetic Dynabeads was performed with minor modifications according to a previously described procedure by Paus and Nustad.16 The procedure in short is described in S2.
2.4 Sample preparation
2.4.1 Human serum samples.
Freeze thawed human serum samples from healthy blood donors (stored at −30 °C) were obtained from the blood bank at Oslo University Hospital, Ullevål (Oslo, Norway). The use of serum from healthy subjects for our research purposes was performed in strict accordance to Norwegian law (“Lov om medisinsk og helsefaglig forskning (helseforskningsloven)”) and have followed the principles outlined in the Declaration of Helsinki for all human experimental investigations. The research project is registered in the in the database for health related research at the Department of Pharmacy, University of Oslo (Oslo, Norway). Informed consent was obtained from all subjects. Methods used to analyze all serum samples were in accordance with relevant guidelines and regulations (mentioned above).
2.4.2 Sample application, extraction, immunocapture and sample clean-up.
Fifteen micro liter sample (proGRP spiked serum) was applied to the sampling device and dried at ambient temperature (the proteins are then digested during drying of the sample). After the serum sample had dried, 10 μL (10 ng mL−1 in 2% FA) internal standard was added and subsequently dried prior to extraction. Extraction of the sampling spots was carried out in ambient temperature with 1000 μL PBS and 20 μL M18 immobilized Dynabeads for 60 min under agitation at 1150 rpm with an Eppendorf (Hamburg, Germany) comfort mixer. After the extraction, the samples were placed in a magnetic rack and the sample matrix was removed. Washing of the mAb beads was further performed by adding and subsequently removing after agitation the following solutions; 500 μL PBS, 500 μL tris-HCL (pH 7.4) and 300 μL 50 mM ABC. Elution of the peptide from the magnetic beads was performed with 30 μL 2% FA and transferred to HPLC-vials. A schematic illustration of the workflow is presented in ESI 3 Fig. S1.†
2.5 LC-MS method
Sample loading (5 μL) on a Thermo Fischer Scientific (Waltham, MA, USA) Acclaim™ PepMap™ 100 C18 trap column (L: 5.0 mm × 300 μm ID, dp: 5.0 μm) was performed over five min at 10.0 μL min−1 (water/ACN/FA (2:98:0.1 v/v)). Mobile phase A (MP A) consisted of water/ACN/FA (95/5/0.1 v/v/v). The chromatographic separation was carried out at 250 nL min−1 over 15 minutes from 5–70% mobile phase B (MP B, ACN/water/FA (95/5/0.1 v/v)) followed by 10 and 15 minutes with 70% MP B and starting conditions, respectively. The chromatography was performed at 35 °C on an Acclaim™ PepMap™ 100 C18 (ID: 75.0 μm, L: 150 mm, dp: 3.0 μm) separation column coupled to a Dionex (Sunnyvale, CA, USA) Ultimate 3000 RSLC nano system. Detection was performed using a Thermo Fischer TSQ Quantiva operated in SRM positive mode nano ESI with 2.25 kV applied voltage. The MS parameters were set as followed: 350 °C ion transfer tube, 35 V collision energy, 1.5 mTorr CID gas, 1 second scan cycle and Q1 and Q3 resolution at 0.7 and 0.7 at FWHM, respectively. Quantification of target ions was carried out by measuring precursor ion m/z 1005.45 and product ion m/z 1028.33 and m/z 1398.53 for the A-peptide (AA-sequence: ALGNQQPSWDSEDSSNFK) and precursor ion m/z 1009.70 and product ion m/z 1036.50 and m/z 1406.70 for the internal standard (AA-sequence: [NH2-ALGNQQPSWDSEDSSNF(K*)-COOH])
3 Results and discussion
3.1 Extraction
3.1.1 When to perform immunocapture.
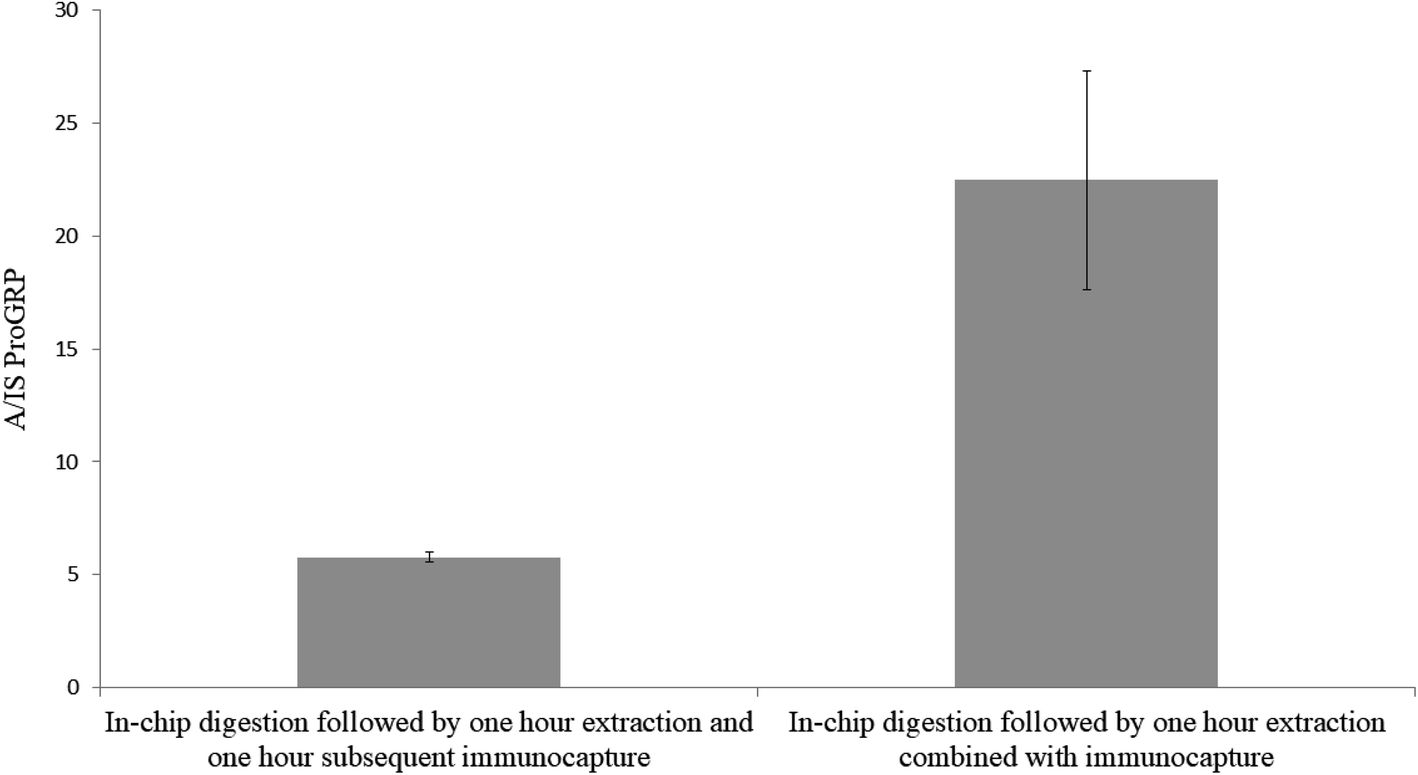
To evaluate if sample extraction after in-device proteolysis could be combined with immunocapture to accelerate the sample preparation, two sample sets (A and B; n = 3 each) were prepared identically (proGRP spiked to serum). After sampling, sample set A was extracted for one hour in PBS and subsequently transferred to a new Eppendorf tube for an additional one hour of immunocapture in the same solution by addition of mAb immobilized beads (the sampling material is not included in the new Eppendorf tube). Sample set B was extracted for one hour in PBS with the immunocapture beads in the extracting solution making for one hour less sample preparation compared to the counterpart. From Fig. 1 it is apparent that it was beneficial to combine sample extraction and immunocapture since it resulted in an approx. four times more intense signals (the absolute intensity for the IS was the same for the two sample sets). However, the relative standard deviation was lower for extraction and immunocapture in separate steps (RSD 4% for sample set A vs. RSD 22% for sample set B). Nevertheless, the combined extraction was chosen for all further experiments given the significant increase in signal intensities. This increase in signal could be related to the one less pipetting step where analyte could be lost from absorption to the pipette tips or new Eppendorf tubes (even though the tubes are of protein low binding material).
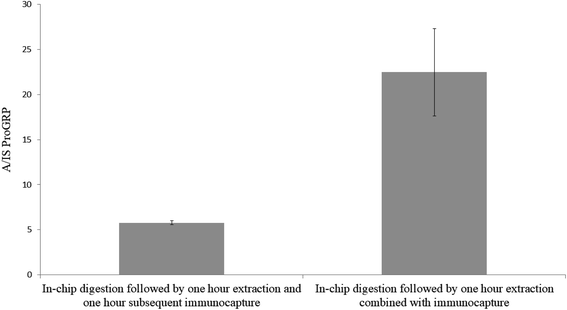 |
| | Fig. 1 In-chip digestion of proGRP spiked serum (2 μg mL−1) with (left bar – sample set A) subsequent immunocapture after 1 hour of extraction in PBS and (right bar – sample set B) one hour extraction combined with immunocapture. The ratio between the signal intensity of the A-peptide and the signal intensity of the internal standard is plotted on the y-axis. n = 3. | |
3.1.2 Extraction solvent.
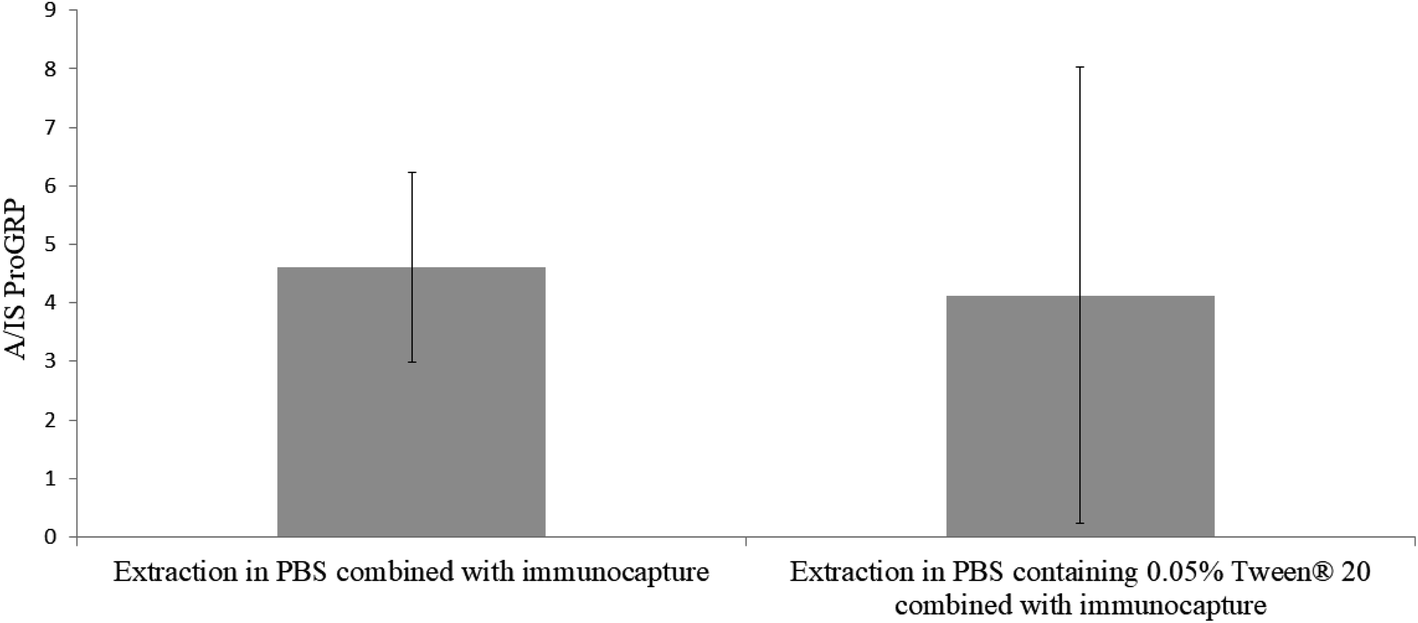
In previous efforts of in-device tryptic digestion sodium deoxycholate (SDC) has been used3 to increase solubility and thus the extraction from the sampling material. This surfactant could however, not be used in combination with the immunocapture beads as SDC is charged and could face unwanted interaction with the antibodies (e.g. inhibit antibody–antigen reaction or denaturation) resulting in low binding efficiency with the target peptide.17–19 As an alternative, the neutral surfactant Tween® (20 and 80) has proven potential for desorbing proteins and other matrix components from both hydrophilic and hydrophobic surfaces.20 Tween 20® has also frequently been used in previous immunocapture efforts at our group.1,21,22 However, the surfactant has not been used directly in the antibody–protein capturing step, but rather afterwards during matrix removal. We wanted to investigate whether it is beneficial to use Tween 20® during the combined extraction and immunocapture to help desorb digested proGRP from the slightly hydrophobic pHEMA–VDM polymer without disturbing the antibody binding efficiency. Two sample sets (n = 3) was prepared with the only difference of 0.05% Tween 20® in the extraction/immunocapture solvent for sample set B. Everything else regarding the sampling and sample preparation was kept identical. The results showed (Fig. 2) that there was no significant difference (p ≫ 0.05) in signal intensity between the two sample sets. However, since the standard deviation for the samples extracted and immunocaptured with Tween 20® present was high, all further experiments was carried out solely with PBS as the combined extraction and capturing solvent. Two additional surfactants were also examined (sodium deoxycholate and sodium deoxysulfate). However, no signals were observed in the samples including either of these surfactant in the combined extraction and capturing solvent. This may resolve from the fact that both of these surfactants are ionic and thus could have a negative effect on the interaction between protein and antibody.23
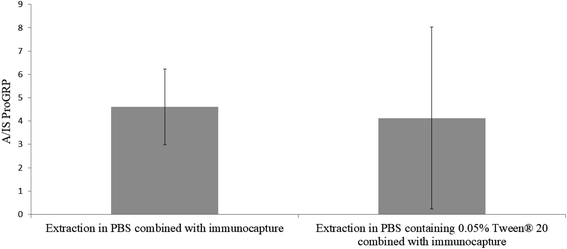 |
| | Fig. 2 In-chip digestion of proGRP spiked serum (2 μg mL−1). Extraction combined with immunocapture carried out for one hour with 1.0 mL solvent with (A) PBS and (B) PBS with 0.05% Tween® 20. The ratio between the signal intensity of the A-peptide and the signal intensity of the internal standard is plotted on the y-axis. n = 3. | |
3.3 Extended tryptic digestion
Since the protein digestion starts instantly in-device while the sample (proGRP spiked serum) is drying, sufficiently long interaction time could potentially be a problem since 15 μL sample dries relatively fast and the proteins will therefore face short digestion time. Extending reaction time potentially gains a complete digestion of the target protein and thus more of the proteotypic peptide of interest. In order to evaluate whether or not the in-device tryptic digestion was sufficient for analysis of protein concentrations in the pg mL−1 to ng mL−1 range, the possibility to extend the protein digestion time was assessed. To achieve this, the following was added subsequently to the sample wells after sampling: (1) PBS in various volumes to extend the drying time (elaborated in Section 3.4) or (2) trypsin immobilized beads in volumes 5 μL, 10 μL and 15 μL to increase the enzyme-to-protein ratio. Addition of buffer to extend in-device reaction time has been elaborated in previous work15 and were not further investigated surpassing 30 μL as elaborated in Section 3.4. The investigation showed (S4, Fig. S2†) that none of the additions of extra trypsin beads served any gains in the production of the A-peptide. Therefore, a tentative conclusion was made that the immobilized pHEMA–VDM sampler yielded sufficient protein digestion as is.
3.4 Sample dilution
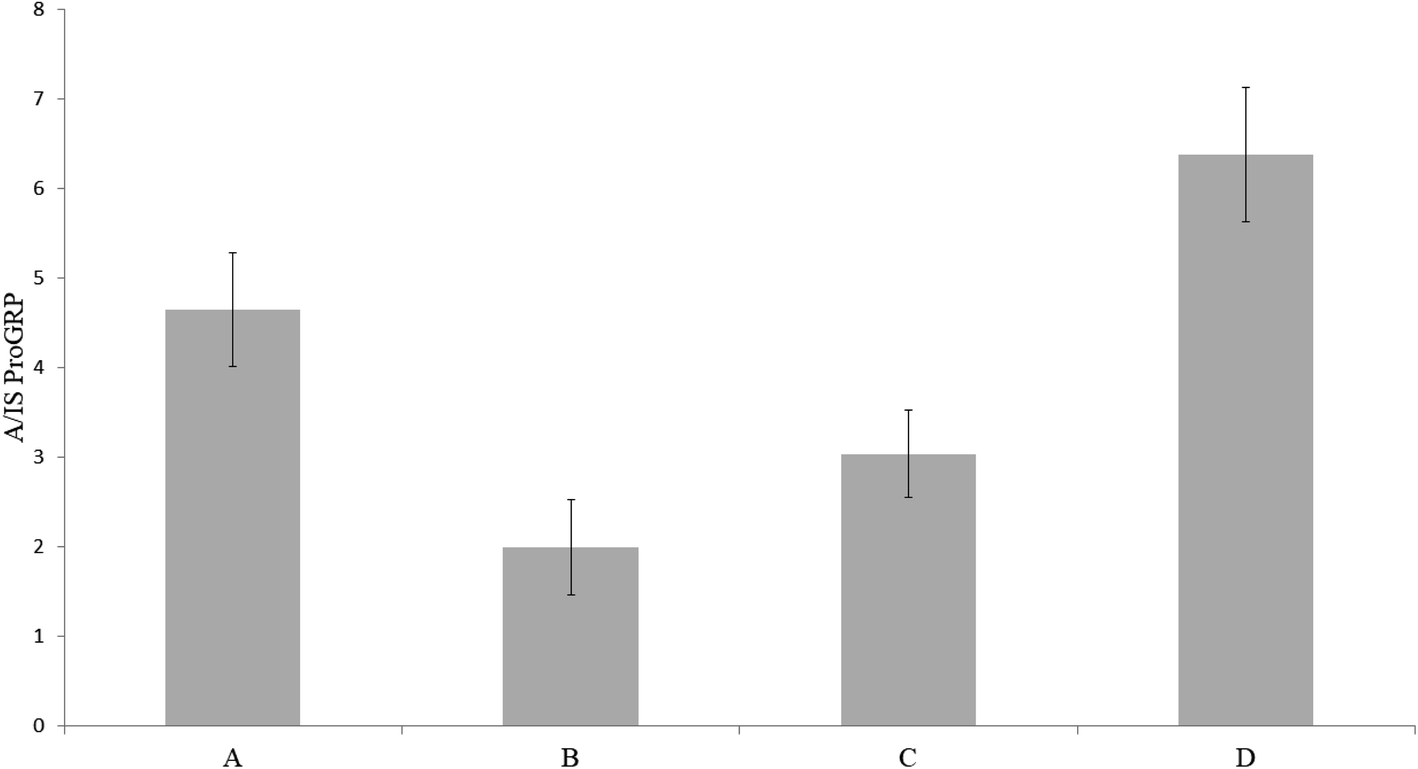
Since all initial experiments had been carried out with proGRP spiked to a matrix composed of 50% serum and 50% PBS, it was of interest to investigate if sample dilution was necessary in order to obtain viable protein digestion. To investigate this, a concentration of 2 μg mL−1 proGRP with an effective sampling volume of 15 μL was chosen. The sampling variants were as following; (A) 30 μL serum:PBS (50:50 v/v), (B) 15 μL serum with a subsequent addition of 15 μL PBS (while the sample spot still was wet), (C) 15 μL serum with an addition of 15 μL PBS after the serum had dried in order to extend the trypsin interaction time and (D) 15 μL serum without additions. There was no gain in either diluting the sample at the time of sampling, after the sample had dried (both to extend the drying time and thus digestion time) or use of a less complex matrix (diluted serum) (Fig. 3). The results indicated that the trypsin-to-protein ratio was sufficiently high for a sufficient digestion within 30 minutes (drying time of 15 μL serum sample). Interestingly, extending the digestion time by adding PBS either as an addition to the serum sample or after the serum had dried had a negative impact on the signal intensity. A possible reason for this might be an additional sample dilution leading to the peptides in the device is being wicked toward parts of the wax printed cartridge to adsorb. If the latter occurred, it is assumed that not all of the sample gets extracted enough for subsequent immunocapture of the A-peptide. These theories were not further investigated. All in all, further experiments were carried out solely with spiked, undiluted serum.
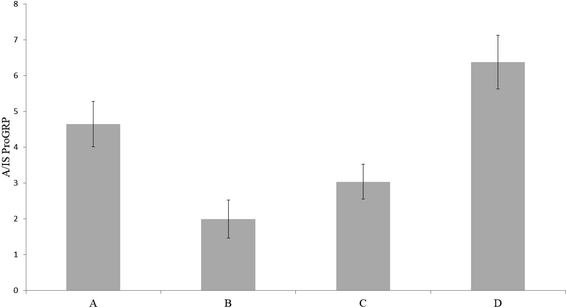 |
| | Fig. 3 Trypsin immobilized pHEMA–VDM sampled with proGRP in (A) 30 μL serum:PBS (50:50 w/w), (B) 15 μL serum with a subsequent addition 15 μL PBS, (C) 15 μL serum with a subsequent addition 15 μL PBS after the serum had dried and (D) 15 μL serum. All serum samples had a concentration of 2 μg mL−1 proGRP isoform 1. The ratio between the signal intensity of the A-peptide and the signal intensity of the internal standard is plotted on the y-axis. | |
3.5 Quantitative performance
3.5.1 Recovery.
Good extraction efficiency and minimal analyte loss are important when low abundant proteins are being analyzed. Additionally, sticky sampling materials such as cellulose1 or sampling materials that provide difficulties (in terms of geometrical attributes) for the analyte to be transferred from sampling material into solution could be critical. Since this concept holds immediate protein digestion in-device obtaining an exact measure on extraction efficiencies is troublesome because it could not be certain if the sample is being digested completely or not. Furthermore, the in-device protein digestion is combined with in-house mAb immobilized magnetic beads targeting only the A-peptide and this capturing step also face potential analyte loss. Therefore, the labeled internal standard was used to determine the extraction recoveries and immunocapture recovery (n = 3), since this was a synthetic peptide should readily go through the sampling and digestion steps without complications. The internal standard was spiked (A) at the time of sampling, (B) at the time of extraction and (C) after extraction and immunocapture (A schematic illustration is presented in S5 Fig. S3†). The experiments were performed using blank serum as sample matrix to avoid unwanted adsorption of the peptide during the analysis. This, because a buffered peptide could face strong adsorption to the sampling material. Serum contains a high concentration of other proteins/peptides that would likely mask the adsorption sites as discussed elsewhere.1 From the investigation the extraction recovery was 78% and the immunocapture recovery was 88%. These recoveries could be seen as comparable to investigations carried out by others.10
3.5.2 Limit of detection and limit of quantification.
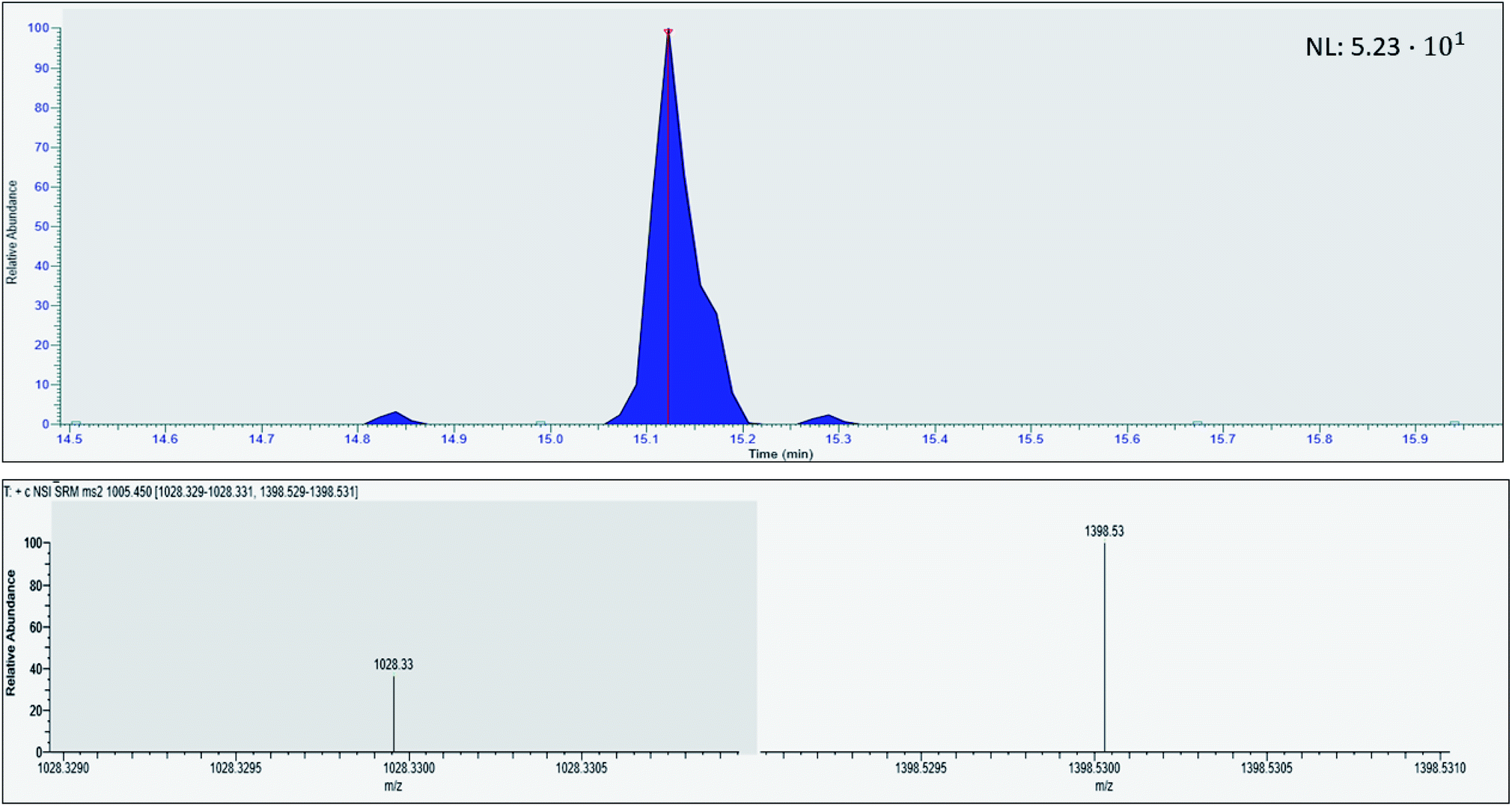
Prior to evaluating the viable concentration range, the LOQ was determined. The LOQ was analyzed on two different days with three sample replicates each (N = 2, n = 3) by sampling and in-device digesting proGRP spiked to serum with subsequent immunocapture in the extracting solution of the A-peptide using the previously optimized conditions. The LOQ (S/N = 10) was obtained with correct ion ratios of the A-peptide's fragments at 500 pg mL−1 (Fig. 4.). The noise was defined as the maximum intensity from the neighboring peaks of the target analyte. From the LOQ at 500 pg mL−1, the LOD was calculated to 150 pg mL−1. Compared to previous efforts on tryptic digestion and subsequent immunocapture of the respective epitope this concept presents a lower detection limit.10 However, slightly higher than what has been performed with in-solution digestion (LOD 54 pg mL−1, 50 μL sampling volume).12 The achieved detection and quantification limit is also comparable to what have been reported by other with more generic sample preparation methods,24,25 especially if sampling volume is taken into consideration. With further optimization the proposed reference level of <60 pg mL−126 in healthy individuals should be achievable.
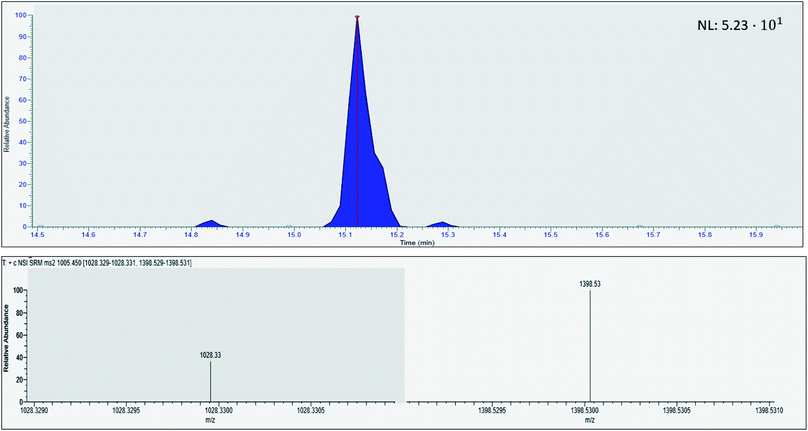 |
| | Fig. 4 Chromatogram of human serum spiked with proGRP (500 pg mL−1) with the respective ion fragment of the proteotypic epitope peptide ALGNQQPSWDSEDSSNFK. | |
3.5.3 Evaluation of quantitative performance.
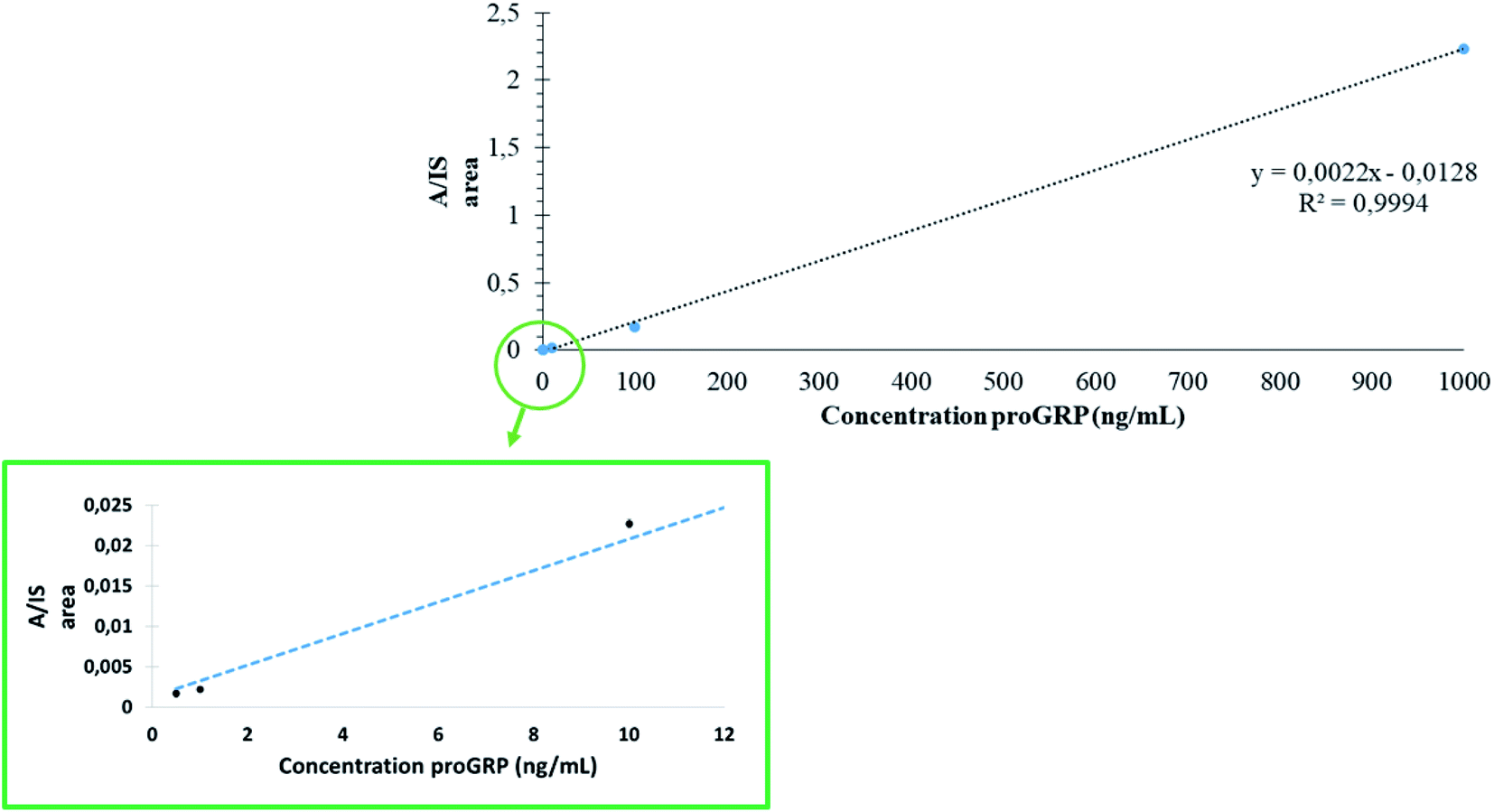
To assess the quantitative performance of in-device digested proGRP spiked serum samples combined with the selective capture of the epitope during extraction linearity were investigated. The concentration range was analyzed at five points (n = 3): 500 pg mL−1 (LOQ), 1 ng mL−1, 10 ng mL−1, 100 ng mL−1 and 1000 ng mL−1. The concentration range was set to demonstrate that the sampling device could readily work over an array of concentrations and was not necessarily set to demonstrate the applicability solely to proGRP specifically. From the analysis (Fig. 5) a satisfactory correlation was obtained (R2 = 0.99) with relative standard deviations also all within 16% (exception of the LOQ at 26%). The RSD values (n = 3) from analysis of proGRP spiked to serum were comparable to the previous work by Levernæs et al.10 (all concentration levels within 13%, here all within 16% except LOQ at 26%) even though the concentration range is broader in the presented work. Precision was in addition measured (n = 6) at 10 ng mL−1 and 1000 ng mL−1 to 20 and 2%, respectively. Compared to more conventional efforts where proteolysis is carried out over-night, the entire workflow including sample drying was on average performed within 2.5 hours (one hour of sample drying, one hour extraction, 20 min of matrix removal and 15 min of elution, based on 12 samples). Reduction and alkylation can then be included as part of the extraction step for proteins where this is necessary as described previously.3
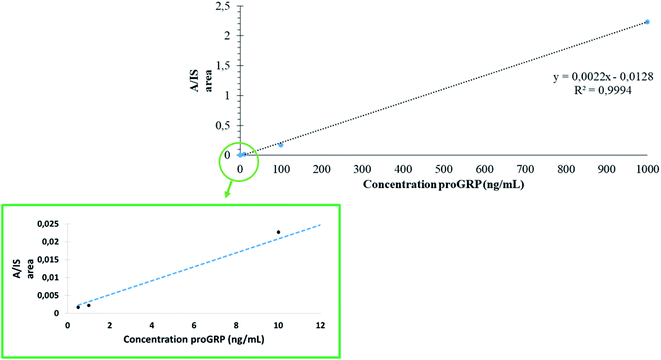 |
| | Fig. 5 Five point standard curve (n = 3) from 0.5 ng mL−1 to 1000 ng mL−1 of proGRP spiked to human serum. The RSD of each concentration level was 26%, 16%, 2%, 8% and 1% for 500 pg mL−1, 1 ng mL−1, 10 ng mL−1, 100 ng mL−1 and 1000 μg mL−1, respectively. | |
4 Conclusion
A simple paper-based sampling device in chip format integrated with in-device proteolysis is here demonstrated in combination with selective proteotypic epitope peptide immunoextraction for simple and accessible sampling and semi-automated sample preparation. The combination showed a high degree of performance from pg mL−1 to μg mL−1 with minimal time manual sample preparing labor for the low abundant serum biomarker proGRP. The entire work flow was performed within 2.5 hours and demonstrated satisfactory correlation and precision. The sampling chips showed promising potential in combination with proteotypic epitope peptide immunoextraction for future clinical applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Elisabeth Paus at Radium Hospitalet, Oslo University Hospital for kindly donating proGRP standards and monoclonal M18 antibodies.
References
- Ø. Skjærvø, E. J. Solbakk, T. G. Halvorsen and L. Reubsaet, Talanta, 2019, 195, 764–770 CrossRef.
- Ø. Skjærvø, C. Rosting, T. G. Halvorsen and L. Reubsaet, Analyst, 2017, 142, 3837–3847 RSC.
- Ø. Skjærvø, T. G. Halvorsen and L. Reubsaet, Analyst, 2018, 143, 3184–3190 RSC.
- Y. Liu, H. Wang, Q. Liu, H. Qu, B. Liu and P. Yang, Lab Chip, 2010, 10, 2887–2893 RSC.
- Y. Liu, W. Zhong, S. Meng, J. Kong, H. Lu, P. Yang, H. H. Girault and B. Liu, Chem.–Eur. J., 2006, 12, 6585–6591 CrossRef CAS.
- C. Zhang, T. Glaros and N. E. Manicke, J. Am. Chem. Soc., 2017, 139, 10996–10999 CrossRef CAS.
- K. Sato, M. Tokeshi, T. Odake, H. Kimura, T. Ooi, M. Nakao and T. Kitamori, Anal. Chem., 2000, 72, 1144–1147 CrossRef CAS PubMed.
- S. Fang, H. Tian, X. Li, D. Jin, X. Li, J. Kong, C. Yang, X. Yang, Y. Lu, Y. Luo, B. Lin, W. Niu and T. Liu, PLoS One, 2017, 12, e0175050 CrossRef.
- B. J. Bills and N. E. Manicke, Clinical Mass Spectrometry, 2016, 2, 18–24 CrossRef.
- M. C. S. Levernæs, O. K. Brandtzaeg, S. F. Amundsen, L. Reubsaet, E. Lundanes, T. G. Halvorsen and S. R. Wilson, Anal. Chem., 2018, 90, 13860–13866 CrossRef.
- R. M. Schoenherr, L. Zhao, R. G. Ivey, U. J. Voytovich, J. Kennedy, P. Yan, C. Lin, J. R. Whiteaker and A. G. Paulovich, Proteomics, 2016, 16, 2141–2145 CrossRef CAS.
-
L. Maren, F. Bassem, O. Inger, A. S. Sazan, P. Elisabeth, R. Léon and H. Trine Grønhaug, Exploring peptide capture by anti-protein antibodies for LC-MS-based biomarker determination, 2018 Search PubMed.
- C. Rossetti, O. G. Ore, B. Sellergren, T. G. Halvorsen and L. Reubsaet, Anal. Bioanal. Chem., 2017, 409, 5631–5643 CrossRef CAS.
- M. Razavi, N. Leigh Anderson, M. E. Pope, R. Yip and T. W. Pearson, New Biotechnol., 2016, 33, 494–502 CrossRef CAS PubMed.
- Ø. Skjærvø, T. G. Halvorsen and L. Reubsaet, Anal. Chim. Acta, 2019, 1089, 56–65 CrossRef.
- E. Paus and K. Nustad, Clin. Chem., 1989, 35, 2034–2038 CAS.
- L. F. Qualtiere, A. G. Anderson and P. Meyers, J. Immunol., 1977, 119, 1645 CAS.
- M. Agarkhed, C. O'Dell, M.-C. Hsieh, J. Zhang, J. Goldstein and A. Srivastava, AAPS PharmSciTech, 2018, 19, 79–92 CrossRef CAS.
- G. J. Dimitriadis, Anal. Biochem., 1979, 98, 445–451 CrossRef CAS.
- R. G. Couston, M. W. Skoda, S. Uddin and C. F. van der Walle, mAbs, 2013, 5, 126–139 CrossRef.
- C. Rossetti, M. C. S. Levernæs, L. Reubsaet and T. G. Halvorsen, J. Chromatogr. A, 2016, 1471, 19–26 CrossRef CAS.
- M. C. S. Levernæs, M. N. Broughton, L. Reubsaet and T. G. Halvorsen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1055–1056, 51–60 CrossRef.
- H. Sjögren, C. A. Ericsson, J. Evenäs and S. Ulvenlund, Biophys. J., 2005, 89, 4219–4233 CrossRef.
- B. Winther, M. Nordlund, E. Paus, L. Reubsaet and T. G. Halvorsen, J. Sep. Sci., 2009, 32, 2937–2943 CrossRef CAS.
- S. B. Torsetnes, M. S. Levernæs, M. N. Broughton, E. Paus, T. G. Halvorsen and L. Reubsaet, Anal. Chem., 2014, 86, 6983–6992 CrossRef CAS PubMed.
- M. S. Nordlund, J. Bjerner, D. J. Warren, K. Nustad and E. Paus, Tumor Biol., 2008, 29, 204–210 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ay01976f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Trine Grønhaug
Halvorsen
,
Trine Grønhaug
Halvorsen