
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection and time-tracking activation of a photosensitiser on live single colorectal cancer cells using Raman spectroscopy†
Julia
Gala de Pablo
 ab,
David R.
Chisholm
cd,
Carrie A.
Ambler
ce,
Sally A.
Peyman
af,
Andrew
Whiting
cd and
Stephen D.
Evans
*af
ab,
David R.
Chisholm
cd,
Carrie A.
Ambler
ce,
Sally A.
Peyman
af,
Andrew
Whiting
cd and
Stephen D.
Evans
*af
aMolecular and Nanoscale Physics Group, School of Physics and Astronomy, University of Leeds, Leeds, UK. E-mail: s.d.evans@leeds.ac.uk
bGoda Lab, School of Science, University of Tokyo, Tokyo, Japan
cLightOx Limited, Wynyard Park House, Wynyard Avenue, Wynyard, Billingham, TS22 5TB, UK
dDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
eDepartment of Biosciences, Durham University, Durham, DH1 3LE, UK
fWelcome Trust Brenner Building, St James's University Hospital, Faculty of Medicine and Health, University of Leeds, Leeds, UK
First published on 7th July 2020
Abstract
Raman spectroscopy has been used to observe uptake, metabolism and response of single-cells to drugs. Photodynamic therapy is based on the use of light, a photosensitiser and oxygen to destroy tumour tissue. Here, we used single-cell Raman spectroscopy to study the uptake and intracellular degradation of a novel photosensitiser with a diphenylacetylene structure, DC473, in live single-cells from colorectal adenocarcinoma cell lines SW480, HT29 and SW620. DC473 was seen to predominantly accumulate in lipid droplets, showing higher accumulation in HT29 and SW620 cells than in SW480 cells, with a broader DC473 peak shifted to higher wavenumbers. DC473 activation and effects were tracked on live single-cells for 5 minutes. Upon exposure to UV light, the DC473 signal intensity dropped, with remaining DC473 shifting towards higher wavenumbers and widening, with a lifetime of approximately 50 seconds. Morphologically, SW480 and SW620 cells showed changes upon photodynamic therapy, whereas HT29 cells showed no changes. Morphological changes correlated with higher remaining DC473 signal after UV exposure. Our research suggests that DC473 forms aggregates within the cells that disaggregate following activation, showing the potential of Raman spectroscopy for the study of time-dependent single-cell pharmacodynamics.
Introduction
Drug efficacy or resistance in vitro is often tracked using single-protein assays. However, this approach may miss the bigger picture and lead to responses that are not consistent with in vivo results.1 Raman spectroscopy provides the molecular signature of a sample using the inelastic scattering of monochromatic light, for the detection of chemical functional groups.2,3 It is a label-free, non-destructive technique, with spatial resolution in the micrometre scale, allowing for single-cell measurements on live cells.4–8 The target compound can be detected within a cell in a semi-quantitative manner and related to the cell response.9–11 Raman has been extensively applied on pharmacological characterization.12The spontaneous Raman signal is weak, making the technique inherently slow.4 Additionally, drug peaks often appear in the cell fingerprint region, merging with bands from components such as proteins, lipids and nucleic acids.13 For tracking a drug within a cell, alternative approaches can be used, such as Resonance Raman scattering (RRS), the use of isotopologues such as deuterium labelling14,15 or biorthogonal Raman tagging such as through alkyne incorporation.13,16–18 RRS has been employed to study cytochrome C at 532 nm,19,20 where a drop in signal intensity, due to oxidation, has been observed at early stages of apoptosis.19 Biorthogonal Raman tagging uses molecules that exhibit vibrational bands in the Raman silent region of cells (1800–2800 cm−1) such as alkynes, enabling more accurate detection of compounds in single cells.16–18,21–26 Alternatively, non-linear RS, such as Coherent anti-Stokes RS (CARS)27 or Stimulated RS (SRS)28 can be used though they require a more complicated setup with multiple laser beams making their translation to medical and biological applications challenging.29
RS has been used to observe both uptake, metabolism and response of single cells to drug treatment,12 and is often carried out post fixation of the cells, thereby marking an end-point to the experiment. For example, Raman microspectroscopic studies were done on the response of cancer cells to tyrosine kinase inhibitors containing –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– or –CN moieties in lung cancer,1 colorectal cancer30–32 and leukaemia,33 and for detecting drug metabolism within the cell.34,35 Yamakoshi et al. used RS to image the different chemical forms of a mitochondrial uncoupler with a nitrile Raman tag on live HeLa cells.18 Spectroscopic studies have also been used to detect cisplatin binding to DNA in the nucleus,36 cell membrane and cytoplasm37 of lung adenocarcinoma cells. Doxorubicin accumulation and the cell's response at different incubation times was measured on fixed cells using RS.38,39 Internalization of biodegradable biocarrier systems in single cells has been extensively studied with Raman microspectroscopy,12,40 with multiple studies on poly(lactide-co-glycolide) beads.41–45 Microspectroscopy studies give insights into the responses to the drugs of interest, however, mapping is time consuming and low throughput, usually in the tens of cells. In addition, using fixed cells precludes the ability to perform time-dependent studies on the same single-cell.
C– or –CN moieties in lung cancer,1 colorectal cancer30–32 and leukaemia,33 and for detecting drug metabolism within the cell.34,35 Yamakoshi et al. used RS to image the different chemical forms of a mitochondrial uncoupler with a nitrile Raman tag on live HeLa cells.18 Spectroscopic studies have also been used to detect cisplatin binding to DNA in the nucleus,36 cell membrane and cytoplasm37 of lung adenocarcinoma cells. Doxorubicin accumulation and the cell's response at different incubation times was measured on fixed cells using RS.38,39 Internalization of biodegradable biocarrier systems in single cells has been extensively studied with Raman microspectroscopy,12,40 with multiple studies on poly(lactide-co-glycolide) beads.41–45 Microspectroscopy studies give insights into the responses to the drugs of interest, however, mapping is time consuming and low throughput, usually in the tens of cells. In addition, using fixed cells precludes the ability to perform time-dependent studies on the same single-cell.
Colorectal cancer (CRC) is the third most common cancer worldwide and second in mortality, with over 1.8 million new cases diagnosed in 2018, and more than 850![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths.46 Approximately half of the patients develop metastasis to the liver47 and resistance to standard chemotherapy.48 Novel therapies for the treatment of CRC are, therefore, needed. Photodynamic Therapy (PDT) is based on the use of light, a photosensitizer (PS) and oxygen to destroy tumour tissue through oxidative damage, vascular shutdown and activation of immune response against cancer cells.49 DC473 is a low molecular weight photosensitiser that can be activated by excitation with UV-A, violet or corresponding near-infrared two-photon wavelengths. DC473 exhibits strong solvatochromic fluorescence and possesses two alkyne groups.25,50,51 Previous in vitro studies have reported epithelial cell death, after UV-A irradiation following incubation with DC473, due to the production of reactive oxygen species.51
000 deaths.46 Approximately half of the patients develop metastasis to the liver47 and resistance to standard chemotherapy.48 Novel therapies for the treatment of CRC are, therefore, needed. Photodynamic Therapy (PDT) is based on the use of light, a photosensitizer (PS) and oxygen to destroy tumour tissue through oxidative damage, vascular shutdown and activation of immune response against cancer cells.49 DC473 is a low molecular weight photosensitiser that can be activated by excitation with UV-A, violet or corresponding near-infrared two-photon wavelengths. DC473 exhibits strong solvatochromic fluorescence and possesses two alkyne groups.25,50,51 Previous in vitro studies have reported epithelial cell death, after UV-A irradiation following incubation with DC473, due to the production of reactive oxygen species.51
One of the first reports of Raman detection of photosensitisers within cells was the publication by Freeman et al. in 1997, detecting zinc phthalocyanines in a fixed human endothelial hybridoma cell line.52 Abramczyk et al. used Raman microspectroscopy and observed preferential accumulation of aluminium phthalocyanine cancerous breast tissue compared to non-cancerous tissue.53,54 Brozek-Pluska et al. used RS to provide a map of lipids and proteins and fluorescence to detect hematoporphyrin and phthalocyanine in healthy and cancerous breast tissue, showing higher accumulation in the latter.55,56In vivo, Bhattacharjee et al. performed RS measurements in breast cancer model rats showing PS accumulation and PDT effects of chlorin photodithazine.57 An alternative approach is to use Surface-Enhanced RS (SERS) to increase the limit of detection of the PS, often done by including metal nanoparticles in theranostic constructs with the PS.58–64 Fales et al., used targeted gold nanostar constructs loaded with PPIX, showing detection and targeting in single cells,65,66 and Farhadi et al. coupled palladium porphyrin with plasmonic nanoparticles to allow SERS detection of the PS, using the same wavelength to excite Raman scattering and to stimulate PDT.67 To our knowledge, there have been no reports in the detection of PS accumulation in live single cells using RS, or in the single-cell pharmacodynamics of the PS upon activation.
In this manuscript, we study both the accumulation and the photoactivation of a novel photosensitiser, DC473, in live single-cells from cell lines representing different stages of colorectal cancer. For our model we used an adenocarcinoma model system comprised by the cell lines SW480, HT29 and SW620, representing Duke's stage B primary, Duke's stage C primary and Duke's stage C secondary cell lines, respectively.20
Methods
Cell preparation
Cells were cultured and measured as in our previous work.20 The SW480, HT29 and SW620 cell lines were cultured in Dulbecco's Modified Eagle Medium (DMEM/F-12, Gibco) supplemented with 10% fetal bovine serum (Sigma), 2 mM Glutamax (Thermo Fisher Scientific) and penicillin 100 units per mL streptomycin 100 μg mL−1 (Sigma). All experiments were done with passage numbers below 50.DC473 incubation
Cells were seeded 2 days in advance of the experiment in 6 well plates at a concentration of 3.5 × 105 cells per well. Before Raman measurements, media was changed and 4 μM of DC473 in a 0.4% DMSO/media solution was added to the cells and incubated for 4 h, whereas the control cells received an equivalent concentation of DMSO (0.4%). Prior to the experiment, cells were washed with Dulbecco's phosphate-buffered saline (DPBS) and gently retrieved from 6-well plates by incubating with TrypLe (Thermo Fisher Scientific) for 5 min, followed by centrifugation and two washes in DPSB (100 gs 1 min) and re-suspension in cell dissociation buffer (Thermo Fisher). When pipetted into the setup, cells sedimented onto the coverslip and showed no visible Brownian motion, remaining in a spherical shape.Raman spectroscopy
Quartz slides (UQG Optics, 75 × 25 × 1 mm) and coverslips (25.4 × 25.4 × 0.15–0.25 mm Alfa Aesar) were sonicated with acetone (VWR Chemicals), 2–5% Decon 90 (VWR Chemicals) and rinsed with MilliQ. All organics were removed by incubation for 20 minutes in Piranha solution (H2O2 30% (Thermo Fisher) and H2SO4 > 95% (Thermo Fisher) in a 3:7 proportion). Spacers were prepared using a 50 μm polyethylene terephthalate film (Goodfellow, UK). A nitrocellulose-based solution was used to bond the coverslip to the slide. The cell solution was pipetted into this chamber immediately before measuring. All experiments were done at room temperature and samples were measured for 1 h.
The Raman system used was an inVia Raman confocal inverted microscope (Renishaw) integrated with a Leica DMi8/SP8 laser scanning confocal microscope system, with a DPSS Diode 532 nm laser (laser power of 22 mW on the sample) and a 785 nm laser (laser power of 45 mW at the sample), with gratings of 1800 l mm−1 and 1200 l mm−1 respectively. Light was collected using either a Newton EMCCD Sensor (DU970P, Andor, 1600 × 200 px) for the visible excitation laser or a near-infrared enhanced CCD array detector (1024 × 256 pixels, Renishaw). Before each experiment a spectrum of a silicon sample was collected using the respective objective, grating and detectors used and the microscope was calibrated to the peak position (520.5 cm−1).
The cell spectra were obtained using a 100× oil objective (HC PL APO CS2 FWD 0.13 mm NA 1.4) and a slit size of either 20 μm (for DC473 accumulation experiments and DC473 off cell activation experiment) or 65 μm (for DC473 PDT experiments). This objective gave a 10.8 μm and 25.7 μm Full-Width Half Maximum axial confocality when tracking the changes of Raman intensity of the 520.5 cm−1 with the distance to a silicon sample for each of the slit openings. The laser spot was defocused by 20% using a beam expander, generating a laser spot of approximately 18 μm diameter, and obtaining the average signal of the cell. Cell spectra for DC473 accumulation were obtained using a step configuration with 1 s exposure time and 5 accumulations in two different windows (300–1800 cm−1 and 1800–3200 cm−1) which gave a total exposure time of 10 s per cell. For the DC473 PDT experiments, the 300–1800 cm−1 region was prioritized to increase the spectral quality and time resolution. The data from multiple experiments were combined for this paper without omitting any outliers, with 135 cells incubated with DC473 and 134–135 cells as control for each cell line. Five background spectra from cell-free regions of the sample measured at the same Z-position as the cells were obtained for each experiment.
Photodynamic activation of DC473
DC473 off-cell activation was done using a 100 μM DC473 solution in DMSO, using a 100× oil immersion objective and 340–380 nm UV light with a light intensity of 131 mW cm−2 and a total dose of 5.2 J cm−2. Acquisition was obtained using the 785 nm excitation before and immediately after activation (1.13 mJ). For tracking the activation of DC473 on live single cells, before cell treatment, a Raman spectrum was acquired setting the system to acquire spectra every 20 s. Then, the PS was activated using 3 s of UV light, for a total dose of 711 mJ cm−2. The following acquired spectra tracked the changes in the cell following the PS activation. Two controls were made, with cells incubated with DMSO exposed to the Raman acquisition light and cells incubated with DMSO exposed to both the Raman acquisition light and the UV light, to understand the phototoxicity due to these light sources in the absence of the PS. Typically, 10–11 cells were measured per group, acquiring 15 spectra per cell, with a total of around 30–35 cells per cell line.Pre-processing of the spectra
The spectra obtained were cosmic ray filtered (WiRE® software) and exported as text files for further analysis using Matlab's Statistics and Machine Learning Toolbox (MathWorks). The Matlab functions used are indicated by italics. The silicon peak of a calibration sample was used to calibrate the wavenumber axis of each spectrum and the spectra were translated vertically, such that the minimum intensity was zero. For each cell spectrum, the average background spectrum of the sample was multiplied by an adjustment factor before being subtracted from the cell spectrum. The spectra were truncated to only consider the regions between 730 cm−1 and 3100 cm−1. The spectra were baseline corrected using the algorithm developed by Koch et al. (2016).68 The regions of the spectra between 1750 cm−1 and 2800 cm−1 were not considered for subsequent analysis. In order to normalize to the protein content, for comparison with biochemical literature data, each spectrum was normalized such that the Amide I peak was unity. For the PDT experiments, where only the fingerprint region was acquired, similar pre-processing was followed.Statistical analysis and classification
Results
Detection of DC473 on live single cells
Fig. 1B shows fluorescent staining of the three cell lines used in this study: SW480, HT29 and SW620. These cell lines belong to progressive stages of colorectal adenocarcinoma (Duke's stage B, C and C metastasis respectively). Nile red emission is shown in green (triglycerides) and red (phospholipids).70 The three cell lines show distinct morphology and the presence of lipid droplets (green), HT29 showed a particularly high quantity of lipid droplets around the nucleus, whereas SW620 showed lipid droplets located near the Poles. SW480 cells showed lipid droplets both around the nucleus and near the Poles. As DC473 is a hydrophobic compound, we expected cells with higher amounts of lipid droplets to have increased uptake of DC473. In a previous study, we showed that DC473 in SW480 cells accumulated in lipid droplets and in the cytosol, presumably on the mitochondrial membrane.25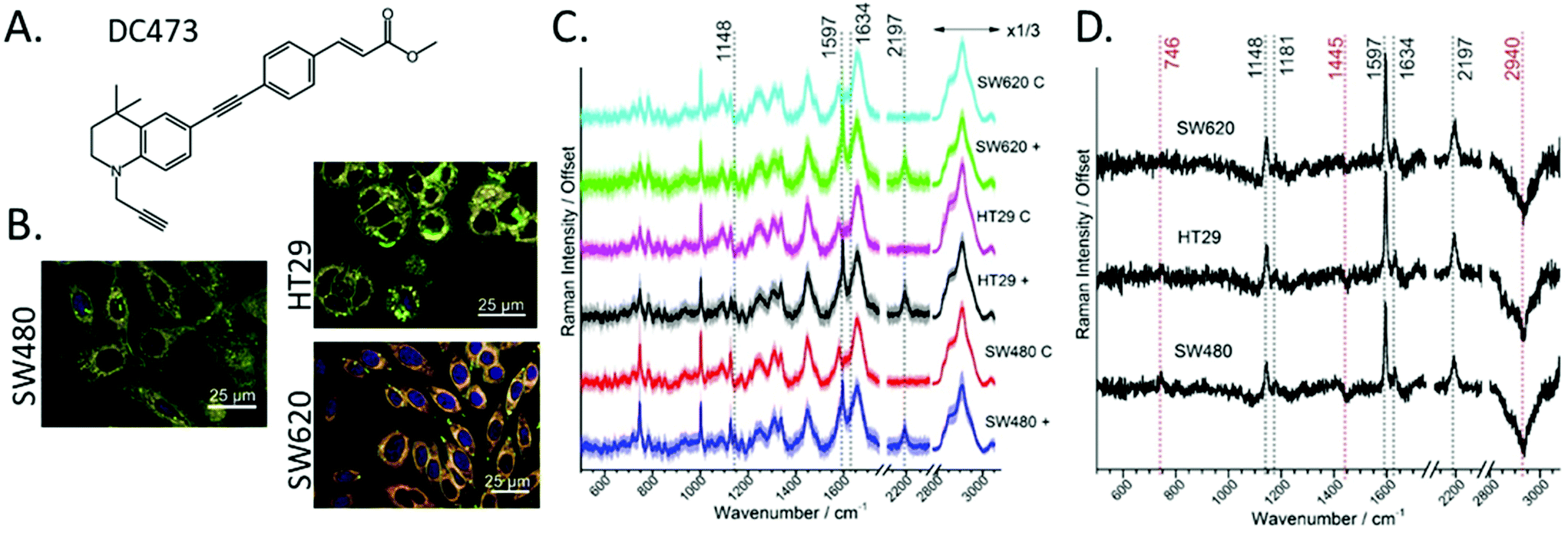 | ||
| Fig. 1 A. Chemical structure of DC473. B. Confocal fluorescence images of cells for each cell line, where blue is DNA, and green and red are two emission channels for Nile Red: triglycerides (green) and phospholipids (red). C. Average spectra of live single cells of the different cell lines, where + indicates cells following incubation with 4 μM of DC473 and C indicates cells with an equivalent volume of DMSO. The area around the curve corresponds to one standard deviation and the main DC473 bands have been indicated. The number of cells in each group is N ≥ 134. D. Difference spectra of the average between DC473 induced cells and control cells for each of the cell lines. All spectra are normalized to the Amide I peak and have been offset for clarity. | ||
Using a beam expanded laser spot allowed the relatively fast acquisition of high-quality averaged spectra per cell. Fig. 1A shows the chemical structure of DC473.25Fig. 1C shows the average spectra for each of the cell lines and comparing the cells following incubation with DC473, or with DMSO control. All spectra are normalized to the Amide I band. The five strongest peaks from the spectrum of the pure DC473 (see Fig. S1 and Table S1 from the ESI†) are identified as: 1141 cm−1 (DC473 phenyl), 1597 cm−1 (DC473 phenyl), 1634 cm−1 (DC473 ester) and 2197 cm−1 (DC473 internal alkyne). The terminal alkyne band that would be expected at lower wavenumbers was not observed71 which is in agreement with general observations that terminal alkynes often show weaker bands than internal and especially conjugated alkynes.23 The average spectra show uptake of the drug for all cell lines, with higher DC473/protein content for HT29 and SW620 cells. DC473 bands were present in all cell lines, where the 1634 cm−1 peak showed as a shoulder on the Amide I band. DMSO was non-detectable in the samples, based on the absence of the C–S νS band at 674 cm−1.
Fig. 1D shows the difference spectra indicating the presence of not only the main DC473 bands, but also a reduction in the CH contribution of both the 1445 cm−1 and the stretching band at 2940 cm−1 following incubation with DC473. This suggests that DC473 could interfere with lipid metabolism and/or regulation. A weak band at 1181 cm−1 was also present in the difference spectra as is associated with the DC473 phenyl peak. We note that incubation with DC473 also correlated with an increase in the 746 cm−1 cytochrome C band, suggesting that DC473 localised in mitochondria and that it may interfere with organelle function even before PS activation.
DC473 band fitting on single cell spectra
To compare the DC473 accumulation in the different cell lines, we used Gaussian Lorentzian sum (GLS) single cell peak fitting of the DC473 spectral peaks at 2195 cm−1 and at 1597 cm−1, as detailed in the Methods section. Example fits are shown in Fig. 2B for the 1594 cm−1 band and Fig. 2D for the 2194 cm−1 band. The results from the fitted amplitude are shown in Fig. 2A and C, where the significance between cell types is also indicated (based on t-test with unequal variances). All cell lines displayed uptake of DC473, but when comparing the different cell lines (normalizing to the Amide I band), SW620 and HT29 exhibited similar levels of uptake and significantly higher accumulation of DC473 than SW480 cells, likely due to the higher presence of lipid droplets (see Fig. 1B).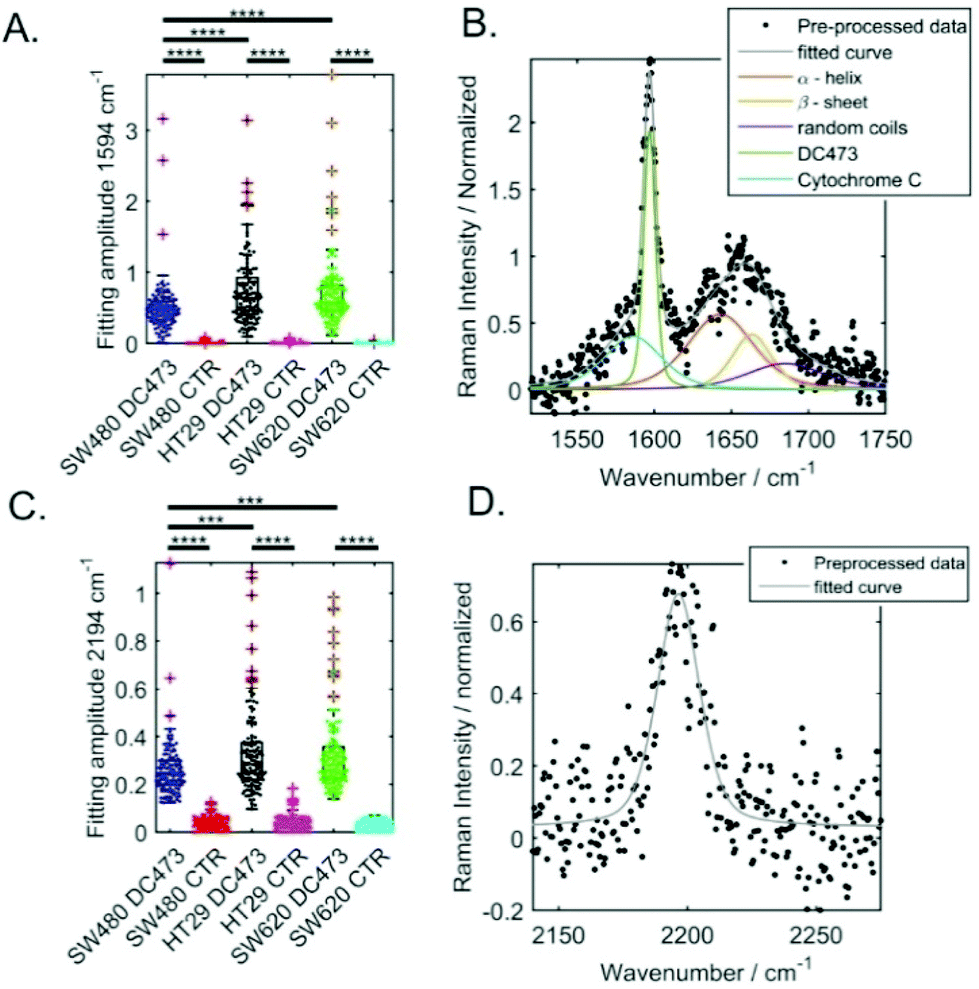 | ||
| Fig. 2 Comparison of the characteristic DC473 signals in different colorectal single cells following incubation with either DC473, or DMSO as a control. A. Bee swarm and box plots of the amplitudes for the 1594 cm−1 peak fitting. B. Peak fitting for a SW620 cell. C. Bee swarm and box plots of the amplitudes for the 2194 cm−1 peak. D. Peak fitting for the alkyne band associated with the DC473 in the same SW60 cell in (B). | ||
For the 2195 cm−1 peak, the position and width of the peak were allowed to vary on a cell by cell basis. Peaks with amplitude lower than 0.15 (normalized to the Amide I band) were excluded. The mean peak amplitude, width and position obtained are shown in Table 1. Cell lines with higher accumulation of DC473 showed broader peaks and a shift towards higher wavenumber. We believe this is related with the local environment where the compound accumulates within the cell, with cell lines with more lipid droplets accumulating more compound in a more hydrophobic environment, with a small shift towards higher wavenumbers.
| N | Amplitude (normalized) | Position (cm−1) | Width (cm−1) | |
|---|---|---|---|---|
| SW480 | 121 | 0.28 ± 0.01 | 2193.9 ± 0.2 | 18.8 ± 0.2 |
| HT29 | 129 | 0.34 ± 0.02 | 2195.2 ± 0.2 | 19.7 ± 0.4 |
| SW620 | 133 | 0.33 ± 0.01 | 2194.8 ± 0.2 | 21.0 ± 0.5 |
The CH stretching band was also analysed at the single cell level, integrating between 2800 and 3080 cm−1 (see Fig. S2 from the ESI†). Data showed a very significant drop in CH-stretching bands intensity for all cell lines (p < 10−4) following incubation with DC473.
Principal component analysis
To further understand the data, PCA was carried out, in which the smoothing of the spectrum was avoided to increase the sensitivity of any band position shifting. Fig. S4A (ESI†) shows the first 4 PCA loadings explaining 21.5% of the total variability, where the main DC473 (black) and cell (red) peaks have been labelled. Fig. S4B (ESI†) shows the scores for those first 4 PCs. PCA results confirmed the increase in cytochrome C band intensity and the overall reduction in the CH stretching intensity following incubation with DC473. This is in line with our previous results showing accumulation in lipid droplets,25 indicating that the compound could be partially disrupting lipid accumulation. The positive correlation of cytochrome C with DC473 suggests that the compound could be interacting with mitochondria.DC473 activation off cell
Following our understanding of the accumulation of the compound in these cell lines, further experiments were made to understand the UV activation mechanisms of DC473. Deep UV light was effective in bleaching the fluorescence and led to the loss of the Raman signal of the compound. A 100 μM solution of DC473 in DMSO was measured before and after exposure to UV light (340–380 nm, 5.2 J cm−2). The spectra acquired using the 785 nm excitation line are shown in Fig. S3 in the ESI,† normalized to the maximum DMSO band. The exposure to UV light caused a significant drop in the main Raman bands associated with the alkyne and phenyl modes, without the appearance of other secondary bands, indicating that the diphenylacetylene structure of DC473 is degraded due to UV light exposure, and the resultant degradation products are too weak to detect or exhibit Raman bands of comparatively weak intensity. It should be noted that even after prolonged UV exposure, not all the DC473 signal disappeared.DC473 PDT on live single cells
To study the effects of the DC473 PS in single cells, the cells were incubated with the compound, then detached from the well plate and resuspended in buffer before analysis. A Raman spectrum in the fingerprint region was acquired before UV treatment. Post UV exposure Raman spectra were acquired every 20 s, for approximately 5 minutes. Two controls without the PS were prepared to separate any potential phototoxicity due to the excitation laser or the UV light used to irradiate the compound. Specifically, cells were either exposed to the same UV dose and the Raman laser or just to the Raman laser excitation. We have already shown that DC473 does not degrade under the 532 nm laser doses used in this experiment, and thus no control with DC473 and no UV light was undertaken. Measurements were typically made on around 10–11 cells per group, with a total of around 30–35 cells per cell line and in each case 15 spectra per cell.An example of the data obtained in such an experiment is shown in Fig. 3A. The DC473 bands at 1597 and at 2195 cm−1 showed a strong contribution in the initial spectrum (t = 0) which decreased rapidly after PS activation. However, not all the DC473 signal disappeared; a small amount of the compound remained according to the cell spectra. This data agrees with the off-cell data, suggesting that DC473 undergoes a significant structural change following UV activation and consequently, loses its characteristic Raman signal.
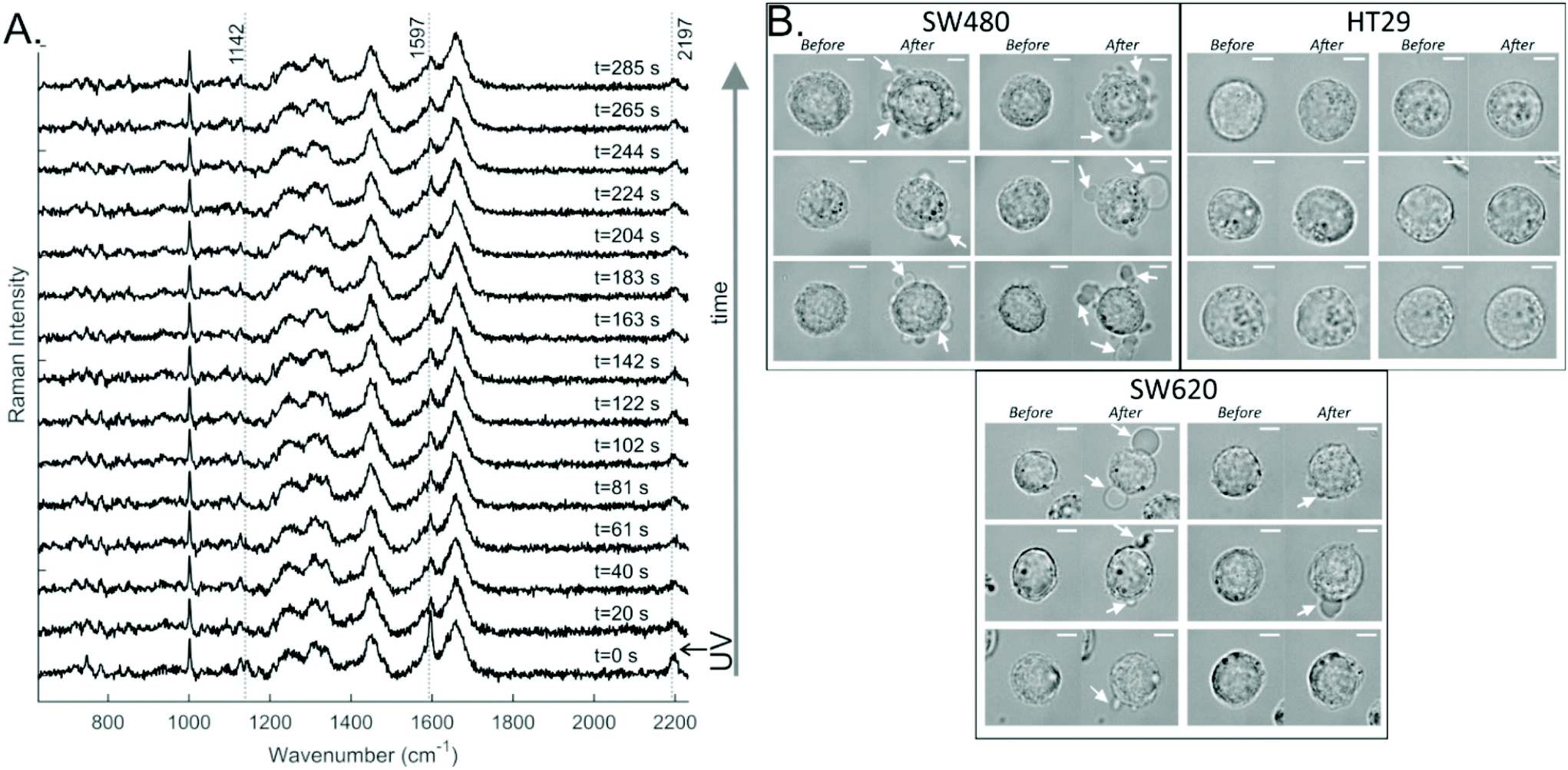 | ||
| Fig. 3 A. Example of a time-dependent single-cell (SW480) response to the DC473 PDT activation with UV light, where the first spectrum was taken before activation of the photosensitiser, and the remaining spectra were collected post activation. The data was pre-processed as described in Methods. B. Bright field images following incubation with DC473 before and after Raman acquisition following PDT treatment with UV light (approx. 5 minutes). Membrane blebbing was observed, as indicated with white arrows. | ||
Interestingly, changes in the morphology of the treated cells following the Raman acquisition were detectable for some cells from the SW480 and SW620 cell lines, as can be seen in Fig. 3B. Cells would undergo blebbing by the end of the acquisition (indicated with white arrows), a sign of cell death and an expected response of PDT. For the controls, only 3% of the cells showed morphology changes, with 2/30 cells for the UV control and 0/30 cells for the Raman laser control. From the DC473-treated cells 64% of SW480 cells, 0% of HT29 cells and 60% of SW620 cells showed morphological changes following PDT. Surprisingly, although the HT29 and SW620 cells displayed very similar DC473 accumulation they had very different responses to UV activation. However, it must be noted that the absence of morphological changes at the time of measurement does not preclude the cells could undergo apoptosis on longer time scales.
To illustrate the differences in cell responses, the average initial and final spectra for each cell are shown in Fig. 4 (Control, Control + UV and DC473 + UV). These spectra were overlaid for each cell line. For the DC473 + UV samples, the main DC473 bands at 1597 and 2195 cm−1 showed a significant reduction in intensity following PDT, but not always down to undetectable levels (see bands at 1143, 1597, shoulder at 1632 and band at 2195 cm−1). In particular, the SW480 and SW620 cells often showed detectable levels of DC473 following UV activation, whilst the HT29 cells showed a drastic reduction of the compound's Raman signal. These results confirmed that the compound was undergoing a chemical modification within the cells. The differences in photodegradation of the compound in different cell lines suggest that the compound may be showing different aggregation states within the cell. Indeed, it has been shown that diphenyacetylene derivatives can form liquid crystalline states in solution and that the highly organised structures formed showed a similar blue shifted absorption band.72 As the compound accumulates both in lipid droplets and in the cytosol, the coexistence of these two states is expected, with the result that these aggregates would be less susceptible to photoactivation due to their absorption band being shifted. When comparing different cell lines, the differences observed in the cell's morphological changes post treatment can also be linked to the degradation of the compound, showing that cell lines with remaining DC473 signal also showed more blebbing, post-treatment. To understand the observed morphology changes, it must be noted that the cells may have different resistances to oxidative stress. SW480 and SW620 cells show KRAS (G12V) mutation, and HT29 cells show BRAF (V600E) mutation,73 however, the relation between these mutations and the redox state is not well understood.74 Furthermore, previous reports showed that lipid droplets can have a protective effect against reactive oxygen species,75 which could explain why HT29 cells showed higher accumulation but no response to the activation of DC473, compared to SW480 and SW620 cells, that showed fewer lipid droplets. Additionally, the location of the compound in each cell line might be different and could have a strong impact in its toxicity following activation. Regarding the controls, whilst the signal remained largely unchanged, the cytochrome C bands were observed to decrease (746 and 1128 cm−1) in all treatments indicating some phototoxicity due to both the PDT, the Raman acquisition laser and UV light dose.
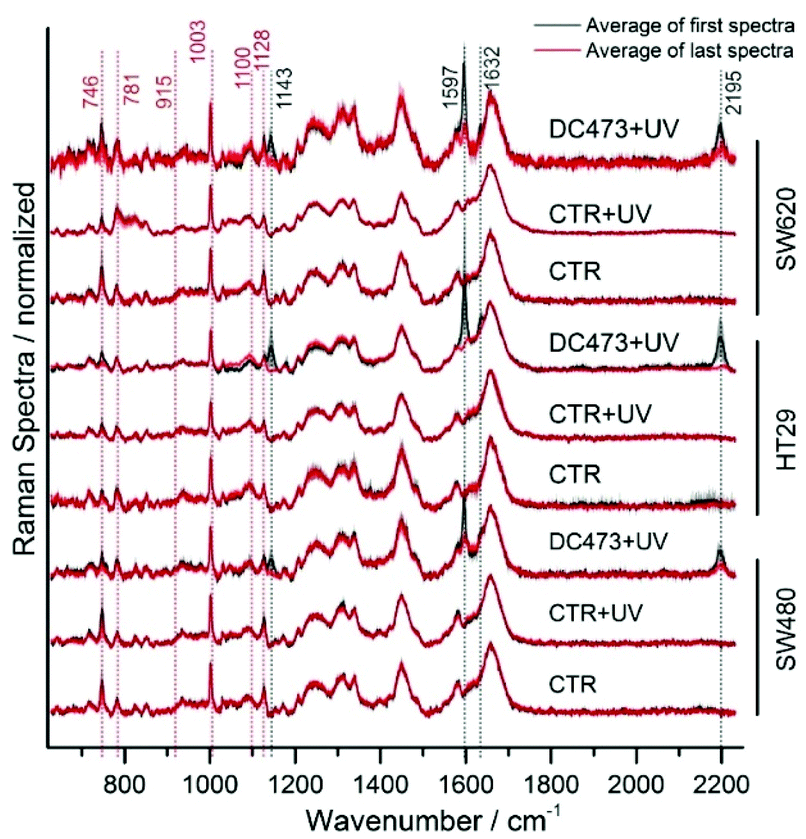 | ||
| Fig. 4 Average of the data before (black) and after (red) each PDT experiment for each of the samples and cell lines, where CRT = Control, CRT + UV = Control exposed to equivalent dose of UV light, DC473 + UV = DC473 sample exposed to UV light. The main bands have been labelled. The area around the curve represents one standard deviation. | ||
When calculating the difference between the average spectra, the main effects of the PDT became evident (Fig. S5, ESI†). Results showed a drop in all DC473 peaks and in cell bands at 1443, 1181, 1597, 1632 and 2195 cm−1. All datasets showed negative peaks corresponding to the cytochrome C progression (746, 1128, 1310 and 1585), indicating phototoxicity of the excitation 532 nm laser light and of the UV light, with also DNA signal loss associated with degradation in some of the samples observed at 781 cm−1. Additional changes were observed in the band at 1338 cm−1, which has mixed contributions from proteins and nucleic acids, in the lipid band at 1445 cm−1 and on the side of the Amide I at 1674 cm−1.
PCA analysis of DC473 PDT on live single cells
To explore these changes further, PCA was performed. Fig. 5A shows the loadings for the first 5 coefficients. Fig. S5 (ESI†) shows the variation of the average scores for each of the conditions and cell lines with time, where the intensity of all traces was aligned to 0, for t = 0 to better see the trends with time. The principal component PC2 was mainly associated with contributions from DC473, whilst PC3 was associated with cytochrome C and PC4 with the unassigned bands at 1314, 1338, 1362 and 1390 cm−1 which showed a negative trend over time, being potential biomarkers of the cell response to DC473 activation. PCA was unable to identify any clear Raman marker for the cell blebbing (morphology changes) observed after PDT, and only showed a strong reduction on PC3 and 4 on all treated cells, in line with the amount of DC473 that the cells accumulated. A 2D plot of the single-cell score traces for PC2 and PC4 is shown in Fig. 5B as a summary of the PCA findings. The initial point for each cell was shown with a marker, and the subsequent points were shown as a line trace. The HT29 control group did not show overall changes with time, whereas SW480 and SW620 cells showed a drop in the PC4 score over time suggesting these cells were more sensitive to the phototoxicity of the 532 nm excitation laser and the UV light. PDT samples showed a dramatic reduction in both coefficients immediately after the UV dose.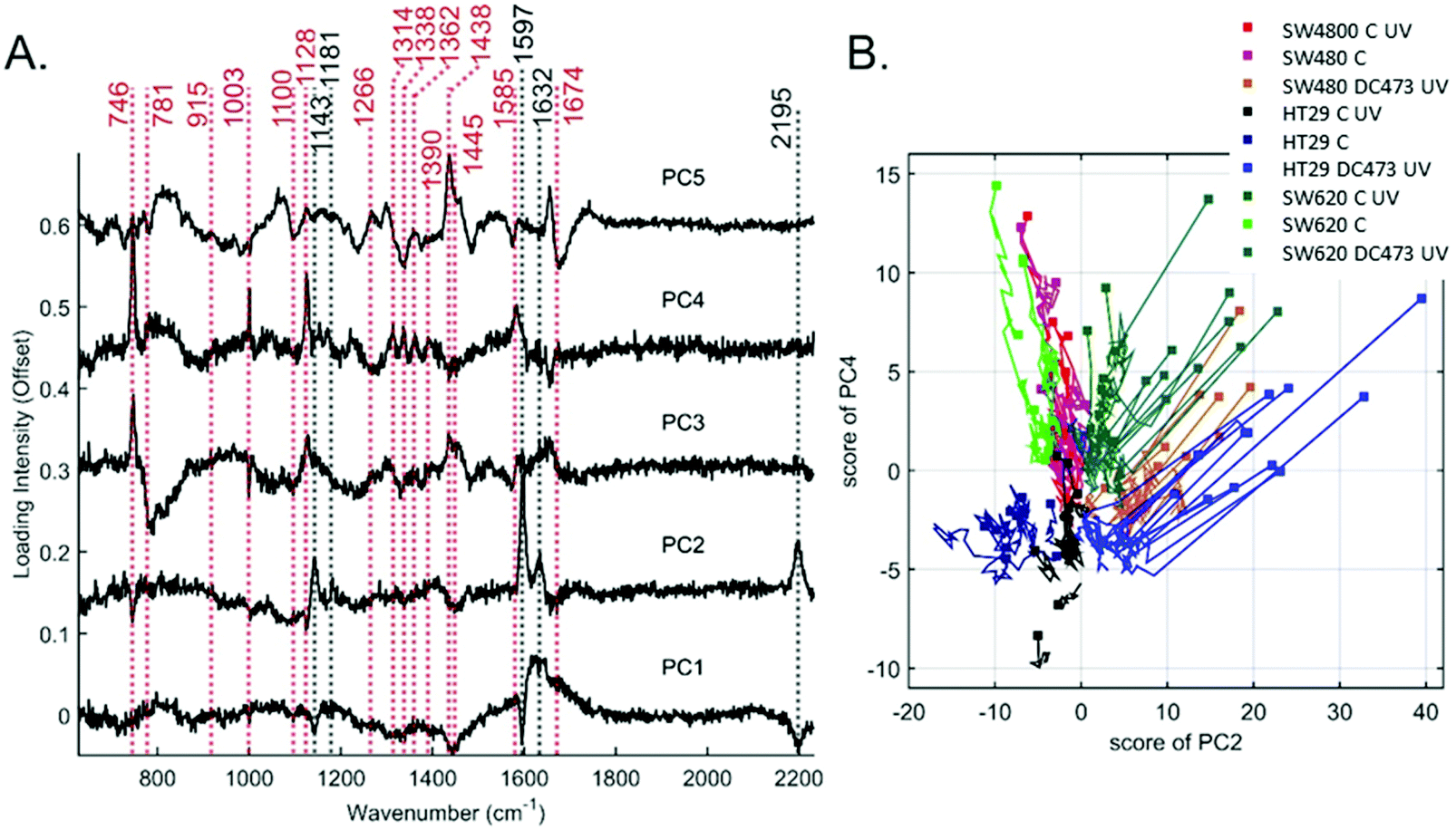 | ||
| Fig. 5 A. PCA loadings for the first 5 PCs for the PDT data. The main cell peaks (red) and DC473 peaks (black) have been labelled. B. PCA traces for PC4 and PC2 for the PDT data, where the first time point is shown as a square, and remaining observations in that cell are shown as a trace. | ||
Single-cell band fitting of DC473 bands following PDT
To better characterize the changes that DC473 underwent following UV light activation, both the 1592 and the 2195 cm−1 bands were fitted to Gaussian Lorentzian Sum (GLS) functions. Fig. 6 shows the average results for all cells after the amplitude filtering on both DC473 bands, where the area around the curves corresponds to one standard deviation, and each data point was an average of at least 20 fittings. As a lot of cells showed too low band intensity for fitting, all cell lines were analysed together. Results showed a sharp drop in the amplitude of both bands caused by the UV irradiation, followed by a plateau. However, the position and widths of the bands showed a slower time-dependent trend post-UV exposure. The four curves were fitted to a sigmoid function over time (y = a/(a + b·exp(−τ·t))), where a is the asymptote of the sigmoid (cm−1), b is the ratio between the asymptotic shift and the initial value (adimensional) and τ (s−1) is the inverse of the lifetime. The values for the parameters of the fitted curves are shown on Table 2 (±SE).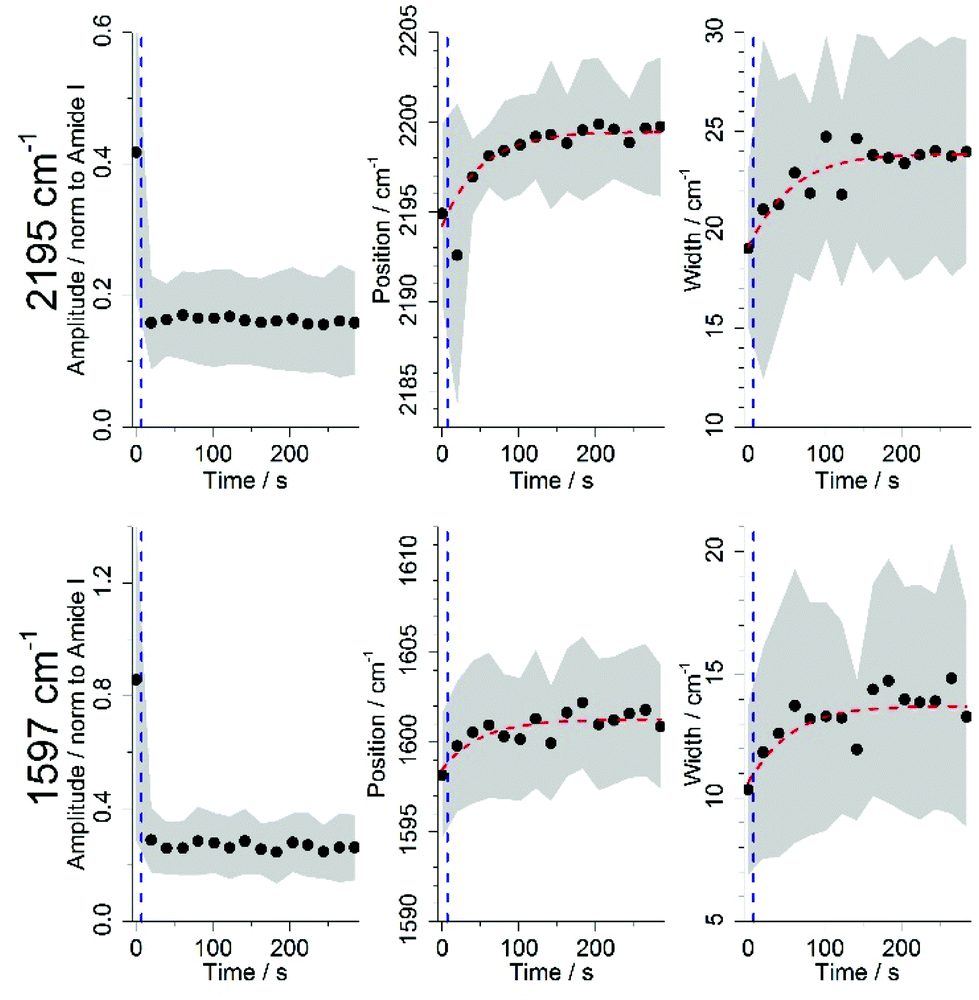 | ||
| Fig. 6 Average time dependence of the fitting parameters for the 2195 and 1597 cm−1 band, for all cell lines incubated with DC473 and treated with UV light after amplitude filtering, where the area behind the curve corresponds to one standard deviation. The point at which the UV dose was applied has been represented with a vertical blue dashed line. Red dashed lines show the sigmoid fittings done to the average data. | ||
| Position/cm−1 | |||
|---|---|---|---|
| Band | a/cm−1 | b | τ/s−1 |
| 2195 | 2199.5 ± 0.2 | 0.0024 ± 0.0003 | 0.020 ± 0.004 |
| 1597 | 1601.3 ± 0.3 | 0.0018 ± 0.0004 | 0.02 ± 0.01 |
| Width/cm−1 | |||
|---|---|---|---|
| a/cm−1 | b | τ/s−1 | |
| 2195 | 23.9 ± 0.4 | 0.25 ± 0.04 | 0.021 ± 0.007 |
| 1597 | 13.7 ± 0.4 | 0.29 ± 0.08 | 0.02 ± 0.01 |
The position of the 2195 cm−1 band shifted towards higher wavenumbers, peaking at 2199.5 cm−1, for a total shift of ∼4.8 cm−1. The 1597 cm−1 band had a slightly smaller shift to 1601.3 cm−1 (∼3.6 cm−1). In terms of the GLS width, the alkyne band showed an increase in width of ∼4.9 cm−1 whilst the sharper phenyl band showed a modest increase in width of around ∼3.0 cm−1. Both bands showed a similar τ constants of 0.02 s−1. The inverse of τ, or the lifetime of these events, is of approximately 50 s. It has previously been reported with phenylacetylene that increased order in a liquid matrix leaded to narrower bands and shifted maximum intensity to lower wavenumbers.76 We believe that PDT causes the DC473 aggregates, which are resistant to photodegradation,72 to disaggregate, causing the Raman band to widen and to shift to higher wavenumbers. Additionally, the biological environment of the drug has a very important role in this process, by either stabilizing or destabilizing these aggregates, and providing substrates for reaction with the compound.
The two most susceptible cell lines to PDT, based on morphological changes, showed residual DC473 post-PDT, whereas the HT29 cells showed an absence of any remaining DC473 post-PDT, in most observations. In our previous publication we observed that DC473 localized in both lipid droplets and in the cytosol.25 We believe that aggregates could be forming with a shifted absorption band compared to the free compound, and thus a lower response to the UV light. Upon PDT, the aggregates would be disrupted, showing a widening and shift of the Raman band to higher wavenumbers. The presence of aggregates anti-correlates with the morphological response of the cells, however, more cell lines need to be studied to confirm this result. Additionally, the inherent concentration of potential substrates for reaction with the activated compound in each cell line is an important factor to explain their different response to PDT. Further experiments are needed to confirm the dual behaviour of the compound and to fully understand the cell toxicity mechanism. Nevertheless, Raman on live cells proves to be a powerful tool to track the time-dynamics to drug responses at the single-cell level.
Conclusions
Our experiments indicate that DC473 can be readily taken up by different CRC cell lines. Raman spectroscopy results indicated that accumulation mainly occurs in lipid droplets and the cytosol, and was significantly higher for more advanced adenocarcinoma HT29 and SW620 cells compared to SW480 cells. This is likely linked to their higher lipid droplet content. However, when activating the compound with UV light, SW480 and SW620 showed clear morphological changes in response to the drug, whereas HT29 cells showed no change. The DC473 Raman signal reduced upon activation, however, some inactivated compound remained for SW480 and SW620 cells, whilst there was minimal compound left in HT29 cells. It is known that diphenylacetylenes can form highly organised aggregates with blue shifted absorption bands,72 which would make them less susceptible to the PDT UV light. Multivariate analysis identified coefficients that showed time-dependent changes in multiple cell peaks over time after DC473 activation, including cytochrome C bands. The main two DC473 peaks at 1597 and 2195 cm−1 showed a significant drop in intensity after irradiation and additional shift to higher wavenumbers and increase in peak width over time at the single cell level. These changes happened with a lifetime of approximately 50 s. These effects possibly indicate a change in the aggregation state of the compound, with disruption of the aggregates causing the widening and shift of the bands to higher wavenumbers.76 Our results indicate that the compound could be suitable for CRC PDT, with higher accumulation on lipid droplet rich cancer stages. However, its effects in lipid metabolism need further understanding and some cells could be resistant to this treatment, as the data for HT29 seems to indicate. The results showed limited phototoxicity due to the UV and Raman excitation light sources and that this may also be different on different cell lines. Finally, the results also highlight heterogeneity both in the accumulation and response to PDT both between and within cell lines, underlining the importance of single cell studies. Further research needs to be done to understand the biological mechanism behind phototoxicity of the compound in these cell lines. These findings confirm the feasibility of doing live single cell drug studies using RS, with sensitivity to cell viability and chemical and environmental changes in the drug over time.Conflicts of interest
AW and CAA are shareholders and directors of LightOx Ltd, a company licensed to pursue commercial applications for the novel compound described in this manuscript.Acknowledgements
JGP thanks EPSRC for the PhD studentship, the University of Leeds Alumni for financial support and JSPS. DC thanks EPSRC, BBSRC and High Force Research Ltd for funding. SDE acknowledges the following funders; MRC (MR/M009084/1) and EPSRC (EP/P023266/1). SDE is supported by the National Institute for Health Research (NIHR) infrastructure at Leeds. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. We acknowledge Catriona Marshal (SJUH) for providing the cell lines and STR validation. We would like to thank Jon Girkin and Valerie Affleck for their input and advice. The data used in the figures of this paper will be available at https://doi.org/10.5518/702.Notes and references
- M. K. Hammoud, H. K. Yosef, T. Lechtonen, K. Aljakouch, M. Schuler, W. Alsaidi, I. Daho, A. Maghnouj, S. Hahn, S. F. El-Mashtoly and K. Gerwert, Sci. Rep., 2018, 8, 15278 CrossRef PubMed.
- I. W. Schie and T. Huser, Appl. Spectrosc., 2013, 67, 813–828 CrossRef CAS PubMed.
- C. Krafft, M. Schmitt, I. W. Schie, D. Cialla-May, C. Matthäus, T. Bocklitz and J. Popp, Angew. Chem., Int. Ed., 2017, 56, 4392–4430 CrossRef CAS PubMed.
- N. K. Das, Y. Dai, P. Liu, C. Hu, L. Tong, X. Chen and Z. J. Smith, Sensors, 2017, 17, 1592 CrossRef PubMed.
- R. Smith, K. L. Wright and L. Ashton, Analyst, 2016, 141, 3590–3600 RSC.
- K. Kong, C. Kendall, N. Stone and I. Notingher, Adv. Drug Delivery Rev., 2015, 89, 121–134 CrossRef CAS PubMed.
- I. Notingher and L. L. Hench, Expert Rev. Med. Devices, 2006, 3, 215–234 CrossRef CAS PubMed.
- I. W. Schie, J. Rüger, A. S. Mondol, A. Ramoji, U. Neugebauer, C. Krafft and J. Popp, Anal. Chem., 2018, 90, 2023–2030 CrossRef CAS PubMed.
- J. Phillip Kennedy, P. Jeffrey Conn and C. W. Lindsley, Bioorg. Med. Chem. Lett., 2009, 19, 3204–3208 CrossRef CAS PubMed.
- M. Diem, A. Mazur, K. Lenau, J. Schubert, B. Bird, M. Miljković, C. Krafft and J. Popp, J. Biophotonics, 2013, 6, 855–886 CrossRef CAS PubMed.
- H. J. Byrne, M. Baranska, G. J. Puppels, N. Stone, B. Wood, K. M. Gough, P. Lasch, P. Heraud, J. Sulé-Suso and G. D. Sockalingum, Analyst, 2015, 140, 2066–2073 RSC.
- G. P. S. Smith, C. M. McGoverin, S. J. Fraser and K. C. Gordon, Adv. Drug Delivery Rev., 2015, 89, 21–41 CrossRef CAS PubMed.
- B. Kann, H. L. Offerhaus, M. Windbergs and C. Otto, Adv. Drug Delivery Rev., 2015, 89, 71–90 CrossRef CAS PubMed.
- C. Matthäus, A. Kale, T. Chernenko, V. Torchilin and M. Diem, Mol. Pharm., 2008, 5, 287–293 CrossRef PubMed.
- F. Hu, L. Wei, C. Zheng, Y. Shen and W. Min, Analyst, 2014, 139, 2312–2317 RSC.
- S. Hong, L. Lin, M. Xiao and X. Chen, Curr. Opin. Chem. Biol., 2015, 24, 91–96 CrossRef CAS PubMed.
- W. J. Tipping, M. Lee, A. Serrels, V. G. Brunton and A. N. Hulme, Chem. Soc. Rev., 2016, 45, 2075–2089 RSC.
- H. Yamakoshi, A. F. Palonpon, K. Dodo, J. Ando, S. Kawata, K. Fujita and M. Sodeoka, Chem. Commun., 2014, 50, 1341–1343 RSC.
- M. Okada, N. I. Smith, A. F. Palonpon, H. Endo, S. Kawata, M. Sodeoka and K. Fujita, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 28–32 CrossRef CAS PubMed.
- J. Gala de Pablo, F. J. Armistead, S. A. Peyman, D. Bonthron, M. Lones, S. Smith and S. D. Evans, J. Raman Spectrosc., 2018, 49, 1323–1332 CrossRef CAS PubMed.
- S. Hong, T. Chen, Y. Zhu, A. Li, Y. Huang and X. Chen, Angew. Chem., Int. Ed., 2014, 53, 5827–5831 CrossRef CAS PubMed.
- H. Yamakoshi, K. Dodo, M. Okada, J. Ando, A. Palonpon, K. Fujita, S. Kawata and M. Sodeoka, J. Am. Chem. Soc., 2011, 133, 6102–6105 CrossRef CAS PubMed.
- H. Yamakoshi, K. Dodo, A. Palonpon, J. Ando, K. Fujita, S. Kawata and M. Sodeoka, J. Am. Chem. Soc., 2012, 134, 20681–20689 CrossRef CAS PubMed.
- F. Hu, C. Zeng, R. Long, Y. Miao, L. Wei, Q. Xu and W. Min, Nat. Methods, 2018, 15, 194–200 CrossRef CAS PubMed.
- J. Gala de Pablo, D. R. Chisholm, A. Steffen, A. K. Nelson, C. Mahler, T. B. Marder, S. A. Peyman, J. M. Girkin, C. A. Ambler, A. Whiting and S. D. Evans, Analyst, 2018, 143, 6113–6120 RSC.
- A. F. Palonpon, M. Sodeoka and K. Fujita, Curr. Opin. Chem. Biol., 2013, 17, 708–715 CrossRef CAS PubMed.
- H. Mikami, C. Lei, N. Nitta, T. Sugimura, T. Ito, Y. Ozeki and K. Goda, Chem, 2018, 4, 2278–2300 CAS.
- M. Li, J. Xu, M. Romero-Gonzalez, S. A. Banwart and W. E. Huang, Curr. Opin. Biotechnol., 2012, 23, 56–63 CrossRef CAS PubMed.
- C. Krafft and J. Popp, Anal. Bioanal. Chem., 2015, 407, 699–717 CrossRef CAS PubMed.
- H. K. Yosef, L. Mavarani, A. Maghnouj, S. Hahn, S. F. El-Mashtoly and K. Gerwert, Anal. Bioanal. Chem., 2015, 407, 8321–8331 CrossRef CAS PubMed.
- H. K. Yosef, T. Frick, M. K. Hammoud, A. Maghnouj, S. Hahn, K. Gerwert and S. F. El-Mashtoly, Analyst, 2018, 143, 6069–6078 RSC.
- S. F. El-Mashtoly, H. K. Yosef, D. Petersen, L. Mavarani, A. Maghnouj, S. Hahn, C. Kötting and K. Gerwert, Anal. Chem., 2015, 87, 7297–7304 CrossRef CAS PubMed.
- D. Fu, J. Zhou, W. S. Zhu, P. W. Manley, Y. K. Wang, T. Hood, A. Wylie and X. S. Xie, Nat. Chem., 2014, 6, 614–622 CrossRef CAS PubMed.
- S. F. El-Mashtoly, D. Petersen, H. K. Yosef, A. Mosig, A. Reinacher-Schick, C. Kötting and K. Gerwert, Analyst, 2014, 139, 1155 RSC.
- K. Aljakouch, T. Lechtonen, H. K. Yosef, M. K. Hammoud, W. Alsaidi, C. Kötting, C. Mügge, R. Kourist, S. F. El-Mashtoly and K. Gerwert, Angew. Chem., Int. Ed., 2018, 57, 7250–7254 CrossRef CAS PubMed.
- H. Nawaz, F. Bonnier, P. Knief, O. Howe, F. M. Lyng, A. D. Meade and H. J. Byrne, Analyst, 2010, 135, 3070 RSC.
- H. Nawaz, F. Bonnier, A. D. Meade, F. M. Lyng and H. J. Byrne, Analyst, 2011, 136, 2450 RSC.
- Z. Farhane, F. Bonnier and H. J. Byrne, Anal. Bioanal. Chem., 2017, 409, 1333–1346 CrossRef CAS PubMed.
- Z. Farhane, F. Bonnier, O. Howe, A. Casey and H. J. Byrne, J. Biophotonics, 2018, 11, 1–14 Search PubMed.
- C. Matthäus, T. Chernenko, C. Stiebing, L. Quintero, M. Miljkovi, L. Milane, A. Kale, M. Amiji, S. Lorkowski and V. Torchilin, Confocal Raman Microscopy, Springer International Publishing, Cham, 2018, vol. 66 Search PubMed.
- A. A. van Apeldoorn, H.-J. van Manen, J. M. Bezemer, J. D. de Bruijn, C. A. van Blitterswijk and C. Otto, J. Am. Chem. Soc., 2004, 126, 13226–13227 CrossRef CAS PubMed.
- P. Xu, E. Gullotti, L. Tong, C. B. Highley, D. R. Errabelli, T. Hasan, J.-X. Cheng, D. S. Kohane and Y. Yeo, Mol. Pharm., 2009, 6, 190–201 CrossRef CAS PubMed.
- G. Romero, I. Estrela-Lopis, J. Zhou, E. Rojas, A. Franco, C. S. Espinel, A. G. Fernandez, C. Gao, E. Donath and S. E. Moya, Biomacromolecules, 2010, 11, 2993–2999 CrossRef CAS PubMed.
- T. Chernenko, C. Matthäus, L. Milane, L. Quintero, M. Amiji and M. Diem, ACS Nano, 2009, 3, 3552–3559 CrossRef CAS PubMed.
- T. Chernenko, F. Buyukozturk, M. Miljkovic, R. Carrier, M. Diem and M. Amiji, Drug Delivery Transl. Res., 2013, 3, 575–586 CrossRef CAS PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, CA-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- C. C. Applegate and M. A. Lane, World J. Gastrointest. Oncol., 2015, 7, 184 CrossRef PubMed.
- R. Abdel-Samad, P. Aouad, H. Gali-Muhtasib, Z. Sweidan, R. Hmadi, H. Kadara, E. L. D'Andrea, A. Fucci, C. Pisano and N. Darwiche, Am. J. Cancer Res., 2018, 8, 39–55 CAS.
- A. Kawczyk-Krupka, A. M. Bugaj, W. Latos, K. Zaremba, K. Wawrzyniec, M. Kucharzewski and A. Sieroń, Photodiagn. Photodyn. Ther., 2016, 13, 158–174 CrossRef CAS PubMed.
- A. Whiting and C. A. Ambler, Synthetic retinoid (in cell killing) UK Patent Application No. GB16137127, 2016 Search PubMed.
- D. R. Chisholm, R. Lamb, T. Pallett, V. Affleck, C. Holden, J. Marrison, P. O'Toole, P. D. Ashton, K. Newling, A. Steffen, A. K. Nelson, C. Mahler, R. Valentine, T. S. Blacker, A. J. Bain, J. Girkin, T. B. Marder, A. Whiting and C. A. Ambler, Chem. Sci., 2019, 10, 4673–4683 RSC.
- T. L. Freeman, S. E. Cope, M. R. Stringer, J. E. Cruse-Sawyer, D. N. Batchelder and S. B. Brown, J. Raman Spectrosc., 1997, 28, 641–643 CrossRef CAS.
- H. Abramczyk, B. Brozek-Pluska, J. Surmacki, J. Musial and R. Kordek, Analyst, 2014, 139, 5547–5559 RSC.
- H. Abramczyk, B. Brozek-Pluska, M. Tondusson and E. Freysz, J. Phys. Chem. C, 2013, 117, 4999–5013 CrossRef CAS.
- B. Brozek-Pluska and M. Kopec, Spectrochim. Acta, Part A, 2016, 169, 182–191 CrossRef CAS PubMed.
- B. Brozek-Pluska, A. Jarota, J. Jablonska-Gajewicz, R. Kordek, W. Czajkowski and H. Abramczyk, Technol. Cancer Res. Treat., 2012, 11, 317–331 CrossRef CAS PubMed.
- T. Bhattacharjee, L. C. Fontana, L. Raniero and J. Ferreira-Strixino, J. Raman Spectrosc., 2018, 49, 786–791 CrossRef CAS.
- G. Lajos, D. Jancura, P. Miskovsky, J. V. García-Ramos and S. Sanchez-Cortes, J. Phys. Chem. C, 2009, 113, 7147–7154 CrossRef CAS.
- Y. K. Kim, H. K. Na, S. Kim, H. Jang, S. J. Chang and D. H. Min, Small, 2015, 11, 2527–2535 CrossRef CAS.
- T. Nagy-Simon, M. Potara, A. M. Craciun, E. Licarete and S. Astilean, J. Colloid Interface Sci., 2018, 517, 239–250 CrossRef CAS.
- N. Nwahara, O. J. Achadu and T. Nyokong, J. Photochem. Photobiol., A, 2018, 359, 131–144 CrossRef CAS.
- T. Simon, M. Potara, A. M. Gabudean, E. Licarete, M. Banciu and S. Astilean, ACS Appl. Mater. Interfaces, 2015, 7, 16191–16201 CrossRef CAS PubMed.
- L. Zhao, T. H. Kim, H. W. Kim, J. C. Ahn and S. Y. Kim, Acta Biomater., 2015, 20, 155–164 CrossRef CAS PubMed.
- Z. Zhou, S. Peng, M. Sui, S. Chen, L. Huang, H. Xu and T. Jiang, Colloids Surf., B, 2018, 161, 394–402 CrossRef CAS PubMed.
- A. M. Fales, H. Yuan and T. Vo-Dinh, Mol. Pharm., 2013, 10, 2291–2298 CrossRef CAS PubMed.
- A. M. Fales, B. M. Crawford and T. Vo-Dinh, ACS Omega, 2016, 1, 730–735 CrossRef CAS PubMed.
- A. Farhadi, Á. Roxin, B. C. Wilson and G. Zheng, Theranostics, 2015, 5, 469–476 CrossRef CAS.
- M. Koch, C. Suhr, B. Roth and M. Meinhardt-Wollweber, J. Raman Spectrosc., 2017, 48, 336–342 CrossRef CAS.
- V. Jain, M. C. Biesinger and M. R. Linford, Appl. Surf. Sci., 2018, 447, 548–553 CrossRef CAS.
- N. Ridgway and R. McLeod, Biochemistry of Lipids, Lipoproteins and Membranes, Elsevier Science, 6th edn, 2015 Search PubMed.
- Y. Chen, J. Q. Ren, X. G. Zhang, D. Y. Wu, A. G. Shen and J. M. Hu, Anal. Chem., 2016, 88, 6115–6119 CrossRef CAS PubMed.
- J. Tong, Y. J. Wang, Z. Wang, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2015, 119, 21875–21881 CrossRef CAS.
- D. Ahmed, P. W. Eide, I. A. Eilertsen, S. A. Danielsen, M. Eknæs, M. Hektoen, G. E. Lind and R. A. Lothe, Oncogenesis, 2013, 2, e71 CrossRef CAS.
- J. K. M. Lim and G. Leprivier, Cell Death Dis., 2019, 10, 955 CrossRef CAS PubMed.
- P. Shyu, X. F. A. Wong, K. Crasta and G. Thibault, Biosci. Rep., 2018, 38, 1–20 CrossRef PubMed.
- H. Abramczyk and G. Waliszewska, J. Phys. Chem. A, 1998, 102, 7765–7771 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an01023e |
| This journal is © The Royal Society of Chemistry 2020 |