
An electrochemical sensor based on a carbon paste electrode for the determination of buserelin
Marjan
Fallah
a,
Mostafa
Rahimnejad
 *a,
Maryam
Asghary
b and
Mehrdad
Mashkour
a
*a,
Maryam
Asghary
b and
Mehrdad
Mashkour
a
aBiofuel & Renewable Energy Research Center, Faculty of Chemical Engineering, Babol Noshirvani University of Technology, Babol, Iran. E-mail: Rahimnejad_mostafa@yahoo.com; Rahimnejad@nit.ac.ir; Fax: +98 11 32334204; Tel: +98 11 32334204
bElectroanalytical Chemistry Research Laboratory, Department of Analytical Chemistry, Faculty of Chemistry, University of Mazandaran, Babolsar, Iran
First published on 19th November 2019
Abstract
This study presents a rapid, simple, sensitive, and selective electrochemical sensor prepared using a carbon paste electrode for the electrochemical determination of buserelin—an anti-prostate cancer drug. Differential pulse voltammetry (DPV) and cyclic voltammetry (CV) techniques were applied to analyze the anodic characterization of the aforementioned drug. Some of the common parameters such as pH, scan rate, and sensitivity have been studied by using the aforementioned electrochemical techniques. The DPV technique showed a remarkable linearity in the concentration range of 1.0 × 10−4 to 6.0 × 10−6 μM with a limit of detection of 0.73 μM. Moreover, the present study could be successfully applied for the determination of buserelin in human serum samples.
Introduction
Cancer is defined as an illness occurring due to the uncontrollable growth of abnormal cells, which have been started by smaller cells with incorrect chromosomal numbers .1,2 According to the World Health Organization, cancer is the second most likely cause of death and has killed about 7.4 million people around the world (including developing countries) by the end of the twentieth century.3,4 The latest report has estimated an alarming increment of about 12 million people by 2030, while by an early detection of cancer, almost 30% of cancer victims can be saved.5,6 Among all types of cancers, prostate cancer is the second most prevalent cause of death in men, particularly the ones above the age of 50.7 Previously, diverse hormone treatments have been evaluated for this type of cancer with the focus of researchers being a substantial decrement in serum androgen levels.8–12 Because of the significant and positive effect of buserelin as a gonadotrophin-releasing hormone in the prostate cancer treatment, a sensitive and simple monitoring system for the precise recognition of buserelin has become an urgent requirement.Recently, some approaches such as ultra-performance liquid chromatography (UPLC), high pressure liquid chromatography (HPLC), and capillary electrophoresis (CE) have been accomplished for the characterization and quantification of buserelin.13
Significant disadvantages of all the previous methods (time and labor consuming, high operation cost, and the usage of large amounts of pure solvents) guided many researchers to find an absolutely easy and cost-effective solution in comparison with the existing approaches to determine buserelin.14 Although numerous researches have been already done to achieve this purpose, none of them have focused on the electrochemical detection of the mentioned drug.
Furthermore, electrochemical methods have been proved to be much more accurate for the determination of organic materials, such as drugs and other related molecules in pharmaceutical formulations, and some biological fluids and their oxidizable components.15–17 Common carbon electrodes, specifically paste electrodes, have been extensively used in electrochemical researches due to their wide potential window, low background current, low cost, suitability, and chemical inertness to detect various biological and organic molecules.18–21
As an example, in 2016, D. Bukkitgar et al. analyzed the electrochemical behavior of the anticancer drug 5-fluorouracil at a bare carbon paste electrode by using the cyclic voltammetry (CV) method.15 Also, in 2015, M. Asghary et al. proposed a genosensor based on the carbon paste electrode (CPE) for studying the interaction between ketamine, used as an anesthetic, and DNA by applying voltammetric techniques, in which the binding mode of DNA and ketamine was clarified by differential pulse voltammetry and UV-vis spectroscopy methods.22 Besides, in 2016, Nikodimos et al. applied CV methods to determine metronidazole of a tablet sample using a CPE, and an irreversible reduction peak at about −0.4 V was found.23
Both electroanalytical studies and electrochemistry benefit from many advantages such as low cost, very low background current, simple surface renewal process, large potential window, and ease of miniaturization of CPE. To the best of our knowledge, this is the first time that the electrochemical behavior of buserelin was investigated by using an electrochemical sensor based on CPE. Thus, in this work, a novel electrochemical sensor based on CPE as a transducer has been developed, which was able to detect low amounts of the target analyte. Hence, in this study, a novel electrochemical sensor was presented to determine buserelin in biological samples using CPE as the transducer. Moreover, the effect of scan rate on cyclic voltammograms of buserelin has been discussed here. The differential pulse voltammetry (DPV) technique has also been developed to determine buserelin in biological samples.
Results and discussion
Electrochemical behaviour of buserelin
In this work, different buffers with different pH values were tested to determine in which buffer the anodic peak is more obvious. The results revealed that buserelin showed an explicit electrochemical activity in the acetate buffer as an optimum solution rather than in other buffer solutions. Fig. 1 shows the CV curve of buserelin (1.0 × 10−4 M) in a solution containing NaCl (0.1 M) and acetate buffer (pH 5.25, 0.5 M). As can be seen, buserelin demonstrated an anodic peak, which appears at around 0.8 V vs. Ag|AgCl|KCl (3.0 M). Depending on the potential range from 0.4 to 1.0, no cathodic peak was shown in the opposite scan of the potential, which confirms that the oxidation of buserelin at the surface of CPE is irreversible. In addition, this oxidation peak can be ascribed to the formation of a hydrogen bond between the diazepine ring of buserelin and the carboxamide group of acetic acid.24 | ||
| Fig. 1 Cyclic voltammograms of buserelin (1.0 × 10−4 M) in the acetate buffer solution 0.5 M (pH 5.25) containing NaCl 0.1 M at a scan rate of 100 mV s−1 (curve a) and blank solution (curve b) recorded at the surface of CPE. | ||
The effect of experimental conditions
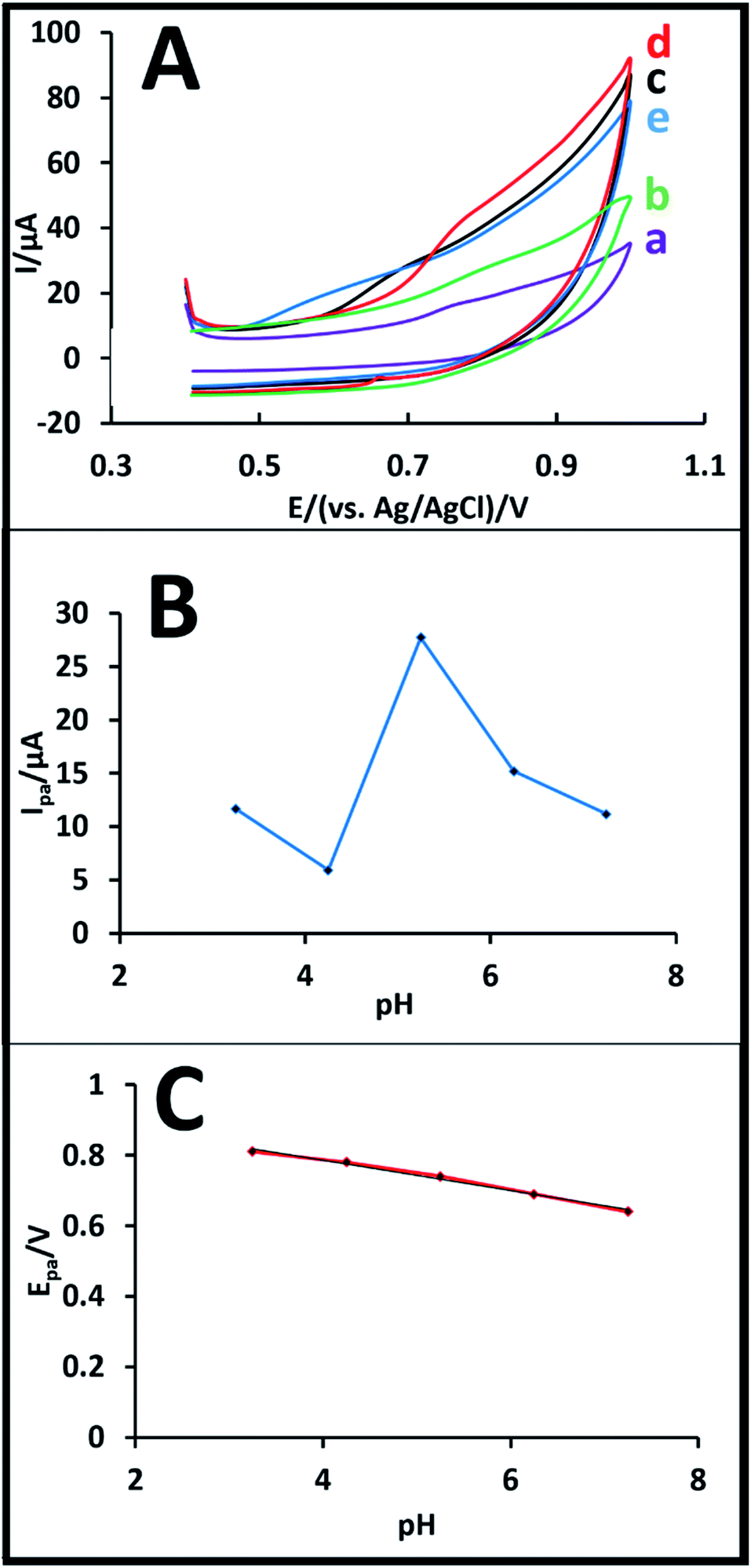 | ||
| Fig. 2 (A) Cyclic voltammograms of buserelin solution (1.0 × 10−4 M) at various pH values of 3.0 (a), 4.0 (b), 5.25 (c), 6.0 (d), and 7.0 (e) keeping other conditions same as that presented in Fig. 1. (B) Plot of the peak current vs. pH and (C) plot of the peak potential vs. pH of buserelin solution (1.0 × 10−4 M) on CPE at a scan rate of 100 mV s−1. | ||
As shown in Fig. 2A, an increase in pH from 3.0 to 5.25 causes a normal increment in the intensity of the anodic peak, while the intensity decreases at pH values of 6.0 and 7.0. In conclusion, both peak current and peak potential were considerably affected by the change in pH, as shown in Fig. 2B and C, respectively. As shown in Fig. 2A, based on the proton production during the oxidation reaction shown in Fig. 3 and the formula pH = log[H3O+], the anodic peak normally increases with the increment in pH from 3.0 to 5.25 and then exhibits a clear decrement at pH 6.0 and 7.0, which is due to the H+ production during the process. Therefore, pH 5.25 was chosen as the optimum pH for all further experiments performed in this study (Fig. 2B). As shown in Fig. 2C, an increase in the pH values leads to a small negative shift in the oxidation peak potential of buserelin, where a linear relationship is observed between the pH and the anodic peak potential at the CPE, which can be explained by the following equation:
| Epa = −0.0581pH + 0.9851, (R2 = 0.9898) | (1) |
 | ||
| Fig. 3 The proposed oxidation mechanism of buserelin at the surface of a CPE. | ||
The slope of this equation is 58.1 mV pH−1, which is nearly close to the expected theoretical value (59.0 mV pH−1) for equal number of electrons and protons transferred in the electrochemical oxidation of buserelin at the surface of CPE.26–28 Based on the results above, the oxidation mechanism of buserelin is suggested in Fig. 3.
| Ipa (μA) = 2.481ν1/2 (mV s−1) + 0/8841, (R2 = 0.996) | (2) |
 | ||
Fig. 4 (A) Plot of Epversus ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ν at a scan rate from 5 to 120 mV s−1 (B) CV response of buserelin (1.0 × 10−4 M) in acetate buffer solution 0.5 M (pH 5.25) at various scan rates from 5 to 120 mV s−1 (C) plot of peak current versus ν (D) plot of peak current versus ν1/2 at the surface of CPE. ν at a scan rate from 5 to 120 mV s−1 (B) CV response of buserelin (1.0 × 10−4 M) in acetate buffer solution 0.5 M (pH 5.25) at various scan rates from 5 to 120 mV s−1 (C) plot of peak current versus ν (D) plot of peak current versus ν1/2 at the surface of CPE. | ||
For the irreversible-diffusion controlled interactions, the electron transfer coefficient α can be calculated by the difference between peak potential (Epa) and the half wave potential (Epa/2) as expressed by the following equation:32
| ΔEpa = Epa − Epa/2 = (47.7/α) mV (diffusion controlled, 298 K) | (3) |
| Epa = E0′ − {RT/F[(1 − α)nF]}ln{RTK0/[(1 − α)nF]} + {[RT/(1 − α)nF]}lnν | (4) |
| Ep = 0.0349lnν + 0.6455, (R2 = 0.9736) | (5) |
The value of α and the number of electrons transferred for an irreversible reaction were calculated and found to be 0.81 and 2, respectively. Moreover, the other parameters such as the standard rate constant (K0) was calculated based on the Lavirone equation (eqn (3)), where R is the gas constant, F is the Faraday constant, and T is the absolute temperature.
The oxidation mechanism of buserelin
In the proposed mechanism for the electro-oxidation of buserelin two protons and two electrons are transferred in the process. The hydroxyl group (–OH) here is appended to the carbon atom (C-*) of the cyclohexane ring of the buserelin. Therefore, buserelin undergoes oxidation at the (C-*) hydroxyl group to form (*-oxo) buserelin. As the first proton gets removed during the electrolysis, oxygen gets a negative charge and the anionic form of buserelin is formed. Stabilizing the anionic form of the buserelin, the hydrogen atom attached to the carbon (C-7) of the cyclohexane is affected by further electro oxidation and then the production of stable 7-oxo buserelin takes place (Fig. 3). A probable mechanism is proposed as shown in Fig. 3.33Evaluating the detection limit of buserelin
DPV experiments were employed as a more sensitive technique compared to CV in order to establish the calibration curve for several concentration values of buserelin in 1 M acetate buffer solution (pH 5.25). Fig. 5A depicts DPV voltammograms of an increasing concentration of buserelin from 1 × 10−4 to 6 × 10−6 M in 0.5 M acetate buffer solution (pH 5.25). Also, the calibration curve of buserelin based on the DPV technique is shown in Fig. 5B. Consequently, the calibration curve of the proposed method demonstrates that there is a linear relation between the oxidation peak current of buserelin and its increasing concentration. In summary, the increase in concentration level results in a great increment in the peak current intensity through an acceptable normal range. | ||
| Fig. 5 (A) The plot of peak current against the concentration of buserelin (B) differential pulse voltammograms of the increasing concentration level of buserelin: a (1 × 10−4) b (5 × 10−4) c (3 × 10−5) d (6 × 10−5) e (3 × 10−6) f (6 × 10−6) M of buserelin solution in 1 M acetate buffer solution (pH 5.25) at a scan rate of 100 mV s−1. | ||
The lowest detection limit can be defined as the lowest analyte concentration likely to be identified from the blank. The ICH recommendation based on both slope of the calibration curve and standard deviation was used to calculate the LOD of buserelin. According to the equation LOD = 3S/m in which m is the slope of the calibration curve and S is the standard deviation of the oxidation peak current the calculated LOD was found to be 0.73 μM.
Analytical performance
The sensitivity and applicability of the designed sensor for the electrochemical determination of buserelin were evaluated by applying CPE in real-life samples using the developed DPV techniques. Recoveries from human serum sample (plasma) were measured by spiking the drug-free serum with specific amounts of buserelin. In this effort, firstly, the serum was diluted 50 times and then known concentrations of buserelin were added to the serum sample. Based on the calibration curve the results obtained are reported in Table 1. Consequently, the low amount of relative standard deviation (RSD) for three measurements (n = 2) clearly proved that the sensor can be potentially used as a precise and sensitive device for diagnosis applications with an allowable reproducibility.| Sample | Added | Found | RSD | Recovery % |
|---|---|---|---|---|
| 1 | 1 | 1.2 | 3.82 | 97 |
| 2 | 10 | 9.6 | 4.53 | 102.81 |
| 3 | 100 | 99.12 | 4.15 | 98.91 |
Experimental
Chemicals
All the employed chemicals were of analytical grade purity. Buserelin was purchased from Cinnagen pharmaceutical Company (Karaj-Iran). All the other chemicals were used without any further purification. By dissolving an appropriate amount of buserelin in distilled water, 0.01 M stock solution was prepared and kept at a low temperature of about 5 °C. The acetate buffer (pH 5.25; 0.5 M) was prepared by mixing acetic acid and sodium acetate anhydrous, which were both purchased from Merck. Britton–Robinson buffer at pH 7.0 and 8.0 was prepared using H3BO3 0.04 M, H3PO4 0.04 M, CH3COOH 0.04 M, and NaOH 0.2 M.Apparatus
All electrochemical experiments were performed by a potentiostat/galvanostat (IVIUM TECHNOLOGY – VERTEX) in a three-electrode electrochemical cell with CPE as the working electrode, a platinum wire as the auxiliary electrode, and Ag|AgCl| as the reference electrode. Also, a pH-meter (METTLER TOLEDO, Switzerland) was used to carry out all the pH measurements.Preparation of CPE
Firstly, a glass tube was well smoothened by using a piece of sand paper. Graphite powder and paraffin oil were mixed well in a ratio of 70:30 producing a homogeneous paste. The resulting paste was packed into the end of the glass tube and connected to a copper wire. Consequently, the electrical contact was established via the copper wire connection. Before each use, the CPE was polished with a piece of clean paper to have a smooth surface in order to avoid electrical background noise during subsequent electrochemical experiments.
Plasma preparation
Human blood samples, which contained sodium citrate and saline solution, were collected in dry and evacuated pipes from a healthy volunteer. The serum sample from a healthy human for real sample analysis was supplied by North Research Center, Razi Institute of Iran (Babol, Iran) under the ethical approval claim no. IR.BNUT.REC13960615. Also, permission was obtained for any experimentation with human subjects. The samples were kept at room temperature and centrifuged with Hermle, Z 326 K for about 10 min at 4 °C and 1500 rpm for the separation of plasma. Then, the samples were moved to polypropylene tubes and kept at 20 °C until analysis. The supernatants were spiked with known amounts of buserelin. Appropriate volumes of this solution were added to ABS (pH 5.25; 0.5 M) as supporting electrolyte to record the voltammograms.Conclusions
This report is the first electrochemical attempt to detect buserelin based on its electro-oxidation reaction using bare CPE. The electrochemical oxidation of buserelin by applying CPE electrode in acetate buffer solution under the optimal pH of 5.25 was analyzed. This means that this study introduced a convenient, rapid and sensitive electrochemical sensor for the detection of buserelin. Low detection limit, remarkable sensitivity, and easy operation are the main reasons that motivated us to develop this sensor for the electrochemical determination of the target analyte. Finally, the proposed procedure can be applied in the analysis of real samples with diverse matrices.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully thank to the Biofuel & Renewable Energy Research Center at Babol Noshirvani University of Technology (BNUT) and to the Danesh Gostar Hamgam Ba Sanat company, for all the financial support associated to the technology, laboratory facilities, and most of all, for their scientific guidance. Also, the authors would like to thank Hoda Ezoji for her valuable help in this work. This research was supported by BNUT (grant number: BNUT/4150033/1395).References
- M. Hejmadi, Introduction to cancer biology, Bookboon, 2009 Search PubMed.
- A. Figueredo, R. Rumble, J. Maroun, C. Earle, B. Cummings, R. McLeod, L. Zuraw and C. Zwaal, BMC Cancer, 2003, 3, 26 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef.
- P. Ravasco, I. Monteiro-Grillo, P. M. Vidal and M. E. Camilo, Support. Care Cancer, 2004, 12, 246–252 CrossRef PubMed.
- W. H. Organization, National cancer control programmes: policies and managerial guidelines, World Health Organization, 2002 Search PubMed.
- B. De Pauw, T. J. Walsh, J. P. Donnelly, D. A. Stevens, J. E. Edwards, T. Calandra, P. G. Pappas, J. Maertens, O. Lortholary and C. A. Kauffman, Clin. Infect. Dis., 2008, 46, 1813–1821 CrossRef.
- A. Qayum, W. Gullick, R. Clayton, K. Sikora and J. Waxman, Br. J. Cancer, 1990, 62, 96–99 CrossRef CAS PubMed.
- H. Lepor, A. Ross and P. Walsh, J. Urol., 1982, 128, 335–340 CrossRef CAS.
- L. Martinerie, Enfances Psy, 2016, 58–65 CrossRef.
- M. Schweizer, Pharmacologic Dose Testosterone to Treat Castration-Resistant Prostate Cancer: Mechanisms of Action and Drivers of Response, University of Washington, Seattle United States, 2017 Search PubMed.
- E. Tamizi and A. Jouyban, Electrophoresis, 2015, 36, 831–858 CrossRef CAS.
- K. Yingsukwattana, S. Puttipipatkhachorn, U. Ruktanonchai and N. Sarisuta, J. Liposome Res., 2016, 26, 69–79 CrossRef CAS.
- H. Wätzig and M. Degenhardt, J. Chromatogr. A, 1998, 817, 239–252 CrossRef.
- H. Wätzig and M. Degenhardt, J. Chromatogr. A, 1998, 817, 239–252 CrossRef.
- S. D. Bukkitgar and P. S. Nagaraj, ChemistrySelect, 2016, 1(4), 771–777 CrossRef CAS.
- M. Asghary, J. B. Raoof, M. Rahimnejad and R. Ojani, J. Iran. Chem. Soc., 2018, 15, 445–453 CrossRef CAS.
- M. Asghary, J. B. Raoof, M. Rahimnejad and R. Ojani, Biosens. Bioelectron., 2016, 82, 173–176 CrossRef CAS.
- L. de Souza Schaumlöffel, J. W. V. Dambros, P. R. B. Fernandes, M. Gutterres and C. M. S. Piatnicki, Fuel, 2019, 236, 803–810 CrossRef.
- F. Ivars-Barceló, A. Zuliani, M. Fallah, M. Mashkour, M. Rahimnejad and R. Luque, Appl. Sci., 2018, 8, 1184 CrossRef.
- H. Ezoji and M. Rahimnejad, Int. J. Appl. Sci. Eng. Res., 2016, 7, 242–246 Search PubMed.
- R. Zokhtareh and M. Rahimnejad, Electroanalysis, 2018, 30, 921–927 CrossRef CAS.
- M. Asghary, J. B. Raoof, R. Ojani and E. Hamidi-Asl, Int. J. Biol. Macromol., 2015, 80, 512–519 CrossRef CAS.
- Y. Nikodimos and M. Amare, J. Anal. Methods Chem., 2016, 2016, 3612943 Search PubMed.
- B. Rezaei, O. Rahmanian and A. A. J. S. Ensafi, Sens. Actuators, B, 2014, 196, 539–545 CrossRef CAS.
- L. u. Švorc, K. Cinková, J. Sochr, M. Vojs, P. Michniak and M. Marton, J. Electroanal. Chem., 2014, 728, 86–93 CrossRef.
- A. M. Bagoji and S. T. Nandibewoor, Phys. Chem., 2016, 3, 65–76 Search PubMed.
- A. Migliore, N. F. Polizzi, M. J. Therien and D. N. Beratan, Chem. Rev., 2014, 114, 3381–3465 CrossRef CAS PubMed.
- J. I. Gowda and S. T. Nandibewoor, Electrochim. Acta, 2014, 116, 326–333 CrossRef CAS.
- H. Purushothama, Y. A. Nayaka, M. Vinay, P. Manjunatha, R. Yathisha and K. Basavarajappa, Journal of Science: Advanced Materials and Devices, 2018, 161–166 Search PubMed.
- M. Rumin, J. Smajdor, B. Paczosa-Bator and R. Piech, ECS J. Solid State Sci. Technol., 2016, 5, M3005–M3011 CrossRef CAS.
- J. Tashkhourian and S. Nami-Ana, Materials Science and Engineering: C, 2015, 52, 103–110 CrossRef CAS.
- H. Ezoji and M. J. S. Rahimnejad, Sens. Actuators, B, 2018, 274, 370–380 CrossRef CAS.
- R. Hegde, B. K. Swamy, B. Sherigara and S. Nandibewoor, Int. J. Electrochem. Sci., 2008, 3, 302–314 CAS.
| This journal is © The Royal Society of Chemistry 2020 |