
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMass spectrometry-based identification of ortho-, meta- and para-isomers using infrared ion spectroscopy†
Rianne E.
van Outersterp
 a,
Jonathan
Martens
a,
Giel
Berden
a,
Valerie
Koppen
b,
Filip
Cuyckens
*b and
Jos
Oomens
*ac
a,
Jonathan
Martens
a,
Giel
Berden
a,
Valerie
Koppen
b,
Filip
Cuyckens
*b and
Jos
Oomens
*ac
aRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7, 6525ED Nijmegen, The Netherlands. E-mail: jos.oomens@ru.nl
bDrug Metabolism & Pharmacokinetics, Janssen R&D, B-2340, Beerse, Belgium. E-mail: fcuycken@its.jnj.com
cvan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 908, 1098XH Amsterdam, The Netherlands
First published on 28th August 2020
Abstract
Distinguishing positional isomers, such as compounds having different substitution patterns on an aromatic ring, presents a significant challenge for mass spectrometric analyses and is a frequently encountered difficulty in, for example, drug metabolism research. In contrast to mass spectrometry, IR spectroscopy is a well-known and powerful tool in the distinction of ortho-, meta- and para-isomers, but is not applicable to low-abundance compounds in complex mixtures such as often targeted in bioanalytical studies. Here, we demonstrate the use of infrared ion spectroscopy (IRIS) as a novel method that facilitates the differentiation between positional isomers of disubstituted phenyl-containing compounds and that can be applied in mass spectrometry-based complex mixture analysis. By analyzing different substitution patterns over several sets of isomeric compounds, we show that IRIS is able to consistently probe the diagnostic CH out-of-plane vibrations that are sensitive to positional isomerism. We show that these modes are largely independent of the chemical functionality contained in the ring substituents and of the type of ionization. We also show that IRIS spectra often identify the positional isomer directly, even in the absence of reference spectra obtained from physical standards or from computational prediction. We foresee that this method will be generally applicable to the identification of disubstituted phenyl-containing compounds.
Introduction
The assignment of the full molecular structure of detected compounds is a common bottleneck in the application of mass spectrometry (MS) in various fields.1–3 Often, isobaric candidate structures exist for a detected m/z and the structural information from LC-MS analysis (such as retention times and MS/MS fragmentation patterns) is insufficient for a full structural identification. Compounds containing a substituted aromatic ring, ubiquitous in therapeutic drugs and their drug metabolites, form a particular challenge to MS, as the positional isomers (ortho, meta, para) that result from a different substitution pattern often have very similar retention times and MS/MS fragmentation spectra. Moreover, chromatographic retention times and MS/MS spectra are merely used as empirical parameters,4–7 which are difficult to predict in silico and thus require libraries derived from physical standards. Synthesis of reference standards is a very time-consuming step in analytical workflows, and may not be possible at all for some compounds.3,8 Ion mobility spectrometry (IMS) may differentiate ortho, meta- and para-isomers, but computational methods to predict collisional cross sections (CCSs) are often not reliable enough to predict the small differences in CCS between isomers; unless all positional isomers are present in the same sample, the use of reference standards is often required.9,10 As a result, analysis of positional isomers by MS most often gives only partially resolved structures (i.e. Markush structures)11 and complete molecular identification must fall back on alternative techniques, such as nuclear magnetic resonance (NMR) spectroscopy, that lack the sensitivity and selectivity of MS and are thus challenging to apply to low-abundance components in complex mixtures.In the field of drug discovery and development, studies on degradation and detoxification of drug compounds in the body are crucial and rely strongly on MS for the detection and identification of drug metabolites in in vitro or in vivo matrices.12,13 While the type of metabolic reaction that occurs can often be derived from the mass difference between the drug and its metabolites as detected by LC-MS, the exact site of the metabolic transformation is often difficult to determine. In particular, establishing the site of oxidation on aromatic moieties in the molecule, the most common phase I metabolic transformation, is a commonly encountered challenge.
In contrast to mass spectrometry, infrared (IR) spectroscopy is well known as a sensitive structural diagnostic, able to differentiate between the positional isomers of substituted phenyl groups.14–16 The 2-(ortho), 3-(meta) and 4-(para)substituted isomers each possess a characteristic pattern of IR absorption bands, particularly in the spectral range between 650 and 900 cm−1. Vibrational bands in this range are attributed to the out-of-plane CH bending modes of the substituted phenyl ring. However, IR spectroscopy is generally not applicable in the identification of low-abundance species in complex mixtures, such as in incubates or body fluids in drug metabolism studies, due to the lack of (mass-)selectivity: spectra are obtained for the complex mixture as a whole rather than for a single molecular component of the mixture. Circumventing this limitation, infrared ion spectroscopy (IRIS) records IR spectra of mass-isolated ions trapped in an ion storage mass spectrometer by using infrared multiple-photon dissociation (IRMPD).17–22 This yields IR information of individual MS-features while maintaining the full sensitivity and selectivity of analytical LC-MS workflows.23–26 Previous studies have shown that IR spectra of ions commonly detected in typical electrospray ionization MS experiments (i.e. protonated, deprotonated, metal adducts etc.) can differentiate closely related isomeric structures.23–33 This enables the identification of small molecules in complex mixtures, such as urine, plasma, cerebral spinal fluid and liver microsomes.23–27
Even in the absence of reference standards, IR spectra can relate to molecular structure by comparison with quantum-chemically computed spectra for candidate structures. In silico prediction of gas-phase IR spectra using density functional theory (DFT) is generally reliable for most small molecules.25 In recent analytical applications of IRIS, identifications have mainly relied on matching of IRIS spectra of unknowns with those of reference standards;23,26 using instead DFT predicted spectra – as is common in fundamental ion-chemistry applications of ion spectroscopy – could mitigate the need for physical standards (and their synthesis), exploiting IRIS to its full potential.25 In this study we address the identification of ortho-, meta- and para-isomers using IRIS and DFT predicted spectra. This provides a new MS-based method to identify low-abundance positional isomers in complex mixtures. Furthermore, we explore the diagnostic CH out-of-plane IR fingerprint of positional isomers in the 650–900 cm−1 spectral range and show that the consistency of this fingerprint across molecular systems makes the method generally applicable to ortho-, meta- and para-isomers and allows to reveal the substitution pattern even without DFT-based analyses.
Experimental
Chemicals and materials
Reference standards of phenylethylpiperidine compounds were synthesized in-house at Janssen R&D (Beerse, Belgium). 2-Fluorobenzyl alcohol, 3-fluorobenzyl alcohol and 4-fluorobenzyl alcohol standards were obtained from Sigma-Aldrich (St Louis, USA). LC-MS grade methanol (MeOH) and water (H2O) were obtained from Merck (Darmstadt, Germany). Cesium chloride salt was obtained from Sigma-Aldrich (St Louis, USA).Infrared ion spectroscopy
A modified quadrupole ion trap mass spectrometer (Bruker, Amazon Speed ETD) coupled to the FELIX IR-FEL was used for all IRMPD experiments. Details of the modifications of this instrument and the synchronization with the infrared laser have been previously reported.34 Solutions of the reference standards of ∼10−7 M (in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 MeOH:H2O for the phenylethylpiperidine compounds and in 50:50 MeOH:H2O + ∼10−7 M CsCl for the fluorobenzyl alcohols) were directly infused into an electrospray ionization source (+ESI) at 120–180 μl h−1 flow rates. Ions of interest were mass-isolated and irradiated with the FELIX IR beam. In order to generate IR spectra of MS/MS fragment ions, mass-isolated precursor ions were first fragmented using collision induced dissociation (CID) and fragment ions of interest were mass-isolated and irradiated.
:50 MeOH:H2O for the phenylethylpiperidine compounds and in 50:50 MeOH:H2O + ∼10−7 M CsCl for the fluorobenzyl alcohols) were directly infused into an electrospray ionization source (+ESI) at 120–180 μl h−1 flow rates. Ions of interest were mass-isolated and irradiated with the FELIX IR beam. In order to generate IR spectra of MS/MS fragment ions, mass-isolated precursor ions were first fragmented using collision induced dissociation (CID) and fragment ions of interest were mass-isolated and irradiated.
The FELIX IR laser was set to produce IR radiation in the form of ∼10 μs macropulses of 50 to 150 mJ at a 10 Hz repetition rate (bandwidth ∼0.4% of the centre frequency). When irradiated ions absorb IR photons (i.e. if the laser frequency matches a vibrational transition), their internal energy increases leading, eventually, to fragmentation. This is observed by recording an MS spectrum after IR irradiation. We recorded IR-induced MS/MS spectra while tuning the frequency of the IR laser through the 650–1750 cm−1 range in steps of 3 cm−1. At each IR laser frequency, 6 MS/MS spectra were recorded and averaged. An IR spectrum was generated by plotting the fractional dissociation (Yield = ∑I(fragment ions)/∑I(precursor + fragment ions)) as a function of IR frequency. IR frequencies were calibrated using a grating spectrometer and the IR yield was linearly corrected for frequency-dependent fluctuations in laser pulse energy. Each experimental spectrum shown in this manuscript is normalized to the most intense peak.
Fourier transform infrared spectroscopy
Gas-phase FTIR spectra were obtained using a Bruker VERTEX 80v FTIR spectrometer and a home-built 15 cm long gas cell with ZnSe windows. The cell was evacuated using a rotary vane vacuum pump and liquid samples were evaporated into the cell by heating the cell to 70 °C. A background subtraction and baseline correction was applied to all spectra.Computational procedure
In order to obtain theoretical IR spectra we performed quantum-chemical calculations using the Gaussian16 software package.35 For the m/z 202 phenylethylpiperidine fragment ions, geometry optimizations and harmonic IR frequency calculations were performed using density functional theory (DFT) at the B3LYP/6-31++G(d,p) level. Input structures for these calculations were generated with a molecular mechanics/molecular dynamics (MM/MD) computational approach using AMBER 12.36 Geometries of initial guess structures were optimized at the B3LYP/6-31++G(d,p) level and, after an initial minimization within AMBER, a simulated annealing procedure up to 1000 K was used to explore the potential energy landscape of the ions. Throughout the procedure, molecular structures were saved periodically as snapshots yielding 500 structures. These structures were clustered based on similarity (yielding ∼10 structures) and used as input structures for the DFT calculations. The results were sorted on the basis of their free energies relative to the ground state conformer and among the lowest-energy structures (relative free energy <2 kJ mol−1) the conformer with the theoretical IR spectrum that showed the best match with the experimental IR spectrum was selected. The selected conformers of the ortho-, meta- and para-isomers have a free energy of 1.5 kJ mol−1, 0.5 kJ mol−1 and 0.1 kJ mol−1 relative to the ground-state conformer, respectively. This is well within the error range expected for energy calculations at this level of theory. Input structures for the cesium adducts of the fluorobenzyl alcohols and neutral fluorobenzyl alcohols were generated using chemical intuition. DFT calculations were performed at the B3LYP/def2-TZVPP and B3LYP/6-31++G(d,p) level of theory for the Cs+ adducts and neutrals, respectively. All vibrational spectra were scaled using a linear scaling factor of 0.975 and the vibrational stick spectra were convoluted with a Gaussian function of 20 cm−1 full width at half maximum to facilitate comparison with the experimental spectra. In general, the resulting spectra were normalized to the most intense peak. However, in the case of the ortho and meta phenylethyl-piperidine fragment ions, the intensity of the highest peak was clearly incorrectly predicted and therefore these spectra were normalized on the second most intense peak to better facilitate the comparison with the experimental spectra.Results and discussion
Our study focuses on a group of 10 compounds sharing a common phenylethylpiperidine substructure (see Fig. 1a), which is analogous to compounds selected previously in an investigation by IMS.9 The compounds differ in the nature of the group R and the site of hydroxylation of the phenyl-ring. The different R-groups are not further specified here as they are not relevant; their purpose is only to show that the disubstituted phenyl ring can be analyzed independently. Five compounds were ortho hydroxy-substituted (with groups R1, R2, R3, R4 and R5), two were meta hydroxy-substituted (with groups R1 and R2) and three were para hydroxy-substituted (with groups R1, R2 and R5). Distinguishing the positional isomers with the same R-group on the basis of MS has been shown previously to be very challenging, even when multiple stages of fragmentation were applied.9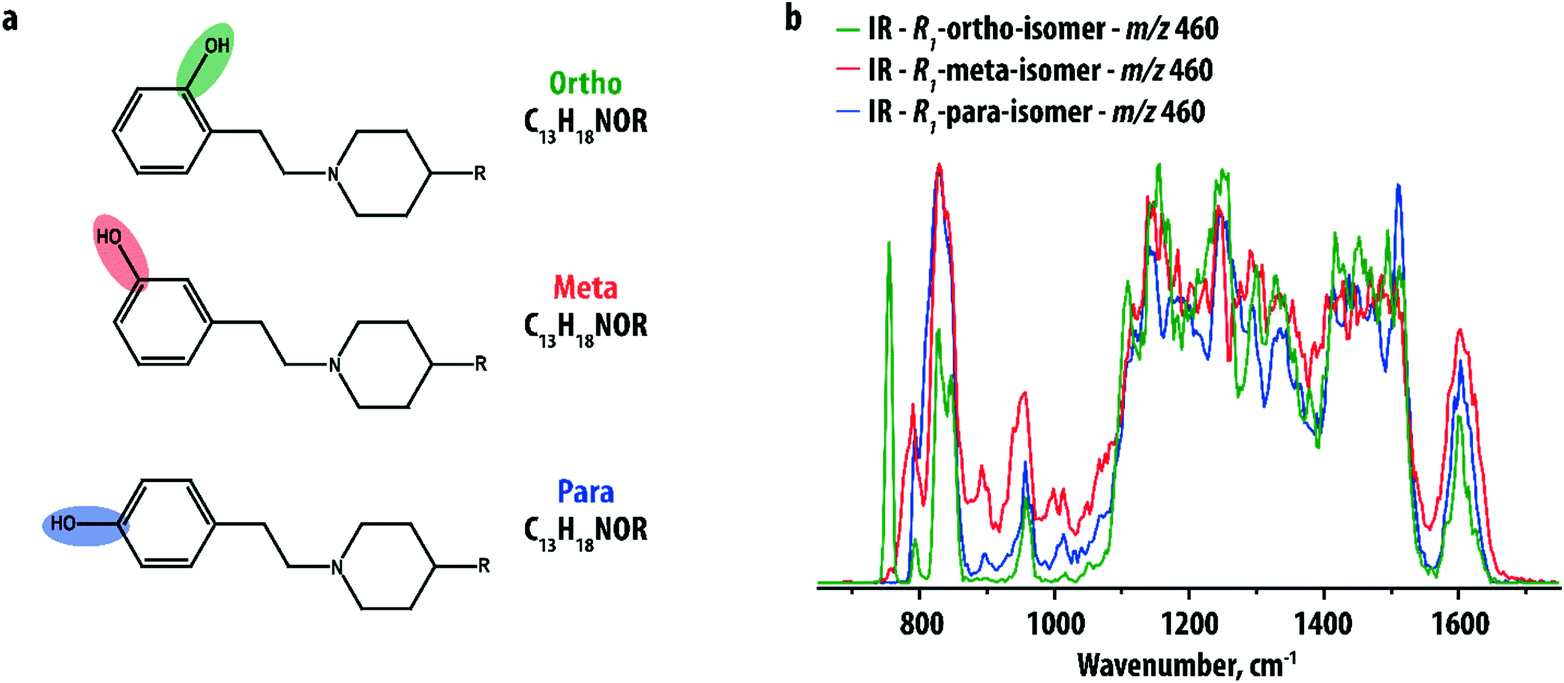 | ||
| Fig. 1 (a) General structure of the selected hydroxy-substituted phenylethylpiperidine derivatives. The compounds share a common structure but differ in the nature of the group R and the hydroxylation-site (ortho, meta or para on the phenyl-ring). (b) Comparison of the IR spectrum of the protonated ortho-, meta- and para-isomers with the R1-group (total m/z 460). | ||
IR spectra were recorded for the [M + H]+-ions of all compounds using IRMPD. Fig. 1b compares the IR spectra of the three isomers (ortho, meta and para-hydroxy substituted) of the R1-substituted compound. The three spectra are indeed distinct, especially in the low-frequency range, but the differences are small. Furthermore, the spectra are crowded and partially unresolved due to the relatively large size of the ions (m/z 460), possessing many partially overlapping IR bands especially in the 1000–1500 cm−1 range. This makes isomer assignment difficult especially on the basis of computed reference spectra.
The extensive MSn capabilities of ion trap mass spectrometers can be used to simplify IR spectra by recording IRIS spectra not for the precursor analyte, but instead for one of its CID MS/MS fragment ions.25,27 We expect that their IR spectra are not only less crowded, but may also contain sharper IR peaks due to reduced conformational heterogeneity. A similar approach to reduce conformational freedom and thereby improve differentiation of drug metabolites was applied using IMS.9 Phase I drug metabolites were shown to be particularly well suited for this strategy as most of the molecular structure is known (i.e. the precursor drug) and only a small part of the structure requires elucidation: the site of the biotransformation.
Fig. 2a shows the CID MS/MS spectrum of the ortho hydroxy-substituted R1-compound, which is very similar to those of the corresponding meta and para hydroxy-substituted compounds shown in Fig. S1 in the ESI.† For all three analytes a fragment ion at m/z 202 is observed, corresponding to the phenylethylpiperidine ion resulting from loss of the R1-group (see Fig. 2b). This fragment retains the hydroxyl-group but lacks the relatively bulky R1-group that is identical for each of the isomers. IR spectra for the three m/z 202 fragment ions are shown in Fig. 2c. Fig. 2d compares the IR spectrum of the m/z 202 fragment of the ortho hydroxy-substituted metabolite with the IR spectrum of its precursor. Although the two spectra have similar overall IR features, the spectrum of the MS/MS fragment indeed shows sharper and better resolved peaks.
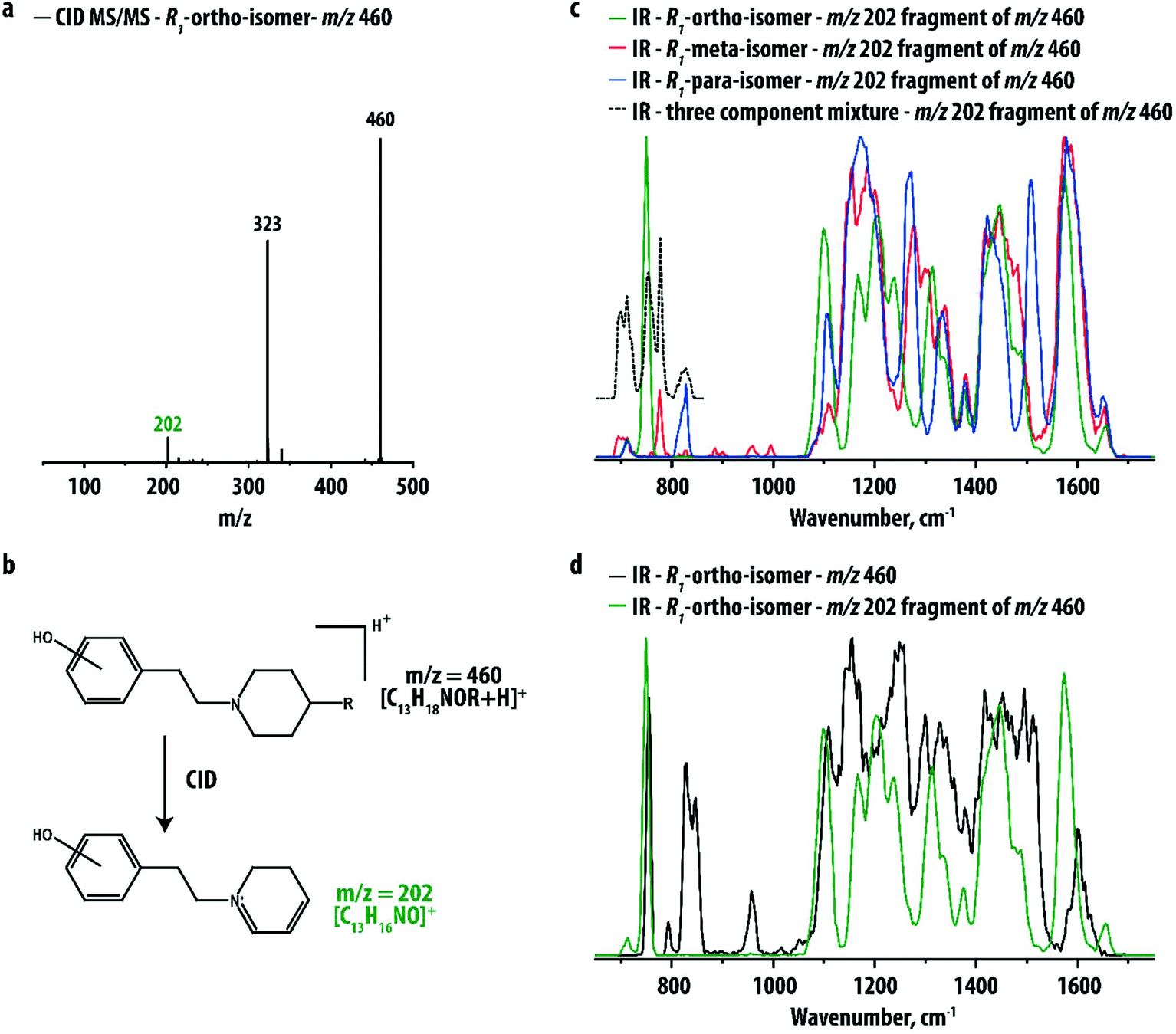 | ||
| Fig. 2 (a) CID MS/MS spectrum showing the fragmentation of the R1-ortho-isomer (m/z 460). (b) Scheme showing that the observed m/z 202 fragment corresponds to the phenylethylpiperidine ion resulting from the loss of the R-group. (c) Comparison of the IR spectra of the m/z 202 fragment of the ortho-, meta- and para-isomers with the R1-group. The dashed line represents the IR spectrum recorded for the m/z 202 fragment ions of a 1:1:1 mixture of ortho-, meta- and para-isomers in the 650–850 cm−1 range. (d) Comparison of the IR spectra of the precursor (m/z 460) and fragment ion (m/z 202) of the R1-ortho-isomer. | ||
IR spectra for the three m/z 202 fragment ions are clearly distinct, particularly in the 600–900 cm−1 range of the spectrum. This spectral region is well-known to be diagnostic for the distinction between ortho-, meta- and para-isomers in conventional (FT)IR spectroscopy. The isolated nature of the IR bands attributed to each of the positional isomers suggests that the three ions may be distinguished from the IR profile over only this approximately 300 cm−1 range of the spectrum, which paves the way towards the development of rapid screening methodologies for pharmaceutical laboratories. Moreover, using these peaks it may be possible to identify the components of a mixture of two or three isomers, for instance in cases where drug metabolism yields more than one of the positional isomers. To verify this idea, an IRMPD spectrum was recorded for the m/z 202 fragment of a three-component mixture. The dashed black trace in Fig. 2c shows that the characteristic peaks of the thee isomers can indeed be individually recognized.
In order to explore the general applicability of this method, compounds with different R-groups were investigated as well. Four compounds, R2 with m/z 460 (ortho, meta, para) and R3 with m/z 347 (ortho), were found to yield the m/z 202 fragment and their hydroxylation-site could be correctly determined from their fragment ion IR spectra, which were identical to the ones shown in Fig. 2c (see Fig. S2 and S3†); slight differences in relative intensity in Fig. S3† are likely caused by small variations in the laser power versus wavelength during the three spectral runs, but no deviations in the band centers are observed, indicating that the structure of the m/z 202 ion is the same, independent of the nature of the R-group in the precursor. Due to its smaller size, the IR spectrum of the R3-precursor ion is much better resolved than the IR spectra of the other precursor ions, probably allowing one to distinguish the positional isomers without CID fragmentation. CID of the remaining three compounds (ortho R4 at m/z 315 and ortho/para R5 at m/z 303) did not yield the m/z 202 fragment ion. However, as a result of the low mass of these ions, the precursor ion IR spectra show well-resolved diagnostic features and the two R5-isomers could be clearly distinguished (see Fig. S4†).
An additional advantage of investigating the smaller fragment ions instead of their precursor ions – especially when the fragmentation pathway is straightforward – relates to the computational prediction of their IR spectra: fragment ion structures are not only smaller but also often found to be more rigid than their precursors, often leading to a better agreement between experiment and theory. The IR spectra of the ortho, meta and para hydroxy-substituted m/z 202 fragment ions were computed using DFT at the B3LYP/6-31++G(d,p) level. Fig. 3a compares the experimental spectra of the m/z 202 fragment ions of the R1-precursors with theoretical IR spectra of the three ions. Although computed band intensities are not entirely in agreement with experimental data, the main spectral features differentiating the three experimental spectra are well reproduced. This suggests that in the absence of reference standards, this method may be used to identify ortho, meta or para hydroxy-substituted positional isomers. As an example, Fig. S5–S7† compare the experimental IR spectra of each of the m/z 202 fragment ions to calculated spectra for all of the three isomers. Clearly, this leads to the assignment of the correct substitution pattern in all cases.
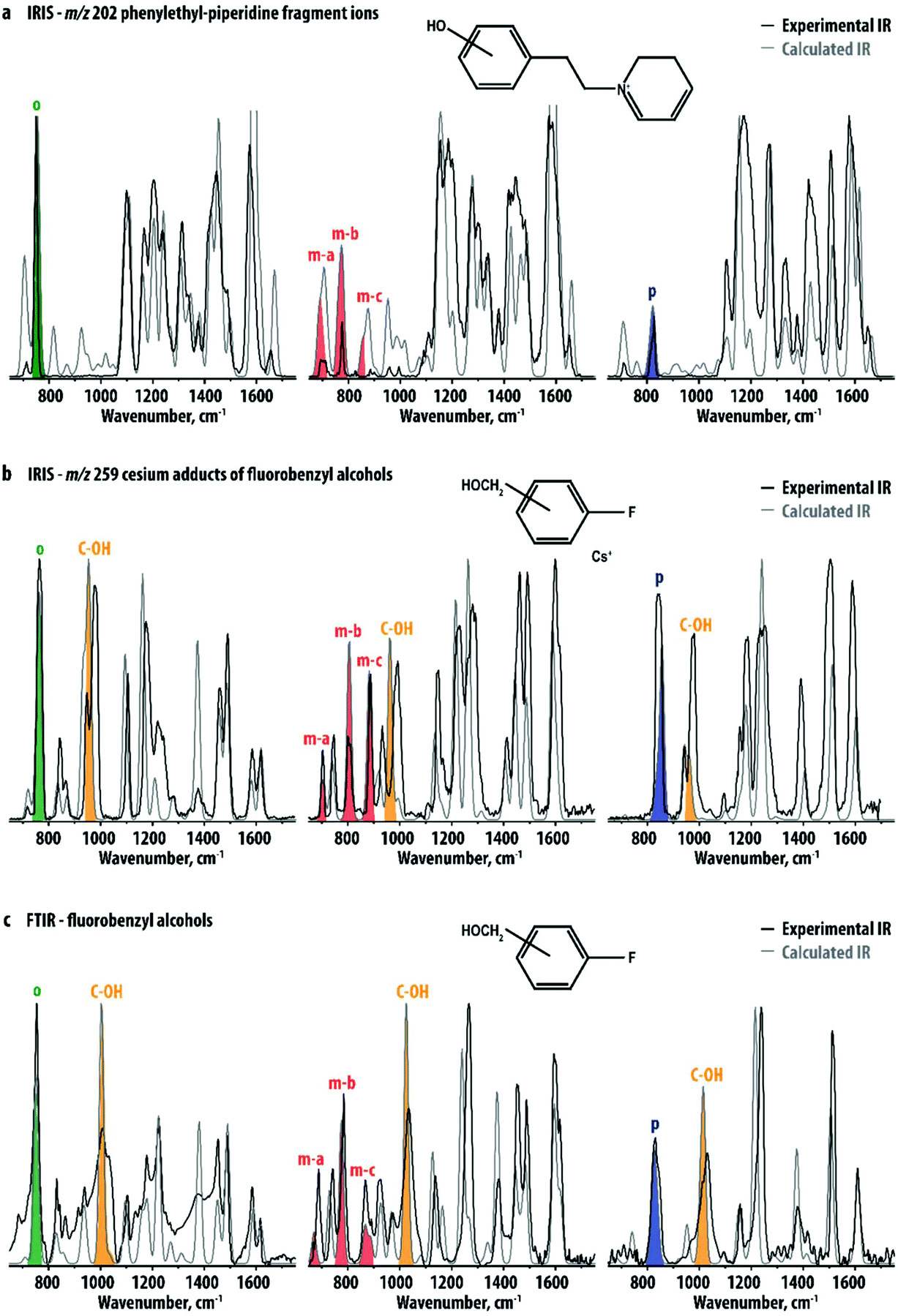 | ||
| Fig. 3 Comparison of experimental (black) and predicted (gray) IR spectra of (a) the m/z 202 fragments of the ortho- (left), meta- (middle) and para-R1-isomers (right), (b) the Cs+ adducts of 2-fluorobenzyl alcohol (left), 3-fluorobenzyl alcohol (middle) and 4-fluorobenzyl alcohol (right) obtained using IRMPD and (c) of the three neutral gaseous fluorobenzyl alcohols obtained using FTIR spectroscopy. The most prominent CH-out-of-plane vibrations in the predicted spectra are indicated in green, red and blue for the ortho-, meta-, and para-isomers, respectively. In panels (b) and (c) the C-OH stretching vibrations are also indicated (yellow). | ||
The quantum-chemical calculations also allow us to assign molecular normal mode vibrations to the experimentally observed IR bands. We focus on the diagnostic CH out-of-plane modes in the 600–900 cm−1 region. The most prominent bands are labelled in Fig. 3a and the corresponding normal modes are visualized in Fig. 4. Their frequencies are listed in Table 1 and compared to tabulated IR group frequencies of out-of-plane CH vibrations of aromatic groups. It is seen that a single vibrational mode involving all CH-oscillators of the phenyl-group is predicted and observed for the ortho- and para-isomers, whereas three bands (involving one or three CH-oscillators) are predicted for the meta-isomer. Two bands, labeled as ma and mb, are clearly observed. The mc-band is not observed, suggesting that its intensity is insufficient to induce dissociation of this (relatively stable) fragment ion. Spectra for the ortho- and para-isomers show some intensity at the position of the low-frequency band of the meta-isomer (ma) as well, but this is due to an overlapping out-of-plane CH bending vibration of the other ring in the molecule. Both the number of these IR bands as well as their frequency positions agree well with known group-frequencies of out-of-plane CH vibrations16 (see Table 1). Note that such an interpretation of the CH vibrations is not possible for the precursor ions themselves due to overlapping bands from the R-group in the 800 cm−1 region.
 | ||
| Fig. 4 DFT optimized molecular structures visualizing the CH out-of-plane normal modes of the m/z 202 phenylethylpiperidine fragment ions (top row), m/z 259 cesium adducts of the fluorobenzyl alcohols (middle row) and neutral fluorobenzyl alcohols (bottom row). | ||
| Frequency (cm−1) | |||||
|---|---|---|---|---|---|
| Ortho | Meta | Para | |||
| a | b | c | |||
| Group frequencies 16 | 740–780 | 680–740 | 750–820 | 860–910 | 800–870 |
| Experimental frequencies | |||||
| Phenylethylpiperidine fragments (IRIS) | 749 | 694 | 775 | — | 828 |
| Fluorbenzyl alcohols (IRIS) | 764 | 704 | 801 | 888 | 845 |
| Fluorobenzyl alcohols (FTIR) | 754 | 688 | 784 | 867/891 | 831 |
| Calculated frequencies | |||||
| Phenylethylpiperidine fragments (IRIS) | 754 | 687 | 777 | 853 | 823 |
| Fluorobenzyl alcohols (IRIS) | 761 | 704 | 807 | 882 | 857 |
| Fluorobenzyl alcohols (FTIR) | 753 | 668 | 773 | 865/886 | 835 |
To further explore the general applicability of structural diagnostics based on the out-of-plane CH-bending vibrations, we selected a set of simple and readily available positional isomers: 2-, 3- and 4-fluorobenzyl alcohol. The relatively high vapor pressure of these compounds allowed gas-phase FTIR spectra to be recorded for the neutral species for comparison. The methoxy- and fluoro-substituents were chosen to assess the effect of the functional groups on the vibrational signatures. For the IRIS measurements in the mass spectrometer, these analytes are not easily protonated or deprotonated by ESI,† but their alkali metal-ion adducts can easily be produced. IRMPD spectra were therefore recorded for the [M + Cs]+ ions of the ortho-, meta- and para-isomers. Cesium has a relatively low binding-energy37–44 as compared to smaller alkali metal ions and [M + Cs]+ adducts should therefore easily undergo IR-induced dissociation. Moreover, the mass of Cs+ (m/z 133) is well above the low-mass cut-off of the ion trap mass spectrometer, which allows for its detection as a fragmentation product upon IR-induced dissociation.
Fig. 3b shows the experimental spectra of the three ions compared with calculated spectra obtained using DFT. Again, the three ions can be spectroscopically differentiated, especially based on their distinctive pattern of vibrational bands in the 600–900 cm−1 region, indicated with colors. Their normal mode displacements are visualized in Fig. 4, showing that they are analogous to the modes of the phenylethylpiperidine fragment ions. Comparing Fig. 3a and b, it is clear that although two rather different classes of molecules are being investigated, the pattern of out-of-plane CH bending modes is very similar for corresponding positional isomers (see also Table 1). This suggests that the band patterns do not strongly depend on the functionality of the substituents, which suggests that this approach is widely applicable. A recent study by Polfer and coworkers used IRIS in the 3000–4000 cm−1 range to identify positional isomers of aminobenzoic acid, where the NH- and OH-stretch frequencies are sensitive to the substitution pattern as a consequence of mesomeric effects.33 In the two sets of systems studied here, differences in inductive effects between positional isomers is minimal and all isomers have nearly identical OH stretch frequencies, as illustrated by the theoretical spectra in Fig. S8.† Therefore, probing the CH out-of-plane bending modes has the advantage of broader applicability as it is not dependent on the presence of NH- or OH-moieties, or of inductive effects of the substituents in general.
In addition, the CH out-of-plane vibrations are especially well modelled by the calculations, suggesting that it is possible to identify the substitution pattern without computational reference spectra, but based on group frequency arguments. We note that other (non CH-bending) vibrations can also appear in the 600–900 cm−1 region and possibly interfere with a direct interpretation of the CH-bending vibrations (see for example the comparison above of precursor/fragment IR spectra in Fig. 2d). Analysis of the IRIS spectra of CID fragment ions can reduce such interferences.
The results shown in Fig. 3b suggest that the interaction of the analyte with Cs+ hardly affect the frequency of the out-of-plane CH-bending vibrations. Moreover, the agreement of the determined band-positions with tabulated group-frequencies of out-of-plane CH vibrations (see Table 1) suggests that the multiple-photon nature of the IRMPD process does not cause strong frequency-shifts of the out-of-plane CH vibrations. In order to directly probe the influence of Cs+-coordination and the multiple-photon nature of IRMPD, we compare these spectra with gas-phase FTIR spectra of the three neutral fluorobenzyl alcohol isomers (Fig. 3c) and their DFT calculated spectra. Fig. S9† provides a direct comparison of the FTIR and IRIS spectra for each of the isomers. The IRIS and FTIR spectra are very similar and mainly differ in the relative intensities of several bands. The most significant effect of the interaction with the Cs+-ion is a small red-shift of the strong band around 1000 cm−1, shaded yellow in Fig. 3b and c. This band is attributed to the C-OH stretching vibration, which shifts to lower frequencies due to the interaction of the O-atom with the Cs+ ion. Experimental and theoretical frequencies of the out-of-plane CH bending modes of the neutral fluorobenzyl alcohols are also listed in Table 1 and the normal modes are visualized in Fig. 4. Generally, these bands shift by 10–15 cm−1 to higher frequencies in the IR spectra recorded with IRMPD. Moreover, in neutral 3-fluorobenzyl alcohol the mc-normal mode is coupled to a nearly degenerate out-of-plane CH bending mode, leading to two, partially overlapping bands (see Fig. 3 and 4). The frequency of this doublet is comparable to the mc-bands in the other compounds. This confirms that the patterns of out-of-plane CH bending modes observed for disubstituted phenyl-isomers are fairly unaffected by the type of analyte probed (neutral, protonated, deprotonated, metal adducts) and the experimental approach. Although experiments on deprotonated positional isomers were not specifically reported here, quantum-chemical calculations for protonated and deprotonated ortho-, meta- and para-aminobenzoic acid indeed suggest a consistent behavior of these modes (see Fig. S10†). An early IRIS study on the isomers of the fluorobenzoate anion also suggest these modes to be characteristic for each isomer.45
Conclusions
We have shown that IRIS spectroscopy is potentially a highly valuable method for the differentiation and identification of positional isomers of disubstituted phenyl-containing compounds in MS-based analysis. The CH out-of-plane normal modes of ortho-, meta- and para-substituted phenyl groups leave distinct signatures in the frequency range of 650 to 900 cm−1. The spectral resolution obtained in the IRMPD spectra was sufficient to resolve these features and to unambiguously assign several sets of positional isomers. Moreover, the diagnostic IR bands appear to be fairly unaffected by the type of substituent and the nature of ionization (protonated, deprotonated, metal adducts). This pattern corresponds well with tabulated IR group frequencies for conventional (FT)IR spectra of neutral molecules with positional isomerism and is well predicted by quantum-chemical calculations. This consistency suggests that IRIS-based identification may be generally applicable in the MS-analysis of ortho, meta and para-isomers, e.g. in the analysis of phase I drug metabolites, but also in a wider range of small-molecule MS applications. The predictability of the patterns of CH out-of-plane bending modes across molecular systems observed here even suggests that it may be possible to directly assign substitution patterns, even in absence of experimental or computed reference IR spectra. This would provide a clear advantage of IR spectroscopy over other MS-hyphenated methods, such as IMS or chromatography, in the identification of positional isomers. Retention times and collisional cross sections usually do not allow one to identify the specific isomer in the absence of accurate reference data obtained from physical standards unless all positional isomers are present in the sample allowing relative ranking; the spectral fingerprint in the 600–900 cm−1 range is in that sense particularly diagnostic and easy to interpret.To resolve issues of spectral congestion for larger molecular systems, we show that IRIS can also be performed on CID fragments of the precursor ion containing the substituted phenyl ring. Moreover, for species with low ionization efficiency that are hard to detect as protonated/deprotonated ions in ESI-MS, IRIS spectra of Cs+-adducts can be recorded. For the systems studied here, their CH out-of-plane vibrational frequencies are similar to those of their gas-phase neutral counterparts. In addition, the relatively low threshold to dissociation of Cs+-adducts, as well as the relatively narrow frequency range in which the diagnostic bands occur, may be exploited in the development of IRIS-based identification strategies using IR laser sources with lower output power, which may eventually extend the applicability of this method to laboratories beyond FEL facilities.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge Rob Vreeken for his contribution to discussions and the Nederlandse Organsiatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. This work was supported by NWO division Natural Sciences (grant numbers VICI 724.011.002, TTW 15769, TKI-LIFT 731.017.419, Rekentijd 2019.062).References
- L. Cui, H. Lu and Y. H. Lee, Mass Spectrom. Rev., 2018, 37, 772–792 CrossRef CAS.
- W. B. Dunn, A. Erban, R. J. M. Weber, D. J. Creek, M. Brown, R. Breitling, T. Hankemeier, R. Goodacre, S. Neumann, J. Kopka and M. R. Viant, Metabolomics, 2013, 9, 44–66 CrossRef CAS.
- F. Hufsky and S. Böcker, Mass Spectrom. Rev., 2017, 36, 624–633 CrossRef CAS.
- J. Bonetti, Forensic Chem., 2018, 9, 50–61 CrossRef CAS.
- S. P. M. Menachery, O. Laprévote, T. P. Nguyen, U. K. Aravind, P. Gopinathan and C. T. Aravindakumar, J. Mass Spectrom., 2015, 50, 944–950 CrossRef CAS.
- M. N. Eberlin, J. Mass Spectrom., 2006, 41, 141–156 CrossRef CAS.
- A. Maas, K. Sydow, B. Madea and C. Hess, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1051, 118–125 CrossRef CAS.
- F. Hufsky, K. Scheubert and S. Böcker, TrAC, Trends Anal. Chem., 2014, 53, 41–48 CrossRef CAS.
- F. Cuyckens, C. Wassvik, R. J. Mortishire-Smith, G. Tresadern, I. Campuzano and J. Claereboudt, Rapid Commun. Mass Spectrom., 2011, 25, 3497–3503 CrossRef CAS.
- C. Lapthorn, F. S. Pullen, B. Z. Chowdhry, P. Wright, G. L. Perkins and Y. Heredia, Analyst, 2015, 140, 6814–6823 RSC.
- T. De Vijlder, D. Valkenborg, F. Lemière, E. P. Romijn, K. Laukens and F. Cuyckens, Mass Spectrom. Rev., 2018, 37, 607–629 CrossRef CAS.
- K. F. Geoghegan and M. A. Kelly, Mass Spectrom. Rev., 2005, 24, 347–366 CrossRef CAS.
- C. Prakash, C. L. Shaffer and A. Nedderman, Mass Spectrom. Rev., 2007, 26, 340–369 CrossRef CAS.
- M. Margoshes and V. A. Fassel, Spectrochim. Acta, 1955, 7, 14–24 CAS.
- B. C. Smith, Infrared spectral interpretation: a systematic approach, CRC press, 1999 Search PubMed.
- D. R. Lide, CRC handbook of chemistry and physics, CRC Press, 2001 Search PubMed.
- J. Lemaire, P. Boissel, M. Heninger, G. Mauclaire, G. Bellec, H. Mestdagh, A. Simon, S. L. Caer, J. M. Ortega, F. Glotin and P. Maitre, Phys. Rev. Lett., 2002, 89, 273002 CrossRef CAS.
- J. Oomens, B. G. Sartakov, G. Meijer and G. von Helden, Int. J. Mass Spectrom., 2006, 254, 1–19 CrossRef CAS.
- T. D. Fridgen, Mass Spectrom. Rev., 2009, 28, 586–607 CrossRef CAS.
- J. Martens, G. Berden and J. Oomens, Anal. Chem., 2016, 88, 6126–6129 CrossRef CAS.
- N. C. Polfer, Chem. Soc. Rev., 2011, 40, 2211–2221 RSC.
- N. C. Polfer and J. Oomens, Mass Spectrom. Rev., 2009, 28, 468–494 CrossRef CAS.
- J. Martens, V. Koppen, G. Berden, F. Cuyckens and J. Oomens, Anal. Chem., 2017, 89, 4359–4362 CrossRef CAS.
- J. Martens, R. E. van Outersterp, R. J. Vreeken, F. Cuyckens, K. L. M. Coene, U. F. Engelke, L. A. J. Kluijtmans, R. A. Wevers, L. M. C. Buydens, B. Redlich, G. Berden and J. Oomens, Anal. Chim. Acta, 2020, 1093, 1–15 CrossRef CAS.
- R. E. van Outersterp, K. J. Houthuijs, G. Berden, U. F. Engelke, L. A. J. Kluijtmans, R. A. Wevers, K. L. M. Coene, J. Oomens and J. Martens, Int. J. Mass Spectrom., 2019, 443, 77–85 CrossRef CAS.
- J. Martens, G. Berden, R. E. van Outersterp, L. A. J. Kluijtmans, U. F. Engelke, C. D. M. van Karnebeek, R. A. Wevers and J. Oomens, Sci. Rep., 2017, 7, 3363 CrossRef.
- J. Martens, G. Berden, H. Bentlage, K. L. M. Coene, U. F. Engelke, D. Wishart, M. van Scherpenzeel, L. A. J. Kluijtmans, R. A. Wevers and J. Oomens, J. Inherited Metab. Dis., 2018, 41, 367–377 CrossRef CAS.
- N. C. Polfer, J. J. Valle, D. T. Moore, J. Oomens, J. R. Eyler and B. Bendiak, Anal. Chem., 2006, 78, 670–679 CrossRef CAS.
- A. P. Cismesia, L. S. Bailey, M. R. Bell, L. F. Tesler and N. C. Polfer, J. Am. Soc. Mass Spectrom., 2016, 27, 757–766 CrossRef CAS.
- B. Schindler, G. Laloy-Borgna, L. Barnes, A.-R. Allouche, E. Bouju, V. Dugas, C. Demesmay and I. Compagnon, Anal. Chem., 2018, 90, 11741–11745 CrossRef CAS.
- O. Gorlova, S. M. Colvin, A. Brathwaite, F. S. Menges, S. M. Craig, S. J. Miller and M. A. Johnson, J. Am. Soc. Mass Spectrom., 2017, 28, 2414–2422 CrossRef CAS.
- F. Berthias, B. Maatoug, G. L. Glish, F. Moussa and P. Maitre, J. Am. Soc. Mass Spectrom., 2018, 29, 752–760 CrossRef CAS.
- A. P. Cismesia, M. R. Bell, L. F. Tesler, M. Alves and N. C. Polfer, Analyst, 2018, 143, 1615–1623 RSC.
- J. Martens, G. Berden, C. R. Gebhardt and J. Oomens, Rev. Sci. Instrum., 2016, 87, 103108 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- B. Yang, R. R. Wu, N. C. Polfer, G. Berden, J. Oomens and M. T. Rodgers, J. Am. Soc. Mass Spectrom., 2013, 24, 1523–1533 CrossRef CAS.
- M. Citir, E. M. S. Stennett, J. Oomens, J. D. Steill, M. T. Rodgers and P. B. Armentrout, Int. J. Mass Spectrom., 2010, 297, 9–17 CrossRef CAS.
- A. Günther, P. Nieto, G. Berden, J. Oomens and O. Dopfer, Phys. Chem. Chem. Phys., 2014, 16, 14161–14171 RSC.
- M. T. Rodgers and P. B. Armentrout, Chem. Rev., 2016, 116, 5642–5687 CrossRef CAS.
- P. B. Armentrout, C. A. Austin and M. T. Rodgers, J. Phys. Chem. A, 2014, 118, 8088–8097 CrossRef CAS.
- R. C. Dunbar, A. C. Hopkinson, J. Oomens, C.-K. Siu, K. W. M. Siu, J. D. Steill, U. H. Verkerk and J. Zhao, J. Phys. Chem. B, 2009, 113, 10403–10408 CrossRef CAS.
- P. B. Armentrout, M. Citir, Y. Chen and M. T. Rodgers, J. Phys. Chem. A, 2012, 116, 11823–11832 CrossRef CAS.
- C. A. Austin, Y. Chen and M. T. Rodgers, Int. J. Mass Spectrom., 2012, 330–332, 27–34 CrossRef CAS.
- J. Oomens and J. D. Steill, J. Phys. Chem. A, 2008, 112, 3281–3283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an01119c |
| This journal is © The Royal Society of Chemistry 2020 |