
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural investigation of oxidized arabinoxylan oligosaccharides by negative ionization HILIC-qToF-MS†
Teresa
Demuth
 ,
Samy
Boulos
and
Laura
Nyström
*
,
Samy
Boulos
and
Laura
Nyström
*
ETH Zurich, Institute of Food, Nutrition and Health, Schmelzbergstrasse 9, 8092 Zurich, Switzerland. E-mail: laura.nystroem@hest.ethz.ch; teresa.demuth@hest.ethz.ch; samy.boulos@hest.ethz.ch; Tel: +41 44 632 91 65
First published on 10th August 2020
Abstract
Owing to the strong structure–function relationship of polysaccharides, the targeted modification of polysaccharides is attracting widespread interest in various fields, such as food industry, nutritional science, and biomedical research. Apart from intended functionalization, polysaccharide degradation mediated by hydroxyl radicals (HO˙) occurs in various industrial processes such as food processing. In particular, the oxidative degradation of feruloylated arabinoxylan (AX), a linearly-branched polysaccharide in cereals, causes chain scissions, and introduces new functional groups in the fiber, which can potentially modify the physicochemical properties and the functionalities of AX. However, the precise characterization of those structural modifications remains challenging due to the diversity of the oxidation products formed, the high molecular weight, and the relatively low quantity of newly formed functional groups. In this paper, selective (TEMPO-mediated) and random (Fenton) oxidations of several commercial xylo- and arabinoxylan oligosaccharides (A)XOS were studied as model systems by hydrophilic interaction UPLC-MS2 in negative ion resolution mode to identify potential oxidation products. An in-depth identification of acidic (A)XOS oxidation products derived from TEMPO-mediated oxidation provided novel insights in the selective functionalization of isomeric oligosaccharides. Furthermore, MS2 enabled the precise localisation of both glycosidic linkages and functional groups in oxidized (A)XOS. An innovative combination of an enzymatic sample preparation combined with a subsequent HILIC-MS2 analysis enabled the unprecedented comprehensive characterization of Fenton-induced oxidation products derived from AX. In future, this holistic analytical approach will enable the characterization of both selective and non-selective AX oxidation procedures in various applications.
1. Introduction
In recent years, polysaccharides and their derived oligosaccharides have aroused great interest in research owing to their widespread biological functionalities.1 The biological activity and the physiochemical properties of polysaccharides are highly affected by their complex structural characteristics such as molecular weight (Mw), varying monosaccharide building blocks, and substitution pattern. Therefore, the chemical modification of polysaccharides is commonly used to modulate specific properties and functionalities in different industrial applications such as drug delivery, materials science, or as functional food.2 Besides this intended functionalization of polysaccharides, food processing modifies the native structure of polysaccharides unspecifically, leading to a significant alteration of their health promoting and technological functionalities.3–5One of the major non-digestible polysaccharides within the human diet is feruloylated arabinoxylan (AX), a partially soluble fiber predominantly found in the cell wall of cereal grains such as rye, wheat, barley, and oat. The polysaccharide is composed of a β-(1→4)-linked D-xylopyranosyl backbone with random substitutions at O-2, O-3, or O-2 and O-3 with α-L-arabinofuranose units. Phenolic acids, primarily ferulic acid, can be esterified on the C(O)-5 position of arabinose units.6 Since cereals usually undergo extensive processing treatments such as grain milling, extrusion, hydrothermal treatments, or baking, AX is prone to degradation induced by those processes.5
This degradation process includes hydrolytic reactions and chemically-induced oxidations. Thereby, the chemically-induced oxidative degradation takes place by hydroxyl radials (HO˙) randomly abstracting hydrogen atoms from any C–H groups within the monomer units, introducing new functional groups and causing – depending on the attack position within the monomers – also chain cleavage of the polymer backbone. HO˙ can be generated through the Fenton reaction (1), mediating the oxidation of AX.7 All in all, a combination of molecular oxygen, metal traces (Fe, Cu), and reducing agents build the catalytic cycle for HO˙ production.8–10
| Fe2+ + H2O2 + H+ → Fe3+ + H2O + HO˙ | (1) |
As a consequence, carbonyl or carboxyl groups are formed, ring openings occur, and the Mw decreases.8 Since such structural features (new functional groups & chain length) are related to the solubility, viscosity and the derived functionalities of AX, the identification of those structural changes is crucial to understand AX's nutritional value.11 However, the analysis of oxidized polysaccharide is challenging due to the high Mw and the relatively small number of newly formed functional groups. Prior research has focused on the application of multiple analytical techniques studying the oxidative state of polysaccharides such as AX. In prior studies, Bagdi et al. and Faure et al. used electron spin resonance spectroscopy (ESR) to monitor the HO˙ formation during polysaccharide oxidation.7,12 In addition, Bagdi et al. studied the influence of AX oxidation on molecular weight, viscosity, bile acid binding, and gel forming capacity. Similar studies have also been performed with β-glucan, in which the main focus was the determination of bulk properties and rheology.13,14 A deeper insight into the molecular level of polysaccharide oxidation was presented by Potthast et al., who quantified carbonyls in polysaccharides through fluorescent labelling by high performance size exclusion chromatography (HPSEC) with multiple detectors.15 In a previous study, oligosaccharide products diagnostic for β-glucan oxidation were investigated by UPLC-MS2 with and without prior functional group labelling, followed by selective enzymatic digestion and solid phase extraction.16 Among the analytical approaches mentioned, liquid chromatography (LC) combined with mass spectrometry (MS), is one of the most powerful techniques to analyse oligosaccharides diagnostic for oxidation.17–19 However, to the best of our knowledge, there is no reported study using this technology for the characterization of Fenton oxidized arabinoxylan, and thus the structural building blocks of oxidized AX have yet to be elucidated.20,21
Hence, this study aims to elucidate the structural modification of diagnostic arabinoxylan oligosaccharides (A)XOS through selective TEMPO-mediated and non-selective Fenton oxidation in a holistic analytical approach and its subsequent application on non-selectively oxidized AX. For this purpose, a detailed characterization of oxidized (A)XOS as model compounds using hydrophilic interaction liquid chromatography (HILIC) coupled with MS2 in negative ion resolution mode was performed. Our work is the first application of this established method for branched heterooligosaccharides. An in-depth identification of acidic (A)XOS oxidation products derived from TEMPO-mediated oxidation provided insights in the selective functionalization of isomeric oligosaccharides. Moreover, this paper presents an unprecedented comprehensive characterization of the radical induced Fenton oxidation of (A)XOS. In a proof of concept experiment, the complex product mixture of Fenton-mediated arabinoxylan oxidation was characterized in detail using an innovative combination of an enzymatic sample preparation strategy and HILIC-MS2 analysis.
2. Materials and methods
2.1. Materials
Linear XOS (X3, X4, X5, and X6), branched AXOS standards (A3X, A2XX, XA3XX, A2+3XX) both >95%, wheat arabinoxylan (medium viscosity; ∼95%), and endo-1,4-β-xylanase from Cellvibrio mixtus (750 U mL−1) were purchased from Megazyme International (Bray, Ireland), ammonium formate (≥99.995% trace metal basis), 25% aqueous ammonia (NH3) and formic acid (both LC-MS grade), 35% hydrogen peroxide (H2O2; purum p.a.), iodine (I2; ≥99.8%), iron(II) sulfate heptahydrate (FeSO4·7H2O; ≥99%), methanol (HPLC-grade), potassium hydroxide (KOH; ≥85%), sodium chlorite (NaClO2; 80%), sodium hydroxide (NaOH; ≥97.0%), sodium hypochlorite solution (NaClO; 10–15%), and 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO; 98%) were purchased from Sigma-Aldrich Chemie GmbH (Germany). L-Ascorbic acid (AA; ≥99.5%) was purchased from Fluka (Germany). Acetonitrile (ACN; ULC/MS grade) was purchased from Biosolve B.V. (Valkenswaard, The Netherlands) and the aqueous solutions were prepared with nanopure Milli-Q® water (H2O; ≥18.2 MΩ cm at 25 °C).2.2. Methods
2.2.3.1. (A)XOS. For the non-selective oxidation of (A)XOS, a reaction mixture of 1.5 mL final volume was prepared containing 1 mM of the respective oligosaccharide, 100 μM FeSO4, 500 μM ascorbic acid (AA), and 10 mM H2O2 on the basis of Bagdi et al. with some modifications.24 The oxidation was performed at room temperature (22–24 °C) with access to air for 24 h. Each oxidation was done in triplicates, including reagent controls that contained only the oligosaccharide in water. SPE fractionation followed as usual (see section 2.2.4.).
2.2.3.2. AX polymer. Arabinoxylan (132 mg) was dissolved in H2O (5 mL) for 1 h at 80 °C and stirred at 20 °C overnight. Subsequently, the reagents were added to give final concentrations of 2% AX (w/v), 50 μM FeSO4, 50 μM AA, and 50 mM H2O2 in this order.24 The oxidation was heated for the first 3 hours to 80 °C, then continued at RT with access to air for 24 h. The oxidation was done in triplicates, including reagent controls, and subsequently freeze dried. The oxidized AX (15 mg) was dissolved in H2O (2.4 mL) for 1 h at 80 °C. Then, sodium phosphate buffer (0.5 M, 60 μL, pH 6.5) was added resulting in a final AX concentration of 0.5% (w/v) and a total volume of 3 mL. Subsequently, the oxidized AX was treated with endo-1,4-β-xylanase from Cellvibrio mixtus (750 U mL−1, 144 μL) for 6 h at 40 °C to decrease the molecular weight.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (v/v) (fraction 1). Following, 1.5 mL of 0.1% (v/v) formic acid in ACN/H2O 1:3 (v/v) was used to elute fraction 2 containing the desired C1 oxidized product. Fractions 2 were collected directly in HPLC vials and analysed by UPLC-MS2.
:3 (v/v) (fraction 1). Following, 1.5 mL of 0.1% (v/v) formic acid in ACN/H2O 1:3 (v/v) was used to elute fraction 2 containing the desired C1 oxidized product. Fractions 2 were collected directly in HPLC vials and analysed by UPLC-MS2.
2.3. UPLC-MS and MS2
:2 and 2:8 (v/v) were used as weak and strong needle wash solutions, respectively. Injection volumes were 5 μL–10 μL of the undiluted and diluted sample (75% ACN) in partial loop mode, respectively. The column was maintained at 35 °C. The first gradient (Gradient I) for an overall analysis of the reaction mixture started with 20% H2O and 80% ACN (both with 0.1% NH3 additive), then increased linearly to 40% H2O in 12 min. After maintaining this for 4 min, it was reequilibrated with the initial conditions for 1 min.
The second gradient was applied for an improved separation of the acidic oxidation products (Gradient II) based on the procedure presented by Westereng et al.25 Eluent A and B were a mixture of ACN/H2O in the ratio of 1:3 and 3:1, both with the addition of 60 mM ammonium formate buffer (NH4HCO2; pH 8). The gradient started isocratically with 100% B for 2 min (= 25% H2O), increased linearly to 34% A for 5.5 min (= 42% H2O), maintained for 1.5 min, and decreased back to 0% A within 1 min and reequilibrated for 2.5 min.
3. Results & discussion
3.1. Identification of selectively oxidized (A)XOS
The obtained MS2 spectra of the TEMPO-mediated oxidized oligosaccharides revealed mainly signals originating from the fragmentation starting from the oxidized downstream end, as shown in Fig. 1 on the example of oxidized pentopentaoses (for -tetraoses and -trioses, see Fig. S1 in the ESI†). This observation is in accordance with negative ion ESI-MS2 data reported for native (A)XOS by Juvonen et al.17 While linear, native XOS exclusively fragment from the reducing end (A, B, C fragments according to the nomenclature of Domon and Costello),28 native AXOS also present fragments which can be assigned to Y fragments originating from the fragmentation starting at the non-reducing end. The Y type fragments are more pronounced in some xylonic acid-bearing oligosaccharides, which may be attributed to the high ionizability of the carboxylate group that is part of all Y fragments in these xylonic acid products.
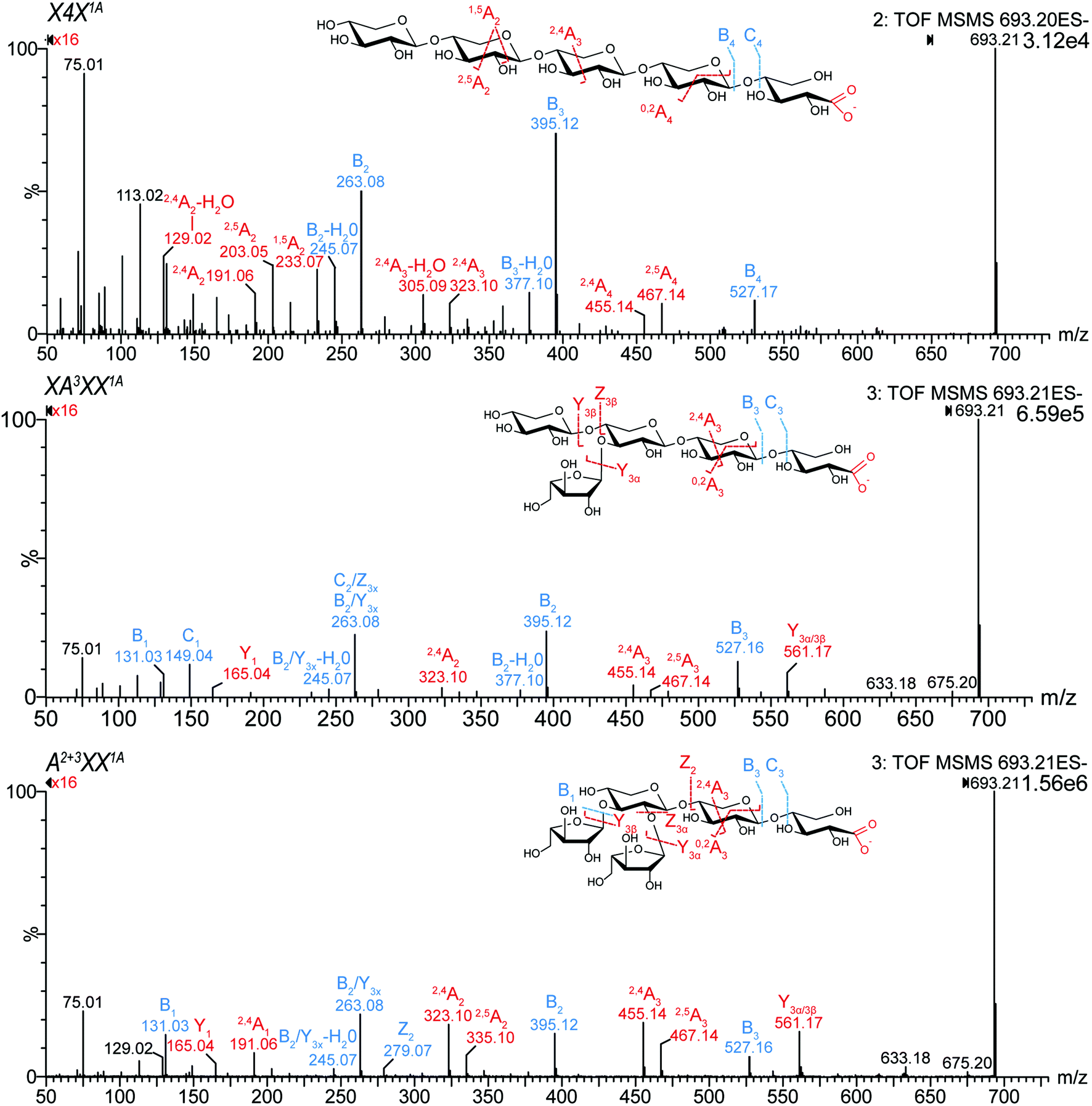 | ||
| Fig. 1 MS2 spectra of isomeric xylonic acid products with m/z 693.21, obtained by TEMPO-mediated oxidation of pentopentaose standards. TEMPO/NaClO/NaClO2 preferentially oxidizes primary alcohols and reducing ends in sugar molecules, which leads among others to the formation of C1 oxidation products.26 Those C1 oxidation products were further confirmed by the selective C1 oxidation using I2/KOH (data not shown). The fragments were named according to the nomenclature of Domon and Costello.28 | ||
All studied isomeric pentose oligosaccharides (Pentn), namely C1-oxidized pentotrioses (X3 and A3X), pentotetraoses (A2XX and X4), and pentopentaoses (XA3XX, A2+3XX, and X5) could be clearly distinguished and linkage positions of arabinose substitution could be easily identified (see Fig. 1, Table 1, and Fig. S1†). Linear XOS xylonic acids (Xyl(n−1)Xyl1A = X(n − 1)X1A) could be mainly elucidated by their dominating B fragments and the absence of Y(n−1) fragment. In contrast, both mono- and disubstituted oligomers presented a Y(n−1) fragment, presumably due to the ease of loss of a side chain arabinose unit. For AXOS, the 1,5A2 fragment (m/z 233.07) was less abundant compared to XOS. Moreover, particular linkages of the arabinose substitution could be identified by the signal ratios of 2,4An/2,5An, namely m/z 455.14/467.14 (for n = 5), 323.10/335.10 (for n ≥ 4), and 191.06/203.06 (for n ≥ 3). While m/z 467.14 (2,5A4) was more abundant in oxidized linear X5 (X4X1A), m/z 455.14 (2,4A4) was dominating in both substituted pentopentaoses (ratio >1). Similar ratios of 2,5A2 (203.06) and 2,4A2 (191.06) were found for pentotrioses. The opposite could be observed for pentotetraoses, where 2,4A2 (323.10) was more abundant in oxidized linear X4 (X3X1A), and 2,5A3 (335.10) was dominating in A2XX1A. Among the pentopentaoses evaluated, only the monosubstituted XA3XX1A formed C1 fragments (m/z 149.04). Therefore, the region below m/z 200 allows for a facilitated differentiation of arabinose substitution pattern.
| Oligosaccharide | Fragment ion (m/z) | Missing fragment ion (m/z) |
|---|---|---|
| Oxidized pentopentaoses | Parent ion (m/z 693.21) | |
| X4X1A | 2,5 A 3 (335.10) | Y3α/β (561.17) |
| 1,5 A 2 (233.07) | ||
| 2,5 A 2 (203.05) | ||
| 2,4 A 2 (191.06) | ||
| C 1 (149.04) | ||
| XA3XX1A | Y 3α/β (561.17) | 2,5 A 3 (467.14) |
| C 1 (149.04) | 2,4 A 3 −H2O (305.09) | |
| 2,5 A 2 (335.10) | ||
| 2,5 A 2 (203.05) | ||
| A2+3XX1A | Y 3α/β (561.17) | B 2 −H2O (377.10) |
| 2,5 A 2 (335.10) | 1,5 A 2 (233.07) | |
| 2,4 A 2 (191.06) | 2,5 A 2 (203.05) | |
| Oxidized pentotetraoses | Parent ion (m/z 561.17) | |
| X3X1A | Y 3 (429.12) | |
| Z 2 (279.07) | ||
| A2XX1A | Y 3 (429.12) | |
| Z 2 (279.07) | ||
| Y 1 (165.04) | ||
| Oxidized pentotrioses | Parent ion (m/z 429.12) | |
| X2X1A | 1,5 A 2 (233.07) | Y 2 (297.08) |
| 2,5 A 2 (203.06) | ||
| A3X1A | Y 2 (297.08) | B 2 −H2O (245.07) |
| 2,5 A 2 (203.06) | ||
| 1,5 A 2 (233.07) | ||
These findings are in line with the previously published MS2 differentiation of C1-oxidized gluco-oligosaccharides with gluconic acid downstream end.21 Consequently, all pento-oligosaccharides with the same m/z revealed unique MS2 fragmentation, which enables a clear identification of isomeric AX, (A)XOS derived oxidation products.
Whereas linear XOS exclusively formed their corresponding xylonic acid product resulting from C1 xylose oxidation, AXOS created a mixture of acidic oxidation products resulting from both the C1 xylose oxidation and the C5 arabinose oxidation (see Fig. 2). Nevertheless, the product bearing the oxidized downstream end remained predominant during the AXOS oxidation as primary oxidation product. A variation of reagent equivalents corroborated the formation of reducing end oxidation prior to further oxidations at other positions (data not shown).21
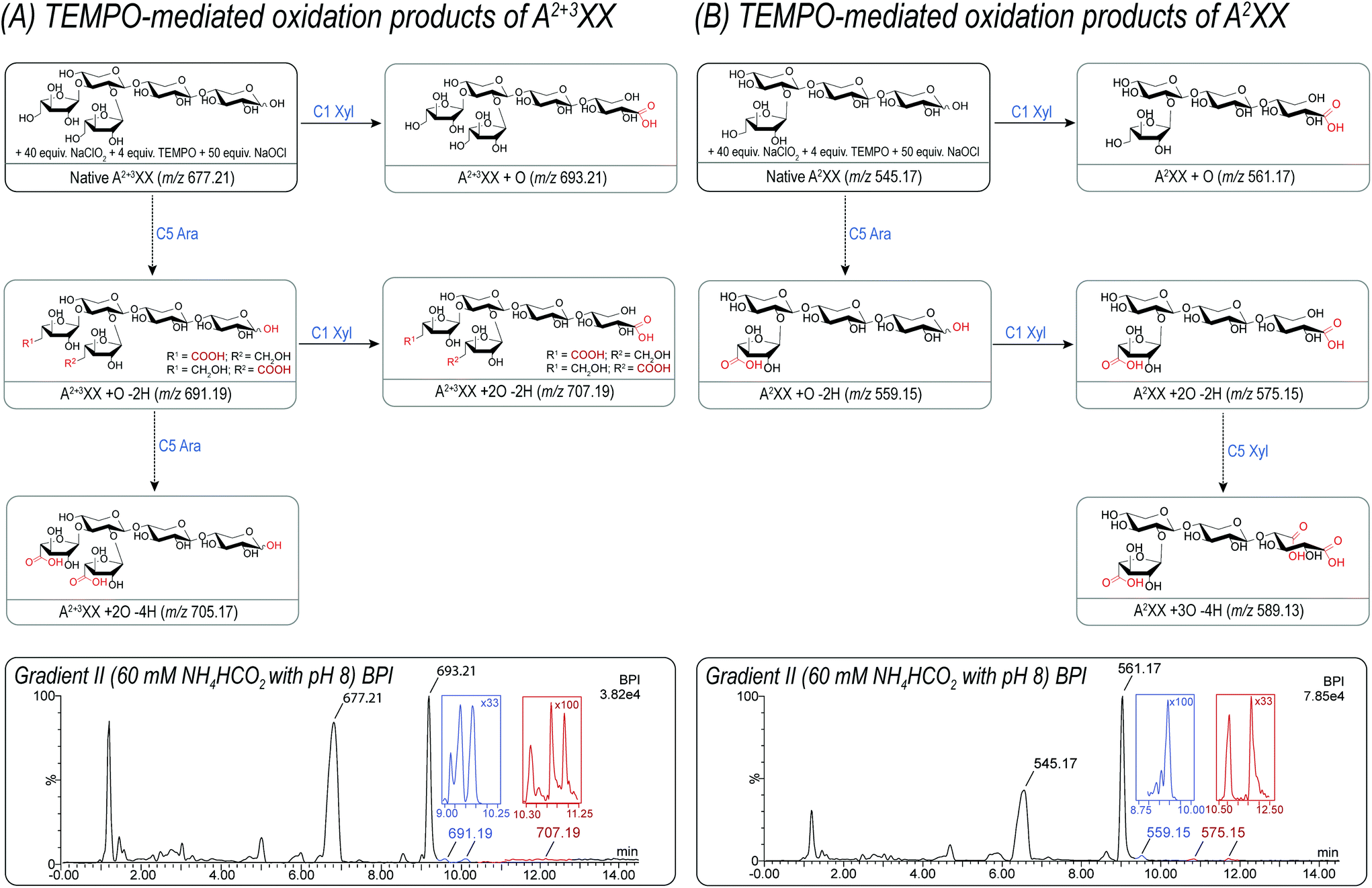 | ||
| Fig. 2 Exemplary overview of TEMPO-mediated oxidation products of A2+3XX and A2XX. The shown m/z in parenthesis represent the deprotonated species [M − H]−. The samples were analyzed by UPLC-MS using a buffered 60 mM ammonium formate eluent system at pH 8 and a BEH Amide column. Several isomeric products were detected for m/z 559.15, 575.15, and 707.19 differing in their oxidation position, e.g. m/z 559.15 can be assigned to a product after C5 oxidation in the arabinose unit as well as to a product after C1 and C5 oxidation of the downstream xylose unit. (A) TEMPO-mediated oxidation products of A2+3XX. (B) TEMPO-mediated oxidation products of A2XX. | ||
Fig. 2 summarizes the successfully identified carboxylic acid-bearing oligosaccharides from A2+3XX and A2XX in the reaction mixture after TEMPO-mediated oxidation using UPLC-MS2 with a buffered ammonium formate eluent (pH 8). Generally, this gradient separated acidic products according to their size and polarity on the UPLC BEH Amide column. The specific products formed could be detected by their unique m/z and even several isomeric products, e.g. of m/z 559.15, 575.15, and 707.19 achieved appropriate separation by the gradient applied. In particular, m/z 559.15 can be assigned to a product after C5 oxidation in the arabinose unit as well as to a product after C1 and C5 oxidation of the downstream xylose unit.
3.2. Identification of non-selective oxidized (A)XOS using Fenton oxidation
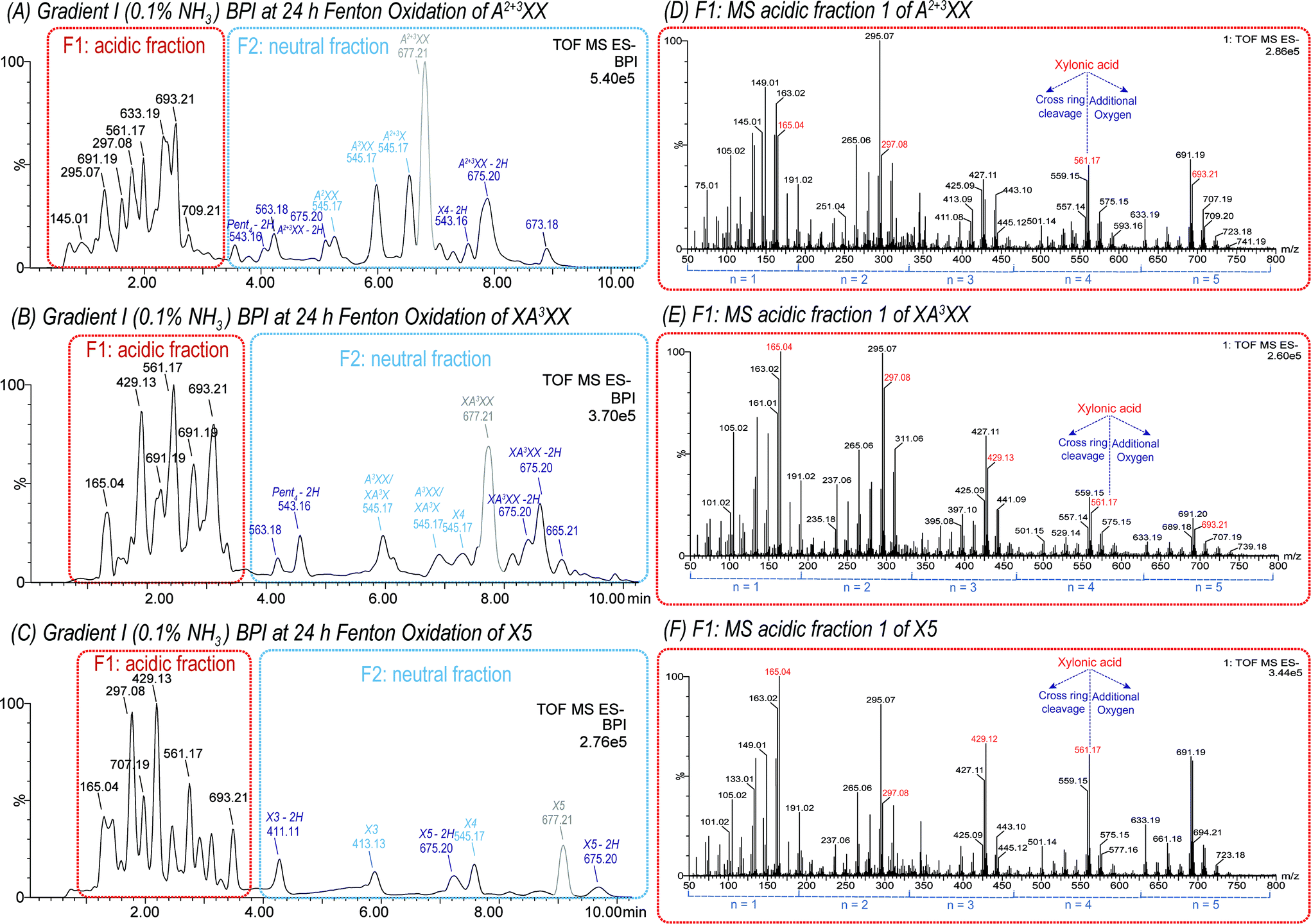 | ||
| Fig. 3 UPLC-MS base peak ion chromatogram (BPI) using a BEH Amide column (ACN/H2O gradient I with NH3 additive) and negative ion mode ESI of 1 mM A2+3XX (A), XA3XX (B), and X5 (C) solution oxidized with 100 μM FeSO4, 100 μM ascorbic acid and 500 mM H2O2 after 24 h at room temperature. Peak area of the starting material is indicated in grey. ESI-MS spectra of acidic fraction (F1) of the respective oligosaccharides (D–F), derived from the red marked area in (A–C). Product assignment was based on elution time of native, commercial oligosaccharides (see Fig. S2†). | ||
The MS spectra from the oxidation of isomeric pentopentaoses (with m/z 677.21) revealed signals clustered around the primary C1-oxidation products with m/z 165.04, 297.08, 429.12, 561.17, and m/z 693.21, representing Pent(n−1)Xyl1A with n = 1, 2, 3, 4, and 5, respectively. Signals with the same number of monosaccharide units n, but higher m/z than their respective pentose (Pent) acid oligomers, such as m/z 707.19 (n = 5), are attributed to products with at least 1 additional oxygen, whereas products with lower m/z refer tetrose-, glyceric-, and glyconic acid oligomers formed by cross-ring cleavages during Fenton oxidation.
As expected, all products formed and identified in the TEMPO-mediated oxidation of oligomers were also present in the complex product mixture of Fenton oxidized XOS and AXOS and were therefore easily identified (see Fig. 3D–F). Furthermore, the product distribution with respect to the degree of polymerization (DP) varies between the oligosaccharides analyzed (see Fig. 3 for A2+3XX, XA3XX, and X5). This observation shows that the fine structure influences the chain scission and oxidation behaviour of oligosaccharides, which is in contrast to previous findings on β-glucan oligomer oxidation, where similar results for all four linear glucotetratose isomers could be observed.21 The product profile obtained from the Fenton oxidation of arabinoxylan will strongly depend on the native fine structure of the polysaccharide.
| n | 5 | 4 | 3 | 2 | 1 |
|---|---|---|---|---|---|
| a Experimental values of m/z (all within ± 2 mDa of theoretical values) with distinct peaks or ≥5% intensity relative to the base peak of the respective fraction (neutral: m/z 663.199; acidic: m/z 193.035). An asterisk denotes products with ≥20% relative signal intensity. b Indicates the number of total sugar units. c The names of the monosaccharides are given according to the oxidized unit of the oligosaccharides. d Starting material. | |||||
| Neutral products | |||||
| Pentosen (Pentn) | 677.21d* | 545.17* | 413.13* | 281.09* | 149.05* |
| Pentn–2H | 675.20* | 543.16* | 411.11* | 279.07* | 147.03* |
| Pentn–CH2O | 647.19 | 515.15 | |||
| Pentn–CH2O–2H | 645.18 | 513.14 | 117.02 | ||
| Pentn–C2H4O2 | 485.15 | 89.03 | |||
| Pentn–C2H4O2–2H | 87.01 | ||||
| Acidic products | |||||
| Pentose acid series: xylonic, xylaric, and arabinuronic acid | |||||
| Pentn + O | 693.20* | 561.16* | 429.13* | 297.08* | 165.04* |
| Pentn + O–2H | 691.19* | 559.15* | 427.12* | 295.07* | 163.03* |
| Pentn + O–4H | 689.17 | 557.13 | 425.09 | 293.05 | 161.01 |
| Pentn + 2O | 709.20 | 577.16* | 445.12 | ||
| Pentn + 2O–2H | 707.18* | 575.14* | 443.10* | 311.06* | |
| Pentn + 2O–4H | 573.13 | 441.09 | 309.05 | ||
| Pentn + 3O–2H | 723.18 | 591.14 | 459.10 | 327.06 | |
| Pentn + 3O–4H | 325.04 | ||||
| Tetrose acid series: onic, uronic, and aric acids | |||||
| Pentn–CH2O + O | 663.19 | 531.15 | 399.11 | 267.07 | 135.03 |
| Pentn–CH2O + O–2H | 661.18 | 529.14 | 397.10 | 265.06 | 133.02 |
| Pentn–CH2O + O–4H | 263.04 | ||||
| Pentn–CH2O + 2O–2H | 677.17 | 545.13 | 413.09 | 281.05 | 149.01* |
| Pentn–CH2O + 2O–4H | 411.08 | 279.04 | |||
| Glyceric acid series | |||||
| Pentn–C2H4O2 + O | 633.18 | 501.14 | 369.10 | 237.06 | 105.20 |
| Pentn–C2H4O2 + 2O | 649.18 | ||||
| Pentn–C2H4O2 + 2O–2H | 647.16 | 515.12 | |||
| Pentn–C2H4O2 + 3O–2H | 267.04 | ||||
| Pentn–C2H4O2 + 3O–4H | 661.14 | ||||
| Glyconic acid series | |||||
| Pentn–C3H6O3 + O | 75.01 | ||||
| Pentn–C3H6O3 + 2O–2H | 353.09 | 89.01 | |||
| Formic acid series | |||||
| Pentn–C4H8O4 + O–2H | 323.06 | ||||
The assembled data suggest that resolution HILIC-MS2 is a powerful technique for the rapid and precise structural identification of neutral and acidic oligomers derived from polysaccharide oxidation. Nevertheless, to characterize the complete product profile of hydroxyl radical-induced degradation, both gradients presented in this paper need to be applied. The basic gradient I enables the investigation of neutral oxidation products in combination with an MS scan of acidic products, whereas the buffered gradient II enables a distinct identification of acidic products and their structural variations.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O location can indicate if it was formed via a lytic or a non-lytic process. In this study, MS2 analysis revealed interesting details about the location of carbonyl groups in the different evaluated oligosaccharides. As an example, the oxidation product m/z 559.15 can be attributed to deprotonated Pent4 + O–2H, and was found in Fenton oxidized X5 and A2+3XX under loss of one pentose unit (for fragmentation pattern, see Fig. 4). Generally, the product contains either
O location can indicate if it was formed via a lytic or a non-lytic process. In this study, MS2 analysis revealed interesting details about the location of carbonyl groups in the different evaluated oligosaccharides. As an example, the oxidation product m/z 559.15 can be attributed to deprotonated Pent4 + O–2H, and was found in Fenton oxidized X5 and A2+3XX under loss of one pentose unit (for fragmentation pattern, see Fig. 4). Generally, the product contains either
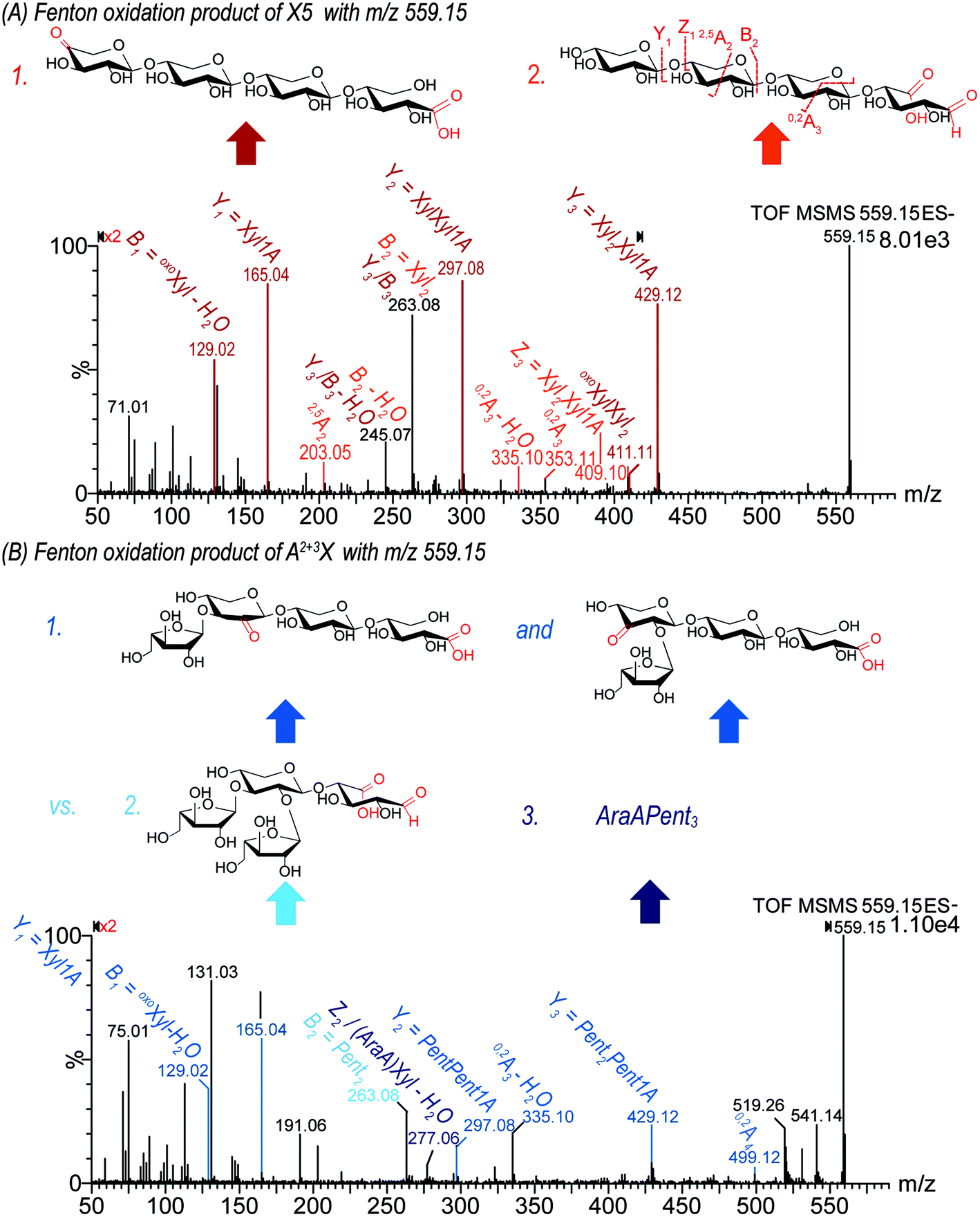 | ||
| Fig. 4 MS2 spectra of the oxidation product Pent4 + O–2H with m/z 559.15, found for Fenton oxidized (A) X5 and (B) A2+3XX to determine the location of carbonyl and carboxylic acid groups. Fragments highlighted in color are identifying the structural unit indicated with an arrow of the same color. | ||
(i) one uronic acid group (from oxidation of one of the pentose units at C5, hence –CH2OH → –COOH), which is then already fully responsible for + O–2H; or
(ii) one carboxylic acid at the downstream end's C1 (forming xylonic acid, hence –CHO → –COOH), plus an additional carbonyl group to account for the –2H. Due to the indiscriminate nature of HO˙, various isomeric products would be expected for this option depending on the order of oxidation, and whether C1-oxidation or carbonyl formation was responsible for the chain scission.
Based on the acidic nature of the possible products, a clear separation of isomeric products could not be achieved by the basic gradient I. However, gradient II impeded acceptable MS2 spectra due to extensive ion suppression caused by the eluent buffer. Only gradient I allowed for sufficient signal abundance for collision induced fragmentation analysis. Consequently, the MS2 pattern of m/z 559.15 from both starting materials X5 and A2+3XX may represent a mixture of isomeric products. However, the fragments found allow for differentiation and identification of formed products. As shown for X5 oxidation, the pattern of Yn fragments (m/z 429.12, 297.08, and 165.04 for n = 3, 2, and 1) produced through the loss of sugar units from the non-reducing end indicated the presence of a carboxylic acid group at the downstream end sugar unit, namely as a xylonic acid (Xyl1A) from C1 oxidation. In addition, these fragments corresponding to Xyl(n−1)Xyl1A (n = 1–3) clearly showed that the additional carbonyl group is not located on the downstream end xylonic acid unit for this product. The neutral loss of −130 Da from the parent ion to the Y3 fragment (m/z 559.15 → 429.12) corresponds to an eliminated oxoXyl unit, which indicates a product with a carbonyl group at the non-reducing end. Furthermore, the fragment m/z 129.02 can be assigned to the mentioned oxoXyl as B1 fragment itself.
There is some indication that the alternative possibility (i), namely a xyluronic acid-bearing oligosaccharide Xyl3XylA, is at least a minor part of the product mixture for X5 oxidation: most notably, the fragment ion m/z 409, which could correspond to Z3 of Xyl3XylA (namely Xyl2XylA–H2O). And some fragments that could also be from oxoXylXyl2Xyl1A, but then need two collision induced scission events to explain them, vs. only one if originating from Xyl3XylA: m/z 263 being B2 for the latter, and B3/Y3 for the former, as well as its dehydrated product m/z 245. Possibly, these fragments have contributions from both product isomers.
In the case of A2+3XX oxidation, the respective product with carbonyl at the non-reducing and C1-oxidized reducing end as found for X5 could be identified due to the occurrence of analogous fragment ions. However, additionally, the MS2 spectrum contained fragment m/z 277.06, which could be assigned to an arabinose unit bearing a carboxylic acid group at C5 (arabinuronic acid, AraA). Hence, for the Fenton-induced oxidation of both X5 and A2+3XX, m/z 559.15 likely represents a mixture of oxoXyl(Pent)2Xyl1A, and Pent3XylA from lytic C5 oxidation at the unit next to the reducing end, whereas for A2+3XX, some C5 oxidized arabinose products (AraA)Pent3 is additionally present. Thus, MS2 analysis is a powerful technique to determine the oxidation site on the oligomers.
Regarding reaction pathways, having the carbonyl group only at the non-reducing end strongly suggests that its formation is part of the lytic step, namely, on the example of X5 oxidation, C4-oxidation with scission of the last β-(1→4)-linkage, leading to 4oxoXylXyl3 (see Fig. 5). Introducing a carbonyl group is, in contrast to C1 oxidation, not selective – unless it is formed through lytic action, then it must be localized at the former glycosidic linkage position, which is C4 for the xylan backbone, and C2 or C3 in the case of Ara sidechains. Under H2O2 conditions, it is known that reducing ends oxidize relatively easily to the corresponding onic acid species.21 This is in accordance with earlier studies, where MS2 analysis was also used to localize the oxidation at the reducing or non-reducing end of glucose oligomers.16,21
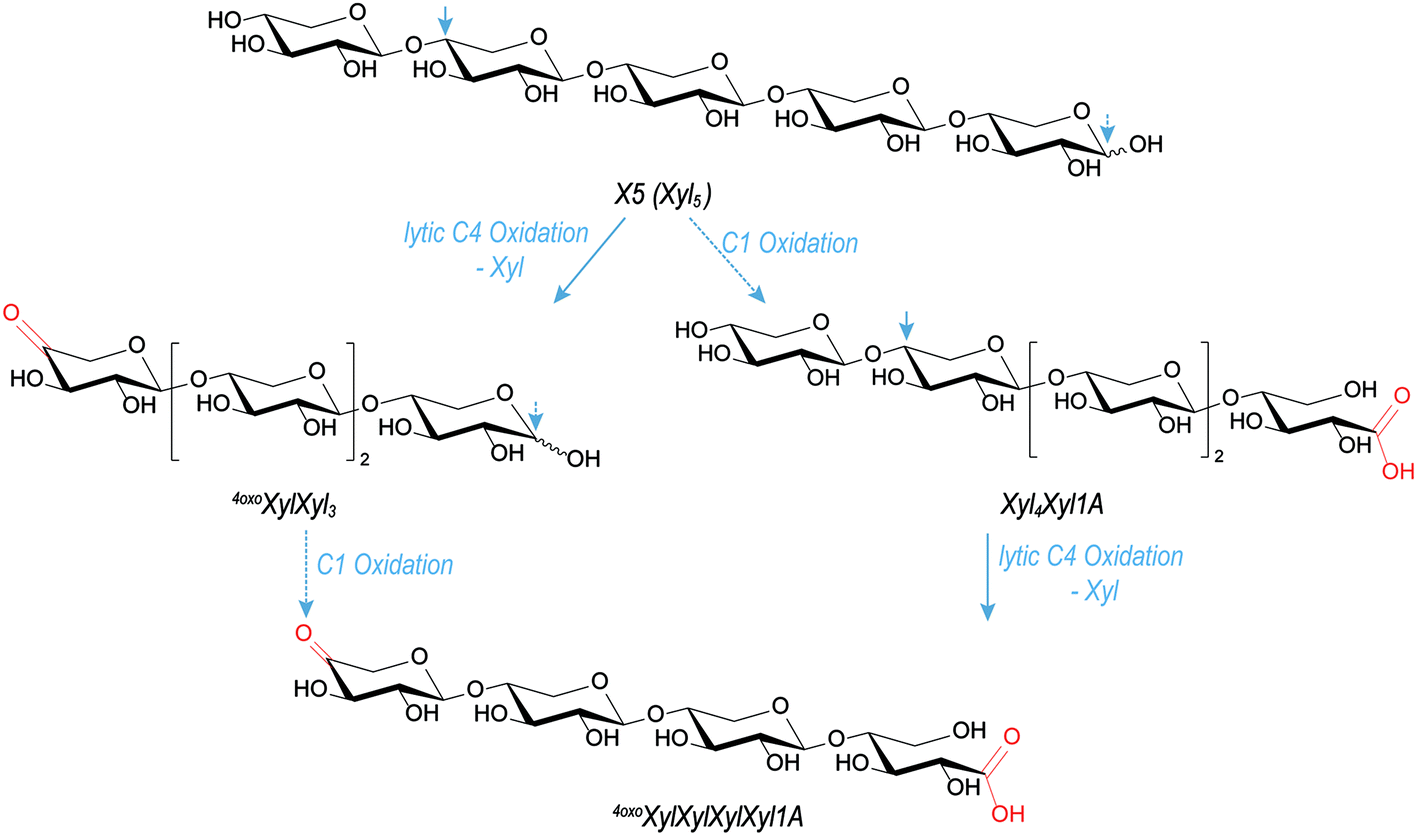 | ||
| Fig. 5 Suggested likely pathways for the formation of m/z 559.15 product from Fenton-induced X5 oxidation through lytic C4 oxidation (HO˙, O2) and reducing end C1 oxidation (H2O2), with loss of the non-reducing end unit, forming 4oxoXylXylXylXyl1A as product. | ||
3.3. Characterization of the complex product mixture from Fenton oxidized AX polymer
Wheat arabinoxylan (AX) was oxidized by Fenton-induced oxidation using 100 μM FeSO4, 500 μM ascorbic acid (AA), and 500 mM H2O2. Subsequently, (A)XOS were produced by endo-1,4-β-xylanase from Cellvibrio mixtus, which selectively hydrolyzes certain 1,4-β-glycosidic bonds within the β-(1→4)-xylan backbone. Consequently, the product profile of oxidized AX derived oligosaccharides was characterized by hydrophilic interaction UPLC-MS2. Scheme 1 provides structures, names, and abbreviations of expected AX oxidation products originating from HO˙-attack on xylose and arabinose carbons C1–5 along the arabinoxylan backbone, based on our results from the (A)XOS model system experiments presented above. Reaction shown are categorized as lytic and non-lytic reactions, where lytic reactions cause chain scission within the polysaccharide backbone and hence depolymerization and loss of viscosity. In this study, loss of Ara sidechains is not classified as lytic, as their loss has a negligible impact on molecular weight and viscosity of the polysaccharide material.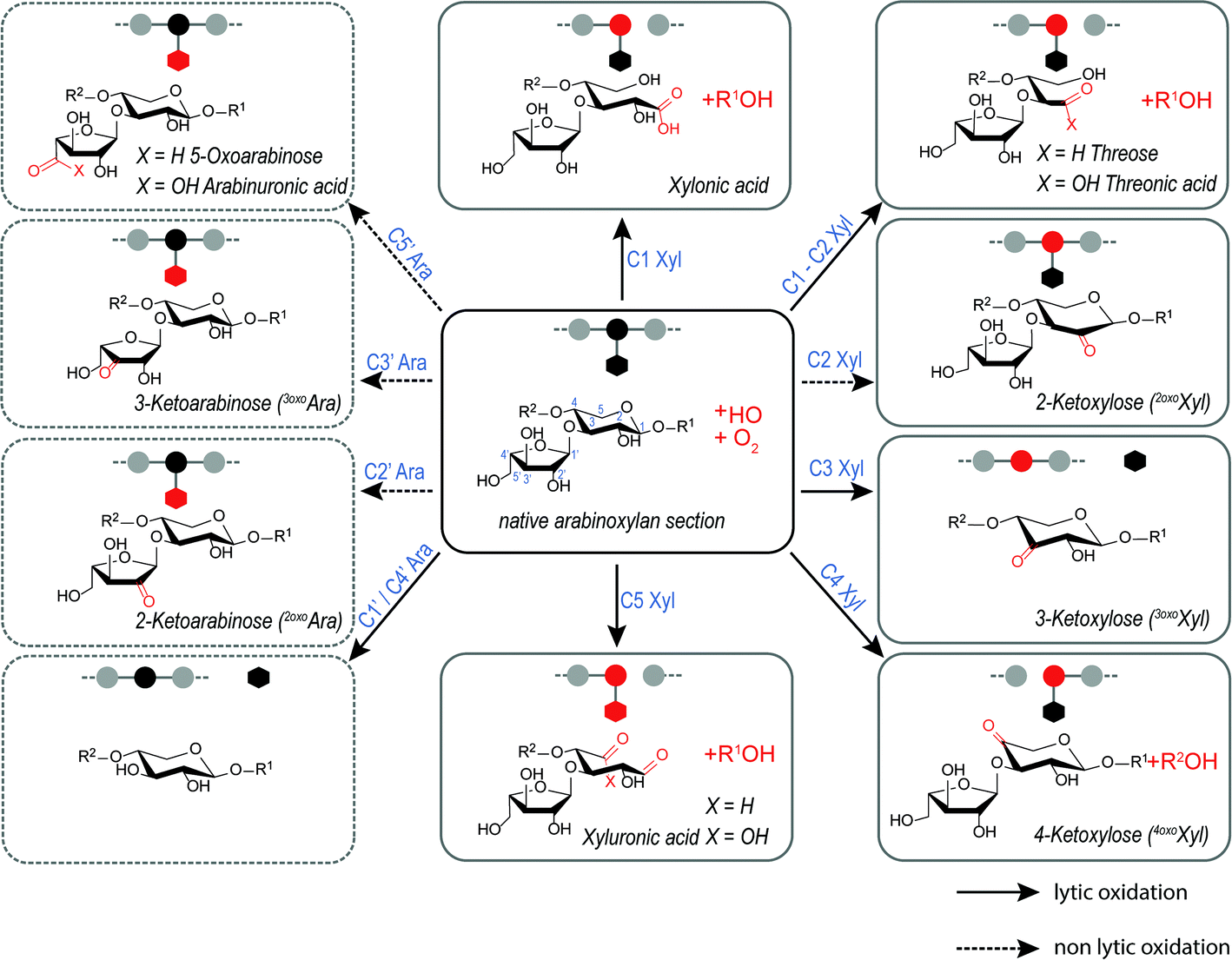 | ||
Scheme 1 Overview of expected AX oxidation products in the presence of molecular oxygen mediated by hydroxyl radicals (HO˙), which attack at any of the five carbons of xylose (plane  ) and arabinose (hashed ) and arabinose (hashed  ). Starting from α-(1→3)-linked L-arabinofuranosyl (Araf) that is part of β-(1→4)-xylan backbone, both lytic (plane arrows), which introduces chain scissions, and non-lytic oxidation (hashed arrows) are shown. Threose can by formed by a lytic cross-ring cleavage (C1–C2). ). Starting from α-(1→3)-linked L-arabinofuranosyl (Araf) that is part of β-(1→4)-xylan backbone, both lytic (plane arrows), which introduces chain scissions, and non-lytic oxidation (hashed arrows) are shown. Threose can by formed by a lytic cross-ring cleavage (C1–C2). | ||
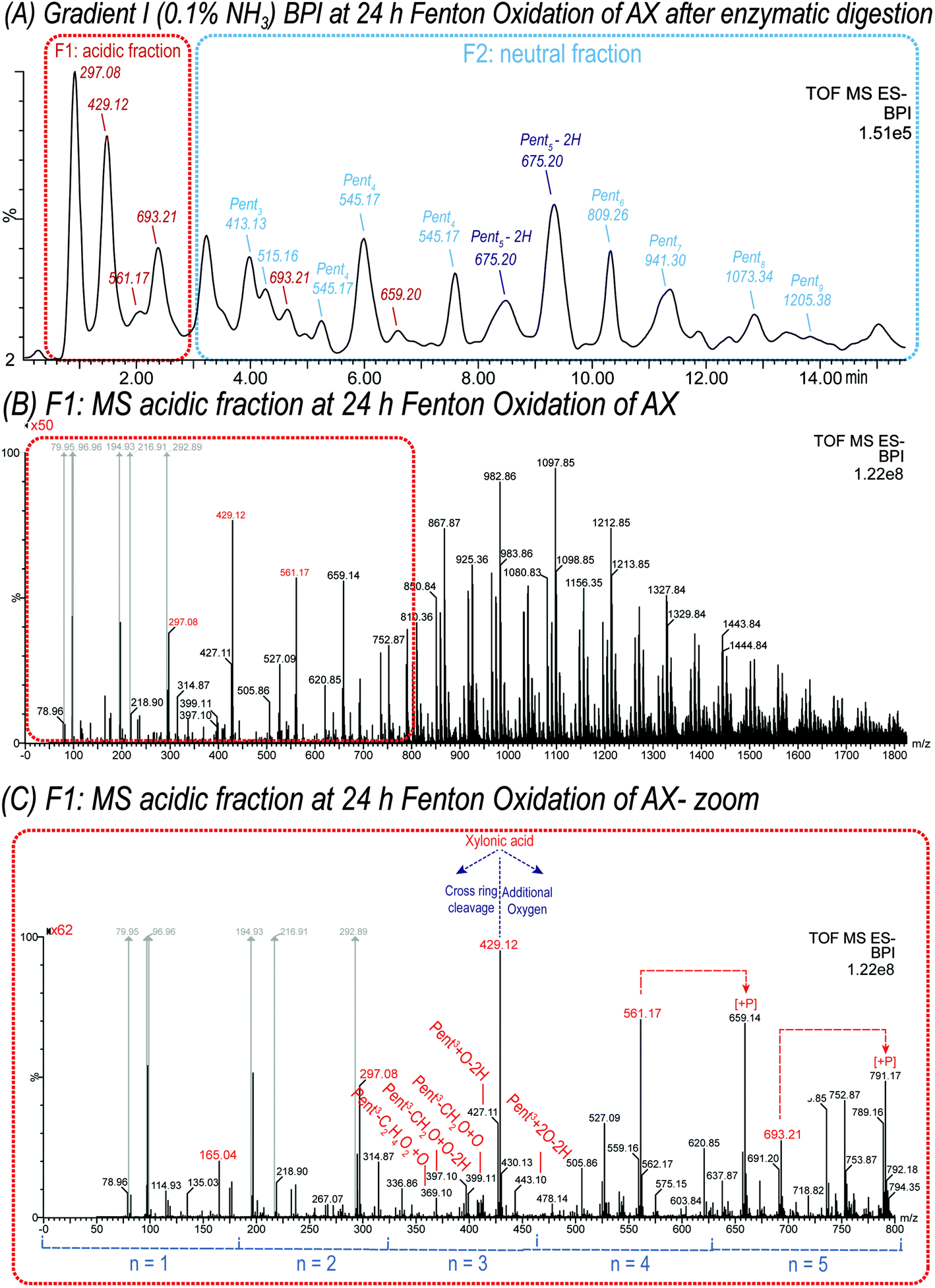 | ||
| Fig. 6 (A) UPLC-MS base peak chromatogram of released oligosaccharides from 1 mM oxidized wheat arabinoxylan solution oxidized with 100 μM FeSO4, 100 μM ascorbic acid and 50 mM H2O2 for 24 h at room temperature. The oxidized AX was digested with endo-1,4-β-xylanase to obtain diagnostic oligosaccharides suitable in Mw for UPLC-MS2 analysis in negative ionization mode using a BEH Amide column and ACN/H2O gradient (0.1% NH3). Peaks are labelled with their respective base peak m/z and with n = number of monosaccharide units in blue for arabinoxylan oligomers Pentn. (B) MS spectrum of acidic products obtained from the highlighted chromatogram region F1 in (A). Signals are labelled with m/z, and carboxylic acid-bearing oligomers are labelled in red. Signals indicated in grey are assigned to common background contamination ions such as buffer salt adducts. (C) MS spectrum of acidic products F1, zoomed in for the range of m/z 50 to 800. Carboxylic acid-bearing oligomer are labelled in red, products with additional oxygens and cross-ring cleavages are indicated. Several Xyl + O products, namely m/z 561.17, 693.21, 825.25, 957.29, 1089.33, and 1221.37 were additionally detected as the respective phosphate adduct (Δm = 97.96 Da) with m/z 659.13, 791.17, 923.21, 1055.25, 1187.29, and 1319.33, respectively (marked with [+P]). | ||
In addition, oxidation products carrying carboxylic acid groups at both C1 + C5 positions along the oligomer could also be detected, as well as the respective acidic cross-ring cleavage products such as tetrose-, glyceric-, and glyconic acid-bearing oligomers (e.g. Pent3–CH2O + 2O–2H with m/z 413.09, and threonic acid Pent2Thr1A with m/z 399.11) (see Fig. 6 and Table 3).
| n | 5 | 4 | 3 | 2 | 1 |
|---|---|---|---|---|---|
| a Experimental values of m/z (all within ± 2 mDa of theoretical values) with distinct peaks or ≥5% intensity relative to the base peak of the respective fraction (neutral: m/z 663.199; acidic: m/z 193.035). An asterisk denotes products with ≥20% relative signal intensity. b Indicates the number of total sugar units. c The names of the monosaccharides are given according to the oxidized unit of the oligosaccharides. | |||||
| Neutral products | |||||
| Pentosen (Pentn) | 677.21* | 545.17* | 413.13* | 281.09 | |
| Pentn–2H | 675.20* | 543.16* | 411.11* | 279.07 | |
| Pentn–CH2O | 515.15* | ||||
| Pentn–CH2O–2H | |||||
| Pentn–C2H4O2 | 617.19 | 485.15 | |||
| Acidic products | |||||
| Pentose acid series: xylonic, xylaric, and arabinuronic acid | |||||
| Pentn + O | 693.20* | 561.16* | 429.13* | 297.08* | 165.04 |
| Pentn + O–2H | 691.19* | 559.15* | 427.12* | 295.07 | 163.03 |
| Pentn + O–4H | 689.17 | 557.13 | 425.09 | 293.05 | |
| Pentn + 2O | |||||
| Pentn + 2O–2H | 707.18 | 575.14 | 443.10 | 311.06 | |
| Pentn + 2O–4H | 441.09 | 309.05 | |||
| Tetrose acid series: onic, uronic, and aric acids | |||||
| Pentn–CH2O + O | 399.11 | ||||
| Pentn–CH2O + O–2H | 661.18 | 529.14 | 397.10 | ||
| Pentn–CH2O + 2O–2H | 413.09* | 281.05 | |||
| Glyceric acid series | |||||
| Pentn–C2H4O2 + 2O–2H | 515.12* | ||||
| Pentn–C2H4O2 + 3O–4H | 661.14 | ||||
| Glyconic acid series | |||||
| Pentn–C3H6O3 + O | 339.09 | ||||
| Pentn–C3H6O3 + O–2H | |||||
| Pentn–C3H6O3 + 2O–2H | 617.17 | 485.13 | |||
The neutral fraction (see Fig. 6A) consisted mainly of pentose-oligosaccharides (DP 1 to DP 9), whereas their respective counterparts with one or more oxidized hydroxyl group (oxo-Pentn) could also be detected, such as oxo-Pent5 isomers (m/z 675.20) separated using basic gradient I (see Fig. 6A).
The degree of polymerization obtained by the enzymatic digestion was ranging from DP1 to DP13, which was generally higher than the DP of oligosaccharides evaluated as oxidation model compounds in this study. A possible explanation for the limited production of smaller diagnostic (A)XOS by endo-1,4-β-xylanase could be the steric hindrance through side groups. This limitation may be eliminated by the implementation of a selective enzyme mixture, which may simplify the analysis. However, crucial structural information may be lost through a more extensive enzymatic degradation.
Furthermore, it could be observed that the non-selective oxidation of AX yielded lower product diversity than the oxidation of (A)XOS. This observation indicates that AX polymer was oxidized to a lesser extent than the oligomers. This is in line with the chosen oxidation conditions if the reagent concentrations are compared relative to the monomer units (AX: 150 mM monomers vs. 50 mM H2O2; (A)XOS: 5 mM monomers vs. 10 mM H2O2). Further oxidation of the AX material might have also produced a similar diversity of products as the (A)XOS.
The assembled data suggest that resolution HILIC-MS2 is a powerful technique for the rapid and precise structural identification of neutral and acidic oligomers derived from arabinoxylan oxidation. Nevertheless, for the complete characterization of the product profile of hydroxyl radical-induced degradation, both gradients presented in this paper need to be applied as was the case in the study for β-glucan oxidation.16,21
4. Conclusion & outlook
A complementary analytical approach using hydrophilic interaction UPLC-MS2 in negative ion mode and a combination of two gradient systems enabled the investigation of oxidized (A)XOS formed through selective TEMPO-mediated oxidation and non-selective Fenton-induced degradation.Overall, our method application on branched heterooligosaccharides broadens the application field of the established method enabling the comprehensive characterization of functionalized branched heterooligo- and heteropolysaccharides. The in-depth analysis of acidic oxidation products synthesized by TEMPO-mediated oxidation provides mechanistic insights in the selective functionalization of oligosaccharides and polysaccharides. The chosen MS2 condition allowed for the differentiation and linkage assignment of constitutionally isomeric native and synthesized C1-oxidation products. The implementation of higher collision energies facilitated the structural elucidation of stable C1-oxidized (A)XOS. Moreover, the localization of newly introduced functional groups by MS2 could be shown on the example of a lytic C4 oxidation combined with reducing end oxidation. This analytical investigation enables the detailed elucidation of reaction pathways occurring during polysaccharide functionalizations such as Fenton oxidation. The implemented enzymatic sample preparation strategy revealed appropriate size distribution and improved signal strength in the HILIC-MS2 analysis enabling the comprehensive characterization of Fenton-induced arabinoxylan oxidation In further findings, MS resolution mode was sufficient for the separation of near isobaric products (Δm = ∼0.05 Da). This feature prevents misassigning oxidation products due to a lack of resolution. In general, the complementary basic and buffer gradient systems yielded appropriate chromatographic resolution in combination with qTOF MS detection. However, to characterize the complex product mixture of hydroxyl radical-induced degradation, both gradients need to be considered. While gradient I enables the characterization of neutral products, gradient II is crucial to investigate acidic products and their isomeric species.
The analytical approach presented enables a molecular screening of selective and non-selective polysaccharide modifications providing valuable mechanistic insight within the broad field of polysaccharide applications.
Abbreviations
| AX | Arabinoxylan |
| AXOS | Arabinoxylan oligosaccharides |
| (A)XOS | Xylo- and arabinoxylan oligosaccharides |
| DP | Degree of polymerization |
| HO˙ | Hydroxyl radicals |
| TEMPO | 2,2,6,6-Tetramethylpiperidine 1-oxyl |
| XOS | Xylooligosaccharides |
Funding source
The study was funded by ETH Zurich and did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank Dr Guido Zeegers for his scientific support in the LC-MS operation, and Jasmin Hirt for the synthesis of oxidized (A)XOS.References
- M. Mendis and S. Simsek, Food Hydrocolloids, 2014, 42, 239–243 CrossRef CAS.
- J. Liu, S. Willför and C. Xu, Bioact. Carbohydr. Diet. Fibre, 2015, 5, 31–61 CrossRef CAS.
- A. Kiszonas, E. Fuerst and C. Morris, Cereal Chem., 2013, 90, 387–395 CrossRef CAS.
- M. R. Cyran, Processing and Impact on Active Components in Food, 2015, 319–328, DOI:10.1016/b978-0-12-404699-3.00038-x.
- T. Demuth, J. Betschart and L. Nyström, Carbohydr. Polym., 2020, 240, 116328 CrossRef CAS PubMed.
- M. S. Izydorczyk, Arabinoxylans, in Handbook of hydrocolloids, Woodhead Publishing, 2009, pp. 653–692 Search PubMed.
- A. Bagdi, S. Tömösközi and L. Nyström, Carbohydr. Polym., 2016, 152, 263–270 CrossRef CAS PubMed.
- R. Kivelä, U. Henniges, T. Sontag-Strohm and A. Potthast, Carbohydr. Polym., 2012, 87, 589–597 CrossRef.
- M. N. Schuchmann and C. Von Sonntag, Int. J. Radiat. Biol., 1977, 34, 397–400 Search PubMed.
- R. Kivelä, T. Sontag-Strohm, J. Loponen, P. Tuomainen and L. Nyström, Carbohydr. Polym., 2011, 85, 645–652 CrossRef.
- L. Saulnier, P.-E. Sado, G. Branlard, G. Charmet and F. Guillon, J. Cereal Sci., 2007, 46, 261–281 CrossRef CAS.
- A. M. Faure, M. L. Andersen and L. Nyström, Carbohydr. Polym., 2012, 87, 2160–2168 CrossRef CAS.
- R. Kivelä, F. Gates and T. Sontag-Strohm, J. Cereal Sci., 2009, 49, 1–3 CrossRef.
- Y. J. Wang, N. Mäkelä, N. H. Maina, A. M. Lampi and T. Sontag-Strohm, Food Chem., 2016, 197(Pt B), 1324–1330 CrossRef CAS PubMed.
- A. Potthast, J. Röhrling, T. Rosenau, A. Borgards, H. Sixta and P. Kosma, Biomacromolecules, 2003, 4, 743–749 CrossRef CAS PubMed.
- S. Boulos and L. Nyström, Front. Chem., 2017, 5, 90 CrossRef PubMed.
- M. Juvonen, M. Kotiranta, J. Jokela, P. Tuomainen and M. Tenkanen, Food Chem., 2019, 275, 176–185 CrossRef CAS PubMed.
- O. Hernandez-Hernandez, I. Calvillo, R. Lebron-Aguilar, F. J. Moreno and M. L. Sanz, J. Chromatogr., A, 2012, 1220, 57–67 CrossRef CAS PubMed.
- B. Westereng, M. O. Arntzen, F. L. Aachmann, A. Varnai, V. G. Eijsink and J. W. Agger, J. Chromatogr., A, 2016, 1445, 46–54 CrossRef CAS PubMed.
- A. S. Moreira, E. V. da Costa, D. V. Evtuguin, M. A. Coimbra, F. M. Nunes and M. R. Domingues, J. Mass Spectrom., 2014, 49, 280–290 CrossRef CAS PubMed.
- S. Boulos and L. Nyström, Analyst, 2016, 141, 6533–6548 RSC.
- K. Hashimoto, S. I. Imanishi, M. Okada and H. Sumitomo, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 1271–1279 CrossRef CAS.
- N. Tamura, M. Hirota, T. Saito and A. Isogai, Carbohydr. Polym., 2010, 81, 592–598 CrossRef CAS.
- A. Bagdi, S. Tömösközi and L. Nyström, Food Hydrocolloids, 2017, 63, 219–225 CrossRef CAS.
- B. Westereng, J. W. Agger, S. J. Horn, G. Vaaje-Kolstad, F. L. Aachmann, Y. H. Stenstrom and V. G. Eijsink, J. Chromatogr., A, 2013, 1271, 144–152 CrossRef CAS PubMed.
- J. Hao, J. Lu, N. Xu, R. J. Linhardt and Z. Zhang, Carbohydr. Polym., 2016, 146, 238–244 CrossRef CAS PubMed.
- A. E. De Nooy, A. C. Besemer and H. van Bekkum, Synthesis, 1996, 1153–1176 CrossRef CAS.
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS.
- A. G. Leijdekkers, M. G. Sanders, H. A. Schols and H. Gruppen, J. Chromatogr., A, 2011, 1218, 9227–9235 CrossRef CAS PubMed.
- J. Simoes, A. S. Moreira, E. da Costa, D. Evtyugin, P. Domingues, F. M. Nunes, M. A. Coimbra and M. R. Domingues, Carbohydr. Polym., 2016, 148, 290–299 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an01110j |
| This journal is © The Royal Society of Chemistry 2020 |