
Identification and quantification of amino acids and related compounds based on Differential Mobility Spectrometry†
Francis
Berthias
 a,
Yali
Wang
a,
Eskander
Alhajji
a,
Bernard
Rieul
a,
Fathi
Moussa
a,
Jean-François
Benoist
bc and
Philippe
Maître
*a
a,
Yali
Wang
a,
Eskander
Alhajji
a,
Bernard
Rieul
a,
Fathi
Moussa
a,
Jean-François
Benoist
bc and
Philippe
Maître
*a
aUniversité Paris-Saclay, CNRS, Institut de Chimie Physique, 91405, Orsay, France. E-mail: philippe.maitre@universite-paris-saclay.fr
bUniversité Paris-Saclay, Lipides, Systèmes Analytiques et Biologiques, 92296, Châtenay-Malabry, France
cService de biochimie hormonologie, Hôpital Universitaire Robert Debré, APHP, 48 Bd Serrurier, 75019 Paris
First published on 21st May 2020
Abstract
Amino acids and related compounds constitute a class of biomarkers which is analyzed for early diagnosis of metabolic diseases (MDs). Protocols based on liquid chromatography hyphenated to tandem mass spectrometry (LC-MS/MS) are routinely used for MD diagnosis. Our ultimate objective is to evaluate the analytical performance of differential mobility spectrometry (DMS) hyphenated to MS/MS, in the perspective of using DMS-MS/MS as an alternative or complementary method for the topics of emergency in metabolic diagnosis and newborn rapid screening. The aim of the present study is to evaluate the robustness of a DMS-MS/MS protocol for the separation, identification, and quantification of amino acids and related compounds. Performance in terms of peak capacity and separation of isobaric and isomeric species is compared to those using drift tube type ion mobility spectrometry instruments. High reproducibility of the measurement of the DMS compensation voltage (CV) of metabolites shows that this CV parameter, or the corresponding electric field, could be used for application in metabolite identification. Multiple measurements show that the CV value of each AA or related compound is stable over a large period of time (6 months). Potential effects of matrix or concentration of the analytes on the DMS identifier are found to be negligible. Quantification of a selected set of metabolites in human plasmas has been carried out. The method linearity, intra-assay and inter-assay precision, detection limit, quantification limit and trueness analysis were assessed as adequate for both physiological and pathological conditions. Concentration levels of metabolites derived with our DMS-MS protocols were found to be in good agreement with those obtained with routine LC-MS/MS protocols used for the diagnosis of MDs at the Hospital Robert Debré (Paris).
Introduction
Metabolomics-based strategies are routinely used for modern clinical research, allowing for more adequate diagnosis and prognosis.1,2 Quantitative and robust methods3 are required for the analysis of biomarkers of metabolic diseases (MDs) involved in a complex network of metabolic pathways. Amino acids (AAs) and related compounds constitute a class of biomarkers which is routinely analyzed for early diagnosis of MDs.4 Separation of these metabolites within, for example, urine or blood samples, has been initially performed by high-performance liquid chromatography (HPLC), which was coupled to spectrophotometric detection.5 For this detection purpose, postcolumn and precolumn derivatization had to be carried out, leading to time-consuming methods. Nowadays, metabolomics profiling is based on chromatographic separation coupled with tandem mass spectrometry (MS/MS).6 Nuclear magnetic resonance (NMR) is also an important tool, but the main advantage of MS/MS is its sensitivity that requires a minimal amount of biological material.7 Identification of the metabolites at their naturally occurring physiological concentrations is routinely achieved by comparing their fragmentation spectra with standard reference spectra.7 Some major drawbacks of the current methods, such as time-consuming procedures, poor reproducibility of the derivatization, and problems of retention have been shown to be overcome using hydrophilic interaction liquid chromatography (HILIC) for the separation. Rapid quantification of underivatized AAs in plasma using a HILIC column,8 and mixed mode stationary phase and a binary gradient of elution,9 coupled with tandem mass-spectrometry have recently been reported. Despite these improvements providing high degree of selectivity, separation and identification of isomeric metabolites remains a difficult task.2 Additional or alternative dimensions of separation could thus be of interest for the separation of isobaric and isomeric metabolites.10–13Ion mobility spectrometry (IMS) is an interesting alternative for the separation of small molecular weight ions such as ionized metabolites.14 Both time- and space-type IMS separation such as drift-time IMS (DT-IMS) and differential mobility spectrometry (DMS, also often referred to as Field Asymmetric Ion Mobility Spectrometry (FAIMS)),15 respectively, have been successfully used for the separation of metabolites. DT-IMS coupled to high resolution time of flight mass spectrometry has been employed for the profiling of human blood metabolome, and separation of 300 isomeric metabolites within various classes of metabolites could be achieved.16 Time separation of IMS (millisecond) allows for the coupling of IMS-MS/MS to chromatographic separation. An LC-IMS-MS/MS protocol has been recently proposed for metabolomics and lipidomics.17
Efforts toward separation of isobaric and isomeric species led to the development of high resolution instruments and/or methodologies. Within a decade, important progresses have been made with new types of time-18,19 and space-mobility20,21 separation instruments. Significant enhancement of the resolving power can also be achieved by introducing a fraction of He22 or polar molecules23 in the DMS carrier gas, as found in particular for AAs.24,25 Separation of diastereoisomeric chiral compounds such as AAs and peptides has been addressed recently. For the separation of D/L AAs, both derivatization26 and formation of a proton bound dimers27 with a chiral agent have been shown to be successful. Mobility separation of peptides with 4–29 residues including a single D-AA at different residues and locations has recently been reported.28 High resolution DMS has also been recently successfully employed for the separation of isobaric peptides containing one or two methylated arginine residues.29
Both space- and time-separation IMS approaches allow for a reduction of the signal-to-noise ratio. In addition, it has been shown that the number of false positive assignments can be reduced using DMS.30 DMS has also been shown to be more orthogonal to MS/MS than DT-IMS or related time-separation methods, in the case of peptides, for example.20,31 Coupled to MS/MS, it was shown that the coverage achieved with the DMS is comparable to that achieved with the chromatographic methods.32 DMS hyphenated to MS/MS has thus been used for analysis of complex biological fluids such as urine and plasma, and especially for the separation of co-eluting isomers.10,24 DMS-MS/MS workflows for lipid analysis has also been shown to overcome some limitation of LC-MS/MS such as low chromatographic resolution and the convolution of mass spectra from isomeric and isobaric species.12 Nevertheless, “despite several success stories concerning DMS in metabolomics, the application range of this technique is not fully understood. Systematic studies investigating the metabolome selectivity of DMS are required”.32
Our aim is to evaluate the analytical performance of DMS hyphenated to MS/MS for the analysis of AAs and related compounds. More precisely, we will focus on the capacity of DMS for providing an additional metabolite identifier. As recently discussed by Barran and coworkers,33 it has been investigated using DT-IMS approaches, and it has been shown that mobility drift-time,34 or even collisional cross sections (CCS),35,36 could be used for identification of pesticides. This idea of using CCS as identifier was further pursued for metabolites.37–40 Using DMS, the additional identification parameter would be the so-called compensation voltage (CV) which has to be tuned for the transmission of a specific metabolite ion.
We first report on the analysis of the 20 common AAs, and the performance in terms of peak-to-peak resolution and peak capacity is compared to those using drift tube type IMS instruments,13–18 in particular for the separation of isobars and isomers. Potential effects of matrix or concentration of the analytes on the CV values are also evaluated, and found to be negligible. An interesting probe of the robustness of the method was also provided by the stability of the CV position for series of DMS-MS experiments carried over 6 months, during which the DMS device was often removed and replaced.
Isotopically labelled internal standards have been used for the quantification41 of a selected set of metabolites in human plasmas. The method linearity, intra-assay and inter-assay precision, detection limit, quantification limit and trueness analysis were found to be adequate in both physiological and pathological conditions. Concentration levels of metabolites derived with our DMS-MS protocols were found to be in good agreement with those obtained with routine LC-MS/MS protocols used for the diagnosis of MDs at the Hospital Robert Debré (Paris).
Experimental methods
Reagents
Methanol and water were used as solvent (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, vol:vol) and acidified with formic acid (0.5%). Methanol was purchased from Merck KGaA (Darmstadt, Germany) and formic acid from Fisher Scientific (Hampton, New Hampshire, USA), both are reagent grade. Deionized water is obtained using Millipore Direct-Q 3 UV (Lilipore corporation, Burlington, Massachusetts, USA). Certified graduated pipettes were used throughout for the sample preparation.
:50, vol:vol) and acidified with formic acid (0.5%). Methanol was purchased from Merck KGaA (Darmstadt, Germany) and formic acid from Fisher Scientific (Hampton, New Hampshire, USA), both are reagent grade. Deionized water is obtained using Millipore Direct-Q 3 UV (Lilipore corporation, Burlington, Massachusetts, USA). Certified graduated pipettes were used throughout for the sample preparation.
Mixture of reference standards
A standard mixture made of 33 AAs and related compounds as listed in Table S1† (left column) was used for calibration. This mixture is based on a commercial AA standard solution (Type B, AN-II purchased from FUJIFILM Wako Pure Chemical Industries, Osaka, Japan). It was complemented by glutamine (Gln), asparagine (Asn), tryptophan (Trp), pipecolic acid, alloisoleucine (Ail), and sulfocysteine which were purchased from Sigma-Aldrich (St Louis, Missouri, USA). Calibration curves were obtained using 6 calibration concentration levels in plasma.Mixture of internal standards
A mixture of stable isotope internal standards (IS) was made using reference compounds from Eurisitop (Saint Aubin, France). The list of these IS as well as their final concentrations in the samples is given in Table S1† (right column). This IS mixture was used for the calibration and quantification protocols.Samples and subjects
Blood samples were obtained from patients suspected of MD based on routine diagnosis. Blood was collected by venous puncture into heparin containing tubes. In practice, blood was centrifuged rapidly after collection. Plasma was subsequently deproteinized by adding sulfosalicylic acid 30% (10:1 v:v) and then stored at −20 °C. Just before analysis, samples were diluted in acidified water:methanol (50:50 v:v) solution, and 20-fold diluted plasma samples were used for electrospray injection.
Four plasma samples (P1, P2, P3, and P4) not specifically collected for validation of this study were analyzed anonymously. The quality control P1 plasma is a pool of plasmas taken from healthy volunteers. Plasma P2 and P3 were collected from patients with Phenylketonuria and Tyrosinemia, respectively. P4 is a random plasma.
Plasma samples were collected for diagnosis purpose as part of the patient's routine care. Informed consent was obtained from patients for the use of leftover samples for analytical developments. The study was approved by the ethics committee of Robert Debré University Hospital (Paris, France), and in compliance with French Public health regulations (Code de la Santé Publique – Article L1121-3, amended by Law no 2011–2012, December 29, 2011 – Article 5).
Concentration of the AAs of interest in these plasmas has been quantified by a liquid chromatography tandem mass spectrometry (LC-MS/MS) method validated according to the ISO 15189 standards. These quantification experiments were performed in an accredited university hospital laboratory where MD diagnoses are routinely performed. In brief, after butylation, compounds are separated by reverse phase liquid chromatography, then ionized by electrospray in positive mode and identified by multiple reaction monitoring (MRM). Concentrations in samples and corresponding uncertainties for the molecules are given in Table S2.†
Differential mobility spectrometry (DMS) coupled to MS/MS instrument
DMS-MS experiments were carried out using a modified Esquire 3000+ quadrupole ion trap mass spectrometer (Bruker-Daltonics, Bremen, Germany) featuring an ElectroSpray Ionization (ESI) source.42 The setup is based on the one developed by Glish and coworkers.43 Our home-built DMS device has been successfully used for the separation and identification of isomers of alanine44 and aminobutyric acids.45 The DMS housing is mounted between the ESI emitter and the glass transfer capillary of the mass spectrometer. It is composed of two parallel electrodes (0.7 × 6 × 20 mm for gap height, width, and length, respectively) between which ions are transported from the atmospheric pressure to a ∼10−3 mbar source region pressure of the mass spectrometer through the glass transfer capillary. In order to enhance the transmission of ions, the standard glass transfer capillary has been replaced with a custom flared glass transfer capillary.33,34 The DMS cell is designed such as to replace the spray shield. The N2 desolvation gas is redirected through the outer housing of the DMS device in such a way that part of the N2 flow (∼0.9 l min−1) is used to transport the ions between the electrodes towards the glass capillary. An NIGen LCMS 40-1 (Claind, Tremezzina, Italy) N2 generator was used to produce the N2 gas with a purity up to 99.99%. Since ion mobility is very sensitive to the composition of the carrier gas, a custom built chamber has been built to accommodate the DMS device, and allowing to adjust the position of the ESI nebulizer needle with respect to the entrance of the DMS device.Identical positive ESI conditions have been used throughout. The voltage of the ESI emitter was set to 4.5 kV and the entrance glass transfer capillary was grounded. The flow rate of the sample was 40 μL h−1.
DMS allows for space separation of the ions, and the working principles can be found, for example, in a textbook46 and a review.15 In practice, a bisinusoidal waveform is applied between the electrodes thus creating an alternating electrical field perpendicularly to the direction of the carrier gas flow. This asymmetric radiofrequency is generated by applying a sinusoidal waveform at 1.7 MHz to one of the DMS electrodes. A second sinusoidal waveform at the second harmonic, phase shifted by approximately 90°, and at 50% of the amplitude of the first sinusoidal waveform is applied to the second electrode. The zero-to-peak voltage (V0p) is referred to dispersion voltage (DV). Ions are transmitted through the DMS device only if the transverse displacements under low and high electric field offset one another. Transmission of ions of interest can be achieved by applying a DC offset to one DMS electrode, referred to as compensation voltage (CV). DMS-filtered ions are then transmitted to the ion trap where they are accumulated before being subjected to MS/MS sequences.
Considering the gap between the electrodes (0.7 mm), and assuming that the carrier gas is near atmospheric pressure and at a temperature around 70 °C (measured temperature of the electrodes using a thermal camera), the reduced electric field value corresponding to a voltage of 1800 V can be estimated to ∼120 Td. In the following, the DMS spectra correspond to extracted ion chromatogram plotted as a function of the CV value, where a 1 V CV value corresponds to an electric field of 1.43 kV m−1 or ∼0.07 Td.
DMS spectra: data processing
DMS spectra are generated by plotting extracted ion chromatograms (XIC) as a function of CV value, which is scanned using a 0.1 V increment. DMS peak centroid (CV) and full width at half-maximum (FWHM) are extracted by fitting each DMS spectrum using Gaussian functions. The fitting procedure is performed using a homebuilt software based on SciPy libraries. An optimization algorithm based on a sequential least squares programming (SLSQP) method is used for the Gaussian fits.DMS spectra of the components of the standard mixture and dispersion plots of individual AA
Unless otherwise stated, DMS spectra presented here were recorded with a DV value set to 1.8 kV. All CV values corresponding to optimal transmission of a particular metabolite given in the text refer to these conditions.Dispersion plots, i.e. evolution of CV value for the optimal transmission of each AA as a function of the DV value, were recorded. DMS spectra were recorded at different DV values ranging from 600 to 1800 V. Solutions of AAs were prepared in methanol:water solvent mixture acidified with formic acid (0.5%) at a final concentration of 100 μM.
Calibration curve and protocol for quantification of metabolites in plasma
The set of standards of AAs and related compounds (Table S1†) was spiked into the P1 plasma at six dilution factors spanning 2 orders of magnitude of concentration (Table S3†). Except for pipecolic acid and allo-isoleucine, which are found at very low concentrations in plasma, the dilution levels were chosen so that the pathophysiological AA concentrations in analyzed plasma lie in the range of the spiked P1 solutions.In practice, the calibration solutions spiked in the P1 plasma sample at a final 1/20 dilution factor were prepared as follow. Following the deproteinization with sulfosalicylic acid, 13.8 μL of the supernatant was mixed with 25 μL of the IS mixture, and diluted in a water:methanol (50:50) solution containing standards and 1.25 μL of formic acid to reach a total volume of 250 μL.
Linear calibration curve parameters were obtained from the plot of the AA/related IS peak area ratio versus the nominal AA concentration. The CV, FWHM, and DMS peak area values were derived from the above mentioned fitting procedure.
Quantification of AAs and related compounds in four plasma samples (P1–P4) was performed. The sample preparation protocol is similar to that used for calibration. 13.8 μL of deproteinized plasma was added to 25 μL of IS mixture and diluted in acidified water:methanol to a final volume of 250 μL. In order to check for potential carry-over, a blank run was systematically performed between two plasma runs. Concentrations of unknown samples were calculated from their corresponding area ratios using the corresponding calibration curve.
Results and discussion
The aim of this work is twofold. First, it is to evaluate whether the CV parameter could provide a means for identification of metabolites. Our second goal is the absolute quantification of AAs and related compounds for targeted metabolomics analysis of plasma samples from healthy or MD patients. In this perspective, performance of DMS for separation of AAs is first evaluated, with a special focus on peak capacity and isobars/isomers resolution. Then, compensation voltage value of the AAs and related compounds within a mixture of standards are determined, in both water:methanol solution and plasma. Based on good robustness and repeatability of measurements, calibration results are then presented. Finally, quantification of a set of AAs and related compounds relevant for the diagnosis of MDs is presented for four sets of plasma samples.
DMS separation of the 20 common AAs
In order to probe the effect of the DV value on peak capacity and resolution, DMS spectra were recorded at DV values ranging from 0.6 kV to 1.8 kV. The set values of flow rate of the N2 desolvation gas and its temperature are 6.5 L min−1 and 220 °C, respectively. This was found to provide a good balance between DMS resolution and ion transmission. Results of separation are reported in Fig. 1. DMS dispersion plots, i.e. evolution of CV as a function of DV for each individual AA, are reported in Fig. 1a. | ||
| Fig. 1 Differential ion mobility data for 20 protonated AAs recorded at different dispersion voltage (DV) using N2 as carrier gas. (a) DMS dispersion plots, or evolution of CV as a function of DV; DMS spectra for each individual AA at DV = 1.4, 1.6, 1.8 kV are given in panels (b), (c), and (d), respectively. A CV value of 1 V corresponds to an electric field of 1.43 kV m−1 or ∼0.07 Td. To facilitate visualization, each DMS peak is individually normalized. | ||
Three classes of AAs can be distinguished based on the evolution of CV as a function of DV (Fig. 1a). These three types of behaviors have been observed for other systems47 and interpretations have been proposed15,46 based on the interactions between ions with the carrier gas. These three types of ions, often referred to as A, B, or C ions, are observed for AAs under the present DMS conditions.47 Protonated asparagine, aspartic acid, threonine, cysteine, proline, serine, alanine and glycine are associated to type A ions, for which ion-molecule clustering and declustering are supposed to occur at low and high electric field, respectively. Type C refers to ions with a decreasing mobility coefficient K as a function of the electric field strength. These ions are supposed to experience hard sphere type collisions with the carrier gas. Protonated arginine and tryptophan were found to be the only two systems of type C under our experimental conditions. For type B, the ion mobility initially increases, reaches a maximum and decreases for larger DV values. As a result, the DMS dispersion curve presents a minimum. It has recently been proposed that the minimum CV values can be correlated to ion-solvent binding energy.48 Protonated lysine, histidine, phenylalanine, tyrosine, methionine, glutamine, isoleucine, leucine, glutamic acid and valine are type B ions. Note that some of these ions have been already found to behave as B type ions, such as protonated leucine,49 and protonated glutamic acid.50
DMS spectra of each individual AA for three DV values, 1.4, 1.6, and 1.8 kV, are given in Fig. 1b–d, respectively. The width (FWHM) of the DMS peaks does not vary with the DV value. A nearly linear increase (R2 = 0.88) of the FWHM value with m/z is observed, the values for the AAs ranging from 0.52 (Gly) to 0.88 V (Trp).
A plot of the CV values of the set of AAs, and also some related compounds which can be found in plasmas, as a function of m/z is given in Fig. 2. Overall, the observed trend is quite similar to that observed for the collision cross section (CCS) values (Å2) derived using time-separation IMS instrument.17,33,38,51 With increasing m/z, the CV value increases from A-, to B-, and C-type protonated AAs ions.
 | ||
| Fig. 2 CV and CCS as a function of m/z for a selected set of AAs and related species (see list in Table S4†). CV values (in V), 1 V corresponding to an electric field of 1.43 kV m−1 or ∼0.07 Td. CCS (in Å2) are taken from (a) ref. 38, (b) ref. 33, (c) ref. 17, and (d) ref. 51. | ||
Separation of AAs: DMS versus IMS peak capacity
Separation of protonated AAs has been investigated using space24,25 and also time separation based ion mobility instrument such as drift-tube52,53 or radio frequency-confining drift cell, i.e. with a Traveling Wave Ion Mobility Spectrometer (TWIMS).51 In the latter study, the effect of the nature (He, Ar, N2, CO2, and N2O) of the drift gas on both peak-to-peak resolution (Rpp) and peak capacity (Pc) has been evaluated. Rpp and Pc are defined in eqn (1) and (2), where CVmin and wmin (CVmax and wmax) are the CV peak position and full width at half maximum (FWHM) values, respectively, corresponding to the minimum (maximum) CV value.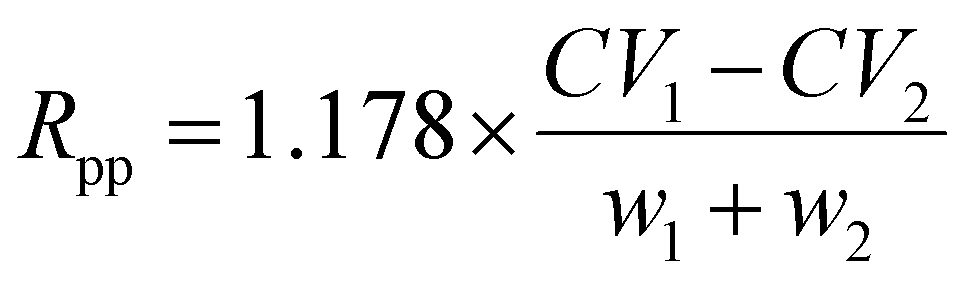 | (1) |
 | (2) |
As can be seen in Fig. 1, Pc increases with DV, from Pc = 18.1 at 1.4 kV to Pc = 23.8 at 1.8 kV. For more details, the evolution of Pc (black curves) and Rpp (blue curve) as a function of the DV value is shown in Fig. S1.† The best separation performance is observed for DV value of 1.8 kV except for few AA pairs. For example, protonated lysine (B type) and protonated tryptophan (C type) are poorly separated at 1.8 kV (Rpp = 0.1), while a better separation is observed at lower DV (Rpp = 0.48 at DV = 1.2 kV). In a consistent manner, the fraction of the pairwise combinations of protonated AAs which are baseline resolved (Rpp = 1.5) increases from 29% at DV = 1 kV to 81% at DV = 1.8 V. It can thus be concluded that these Rpp and Pc values are very close to those estimated (Pc ∼ 15 and ∼80% resolved AAs (Rpp > 1.0)) for a high performance drift tube instrument using N2 as a carrier gas.51
CV as metabolite identifier: no matrix, nor concentration effects
DMS data recorded for a selected set of AAs and related metabolites in two matrices, plasma and water:methanol, are reported in Fig. 3. 2D plot of m/z as a function of CV values for a mixture of standards diluted in water:methanol is given in Fig. 3a. The corresponding 2D plot for the pool of plasma sample (P1) containing IS is given in panel b. In each case, the mass spectrum integrated over the whole CV range is given on the right hand side in panels (d) and (e), respectively. XIC for a selected set of m/z values specific of molecules of interest for plasma study are given in Fig. 3c. The blue and red lines correspond to standards in water:methanol solution and plasma sample, respectively. All the curves are normalized to maximum and smoothed using a rolling average over two points.
 | ||
| Fig. 3 DMS data recorded for a set of standards (AAs and related compounds) in water:methanol and for a plasma sample (P1) containing IS. The corresponding 2D plots, with m/z as a function of CV, are given in panels (a) and (b), respectively. Extracted ion chromatograms (XIC) of a selected set of metabolites of interest are given in panel (c) where data for the plasma (red) and standards in water:methanol solution (blue) are superimposed. Mass spectra integrated over the CV range corresponding to 2D plots of panels (a) and (b) are given in panels (d) and (e), respectively. Peak assignment in the XIC panel (c) is provided. 3-Letter nomenclature symbols are used for AAs, as well as for Aminoadipic acid (AAA), Aminobutyric acid (ABA), Citrulline (Cit), Ornithine (Orn), and Sarcosine (Sar). Peaks corresponding to post-DMS (in-source) fragments or also first and second isotopic peaks are labelled with an “#” and an “&” symbol, respectively. Unassigned DMS peaks are symbolized by a “*” mark. A CV value of 1 V corresponds to an electric field of 1.43 kV m−1 or ∼0.07 Td. | ||
As expected, a higher density of contributions both in CV and m/z dimensions is observed in plasma. Nethertheless, the 2D plots of standards and plasma show common features. As can be seen in Fig. 3c, no significant matrix effect on CV position nor on the DMS peak width are observed. This is confirmed through the comparison of the CV values determined for a selected set of metabolites in a plasma or diluted in water:methanol solution (Table 1). The standard deviation (SD) derived using a set of 36 plasma measurements is smaller or equal to 0.1 V, except for glycine (0.2 V), alanine (0.3 V), and serine (0.2 V). Considering the SD values, there is an excellent agreement between the two sets of values, and thus no evidence of matrix effects.
:methanol solution. Mean CV and the corresponding standard deviation (SD) values are given in Volt unit. Day-to-day reproducibility of the DMS peaks and correspondence between CV and electric field values are given in Table S6.†
| Plasma | Water:MeOH |
|||
|---|---|---|---|---|
| CV | SD | CV | SD | |
| Glycine | −10.7 | 0.2 | ||
| Alanine | −7.6 | 0.3 | −7.9 | 0.3 |
| Serine | −6.6 | 0.2 | −6.8 | 0.2 |
| Proline | −5.3 | 0.1 | −5.4 | 0.2 |
| Valine | −3.7 | 0.1 | −3.9 | 0.2 |
| Threonine | −4.8 | 0.1 | −4.9 | 0.2 |
| Asparagine | −4.2 | 0.1 | −4.3 | 0.1 |
| Glutamine | −2.7 | 0.1 | −2.8 | 0.1 |
| Lysine | −0.3 | <0.1 | −0.3 | 0.2 |
| Methionine | −2.3 | 0.1 | −2.4 | 0.2 |
| Histidine | −1.5 | 0.1 | −1.5 | 0.2 |
| Phenylalanine | −1.3 | 0.1 | −1.4 | 0.2 |
| Arginine | 0.9 | <0.1 | 1 | 0.2 |
| Citrulline | −0.7 | <0.1 | −0.7 | 0.2 |
| Tyrosine | −1.4 | 0.1 | −1.4 | <0.1 |
| Tryptophan | 0.2 | 0.1 | 0.2 | 0.1 |
Concentration effects on DMS peak width and position could also be expected. DMS peaks were characterized for a set of AAs and related compounds when spiked in plasma P1 using six dilution factors spanning two orders of magnitude of concentration (1–100 μmol). Overall, both FWHM and position of DMS peaks were found constant over these experiments. For the molecules of interest here, the mean value of CV standard deviation was 0.1 V. The maximum CV standard deviation (0.18 V) was found for protonated glycine. Similarly, the peak width was also found not to be correlated with the concentration value. The mean FWHM value was 0.72 V, and the standard deviation was 0.1 V in the concentration range examined.
A full assignment of the XIC is out of the scope of this study, and unassigned peaks are labeled with a “*” symbol in Fig. 3c. DMS peaks associated to positive CV values, however, have to be analyzed carefully. It is indeed well-known that protonated homo- and hetero-dimers are typically transmitted at positive CV values.54 And these dimers are partially fragmented in the source region, at the exit of the glass capillary, just downstream of the DMS device.54 Due to this so-called “post-DMS fragmentation”, their corresponding protonated monomers are observed at the corresponding transmission CV of the dimers.55 Hence, in addition to the DMS peak of a given protonated monomer in a given m/z XIC, peaks are also observed at positive CV values corresponding to maximum transmission of their proton bound dimers. This is evidenced in Fig. 3a and b where the 2D plots in the positive CV range do not significantly depend on the m/z value.
In addition, a slight fragmentation of some protonated monomer of standards may also occur. This is the case for phenylalanine (m/z 166 → 120), and methionine (m/z 150 → 104) as shown in Fig. 3c with “#” marks. These fragments can be easily assigned because they are generated through post-DMS dissociation and are thus found at the CV position of the corresponding precursor ion. Finally, one must also pay attention to isotopic peaks (see “&” in Fig. 3c). In the case of arginine (MH+m/z 175), which is abundant in the plasma and observed at 0.95 V, a significant contribution associated with the first isotopic peak is also observed in the m/z 176 XIC. Similarly, contributions of the first and second isotopic peaks of leucine/isoleucine are found in the m/z 133 and 134 XIC, respectively.
Separation of isobaric and isomeric AAs and related species
Considering the limited mass-resolution of our quadrupole ion trap instrument, the separation of isobaric species is an important issue. The performance of DMS relative to drift-tube instruments can be assessed by inspecting Fig. 2 where CV and CCS are plotted as a function of m/z. The corresponding values for a selected set of isobaric and/or isomeric species discussed below are given in Table S4.†Apparent disagreements in terms of DMS peak position are in fact the result of the different relative concentrations of (partially) unresolved isomeric compounds in the two matrices (water:methanol and plasma). This is the case for example for the broad DMS peak observed near −7.5 V for m/z 90 ions (Fig. 3c). It results from contribution of α- and β-alanine,44 the latter being observed at slightly more negative (−0.3 V) CV value than the former. The difference between the two m/z 90 XIC near −7.5 V (Fig. 3c) is thus simply due to the fact that β-alanine is only found as trace amounts in the plasma while it is a component of our standard mixture. More important is the issue of the separation of α-alanine and sarcosine which cannot be distinguished based on their fragmentation mass spectra. These two ions were found to be baseline separated, the sarcosine peak being observed at −10.6 V. This DMS peak observed for the standard mixture, but not for the plasma, where sarcosine is expected at a very low concentration.
A similar issue occurs in the XIC at m/z 132 (Fig. 3c). Indeed, leucine/isoleucine have similar concentrations in the standard mixture, but not in the plasma. As a result, the shape of the unresolved DMS peak in the XIC at m/z 132 (Fig. 3c) changes accordingly. In addition, note that Allo-isoleucine (Ail) is also included in the standard mixture, while it is not expected in plasma P1 which is from healthy volunteers. Measurements on each individual Leu isomer were performed, and the CV values of Leu, Ile, and Ail were found to be −3.0, −2.8, and −2.9 V, respectively. Better peak-to-peak resolution is obtained with isobaric cis- and trans-hydroxyproline, which were found to be transmitted at −3.6 and −4.0 V, respectively.
For m/z 133, three baseline separated peaks were observed at −4.0, −2.6, and −1.4 V. Based on measurements on individual metabolites, the peaks at −4.1 and −1.3 V can be assigned to Asn and Orn, respectively. As can be seen in Fig. 2, the DMS resolution is better than that obtained with a drift-tube instrument. For example, Orn and Asn measured CCS were found to be 130 and 132 Å2, respectively.38
Similarly, protonated glutamine and lysine were found to be baseline resolved and found at −2.7 and −0.3 V, respectively. Their measured CCS, however, were found very close (133 and 132 Å2, respectively).38 DMS performance in terms of separation of isomeric species can be illustrated with 1- and 3-methylhistidine. DMS measurement on individual isomers show that they are found at −0.2 and 0.4 V.
CV as metabolite identifier: reproducibility and robustness
The robustness of the method in terms of accuracy on the peak position over time is also an important issue in the perspective of using CV as a metabolite identifier. Fig. 4 illustrates the stability of DMS peak position for a selected set of six AAs over a period of 6 months. These DMS spectra include experiments performed with plasma (filled circles) and standards in water:methanol solutions (open circles). The mean CV and associated standard deviation values of the DMS peaks given in Fig. 4 can be found in Table 1. The maximum standard deviations were found for serine (0.15 V) and proline (0.12 V) while other standard deviation values are smaller than the CV scan step (0.1 V).
 | ||
| Fig. 4 Selected DMS spectra recorded for serine (Ser), proline (Pro), glutamine (Gln), phenylalanine (Phe), lysine (Lys) and arginine (Arg) over a period of 6 months. Experiments were performed with plasma samples (filled circles) and standards of AAs and related compounds in water:methanol solutions (open circles). The mean CV and associated standard deviation values are given in Table 1. A CV value of 1 V corresponds to an electric field of 1.43 kV m−1 or ∼0.07 Td. | ||
DMS specificity
The filtering effect of DMS over a short range of m/z values containing protonated proline (m/z 116) and valine (m/z 118) is illustrated in Fig. 5. The mass spectrum of a pool of plasmas (P1 sample) recorded with RF off and CV = 0 (DMS Off or transparent mode) is given in black in Fig. 5a. A significant reduction of background signal is observed when DMS is turned on with the CV value set to −3.7 and −5.3 V for optimal transmission of protonated valine (blue) and proline (red) as illustrated in Fig. 5. The mass spectra in panel (b) are those recorded at the optimal CV value, while mass spectra integrated over the targeted DMS peak are given in panel (c). | ||
| Fig. 5 Mass spectra recorded with DMS-off (black) and DMS-on (color) for a pool of plasmas sample P1. (a) DMS off and CV set at 0 V. (b) CV set at −5.3 V (red) and −3.7 V (blue) for optimal transmission of protonated proline (m/z 116) and valine (m/z 118), respectively. (c) Mass spectra integrated over the DMS peaks of protonated valine (dark blue) and proline (dark red). A CV value of 1 V corresponds to an electric field of 1.43 kV m−1 or ∼0.07 Td. | ||
The DMS specificity is also illustrated in Fig. 5. When the DMS filtering is turned on, only the targeted ions are transmitted, as well as their corresponding IS, protonated Val-d8 and Pro-d7. Note that the maximum transmission of these IS is achieved at a CV value slightly different from that of the corresponding standard. This explains the fact that the signal at m/z 123 and 126 is lower when the DMS filtering mode is turned on than under transparent mode. The MS instrument parameters were tuned so as to avoid saturation of the trap. This is the reason why the peak intensity of protonated valine (m/z 118) and proline (m/z 116) are the same using either DMS-off or DMS-on mode. If the mass spectra integrated over the DMS peaks are considered (Fig. 5c), a ∼5-fold intensity increase is observed as compared to DMS off. A similar discussion the DMS specificity has been reported by Chen and coworkers in a study of two radiation biomarkers in human and non-human plasmas using a triple-quadrupole instrument.56
Validation of the calibration method
Calibration curves were constructed by plotting the ratio of the peak area of each reference compounds and its corresponding IS versus the corresponding reference compound concentration. Six concentration levels of the reference compounds spiked in P1 plasma were used. The results given in Table S5† were derived from 4 independent experiments performed over 6 months. In most cases, linear response profiles with R2 > 0.99 were obtained in the concentration ranges of 1–300 μM after sample dilution. This concentration range covers the expected pathophysiological range for this class of molecules.Limit of detection and limit of quantification
Limits of detection (LOD) and of quantification (LOQ) were derived from the linear regressions discussed above, where LOD can be defined as 3 × SDb/S, where S is the slope and SDb is the standard deviation of the intercepts of the regression lines. This method is an alternative to the well-known signal-to-noise method when the MS analysis method does not involve background noise.57 LOD values for targeted AAs and related compounds are given in Table S5.†Quantification of AA levels in plasmas
Plasma samples were analyzed, and concentration levels of metabolites of interest are presented in Fig. 6. Results are given for 13 AAs and related compounds. Quantification results obtained with LC-MS/MS for a purpose of diagnosis of MD at the hospital are given in blue bars. Results obtained with the present method are given in red bars. Inter-day quantification experiments were repeated at least three times. A calibration experiment was performed before each quantification experiment. | ||
| Fig. 6 Concentration (μM) of 13 selected metabolites in three plasmas samples: quality control pool of plasmas P1 (a), and plasmas P2 and P3 collected from patients with Phenylketonuria (b) and Tyrosinemia (c), respectively. DMS-MS experiments (red bars) were performed on different days, and standard deviations are indicated as error bars. LC-MRM data are shown as blue bars, and error bars correspond to RSD calculated over 5 months. | ||
Concentration levels determined for plasma P1 from healthy patients are given in panel a. Overall, our method performs well compared to LC-MS/MS except in the case of Ala. This issue has been discussed above, and is probably related to the presence of β-alanine in our standard mixture used for the calibration procedure. One can also expect similar issue for the quantification of leucine, isoleucine, and allo-isoleucine (not presented). While the two formers can be separated using helium-rich carrier gas,22 these three isomers are poorly separated using N2 as a carrier gas.
Error bars for the LC-MS/MS protocol were derived for each compound from the corresponding inter-day standard deviation (SD) of an equivalent concentration calculated from plasma controls daily analyzed over a period of 5 months. For the present DMS-MS method, error bars correspond to standard deviations over the series of inter-day experiments. Taking into account the SD values, whose mean value is 18%, there is a good quantitative agreement between the two approaches in 77% of the cases. As already discussed, concentration of alanine cannot be accurately determined. In the cases of lysine and methionine, concentrations are slightly overestimated using our DMS-MS approach as compared to LC-MS/MS.
Inter-day quantification experiments were also carried out on two plasmas from MD patients in order to analyze AA profiles for diagnosis of known MD. Results are given for plasmas P2 and P3 (see panels b and c, respectively) from patients with Phenylketonuria and Tyrosinemia diseases, respectively. Overall, qualitative observations are the same as for plasma P1. More importantly, the concentration increase of the biomarker for each MD disease determined by the DMS-MS method is in excellent agreement with that obtained upon LC-MS/MS diagnosis. In the case of Phenylketonuria P2 plasma, phenylalanine concentration was calculated at 449 ± 8 μM with our method, compared to 423 ± 24 μM with an LC-MS/MS protocol used in routine for MD diagnosis. In the case of the plasma P3, tyrosine concentration was found to be 448 ± 25 μM with DMS-MS, compared to 436 ± 24 μM with LC-MS/MS.
Intra-day quantification experiments were performed on random plasma (P4). The concentration levels determined by our method and LC-MS/MS are given in Fig. 7. AAs and related metabolites profiles were analyzed 6 times, and a blank was recorded between two plasma analyses. Overall, there is a good qualitative agreement between the two sets of concentration derived by the two methods. Compared to the pool of plasmas (P1), a concentration increase of 4 out of the 13 selected metabolites (Lysine, threonine, tyrosine, methionine) is found with DMS-MS, in good agreement with LC-MS/MS. In the case of Met, however, the increase is slightly overestimated using the DMS-MS protocol. For the three other biomarkers (Lysine, threonine, tyrosine), there is a good quantitative agreement between the DMS-MS results (363 ± 91, 385 ± 73, 205 ± 45, respectively) and the LC-MS/MS ones (397 ± 24, 412 ± 26, 184 ± 10, respectively).
 | ||
| Fig. 7 Concentration (μM) of selected metabolites in random plasma (P4). DMS-MS data (red bars) correspond to the mean value derived from 6 intra-day experiments. Standard deviations are indicated as error bars. LC-MRM data are shown as blue bars, and error bars correspond to RSD calculated over 5 months. | ||
The CV values of the different metabolites were found stable. The mean value of the CV standard deviations was 0.05 V, lower than the overall value (0.1 V) derived over 6 month experiments. A similar value (0.05 V) was found for the mean standard deviation value of the IS. It is worth to note that the CV positions of standard metabolites are found to be slightly different from that of their corresponding IS. The latter were (almost) systematically found to be more positive by 0.09 V (mean value). Such isotopic shifts have been observed by Guevremont and coworkers for small ions such as 37Cl− and 35Cl−.58 In recent studies performed on high-definition DMS by Shvartsburg and coworkers, these isotopic CV shifts have been found to allow for identification of isomers of dichloroanilines,59 and to be structurally informative for dibromoanilines and tribromoanilines.60
Conclusions and perspectives
We herein provide evidence that DMS hyphenated to an MS/MS instrument is a valuable alternative approach for the separation, identification, and quantification of AAs and related compounds. As retention time in LC-MS/MS, compensation voltage (CV) value allowing for the maximum transmission of a specific metabolite could allow for its identification. It is thus important to stress that the position of the DMS peak position values of metabolites of interest do not depend on concentration and/or matrix effects. Variations of CV positions were found to be stable within ±0.1 V, as compared to peak width (FWHM) of 0.72 V. For a set of AAs and related compounds, we show here that CV values are reproducible over a large period of time (6 months). The proposed DMS method itself is robust since these experiments were carried out on an open MS/MS platform, and the DMS device thus had to be mounted and unmounted several times.DMS could thus be an alternative separation method for MS/MS based targeted metabolomics for the quantification of biomarkers. In particular, two applications dedicated to MD diagnosis could be performed in order to address the topics of emergency in metabolic diagnosis and newborn rapid screening. Biomarker analysis time could be significantly reduced using DMS-MS/MS compared to LC-MS/MS approaches. Even using an ion-trap as in the present study, a full CV scan can be performed within ∼5 minutes. Furthermore, a full CV scan is no longer needed once the CV values are known for the targeted metabolites/biomarkers. Quantification of these biomarkers could be achieved by recording mass spectra at selected identified CV values. Finally, as discussed in the recent literature DMS can also be hyphenated to LC-MS/MS.11,61–64 Two dimensional separation LC-IMS-MS/MS method allows for resolving chromatographic peaks, and an increase of signal-to-noise ratio.11
In the perspective of using DMS for identification purpose, the addition of selected organic molecules to the carrier gas is undoubtedly the approach of choice for improving DMS specificity. Drastic improvement of ion, and especially isomeric ions, separation has beenachieved by adding alcohol or other polar and/or proton-acceptor molecules in the buffer gas,23,65–67 and in the case of AAs in particular.24,25 A systematic study of the effect of different modifiers on the separation of AAs and related compounds is under study in our group. Using methanol as an added modifier to N2 as carrier gas, we already showed baseline separation of isomeric ions could be obtained in the case of sarcosine, α- and β-alanine,44 as well as for the α-, β-, and γ-aminobutyric acid isomers.45 As reported in the literature, adding of a fraction of He to the carrier gas20,22,29 or increasing the waveform amplitude21 should also be considered for high resolution DMS.
Quantification of AAs and related compounds were achieved using our 3D quadrupole ion trap despite the intrinsic limitations of these instruments in terms of sensitivity and linear dynamic range. However, FT-ICR or QIT instruments are interesting for the coupling with IR lasers for DMS-peak assignment using IRMPD spectroscopy.42,44,45 Ultimately, however, faster quantification will be performed with DMS hyphenated to a triple quadrupole instrument.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Agence Nationale de la Recherche (ANR-19-CE29-0016-01, ANR-10-LABX-0039) is gratefully acknowledged. YW would also like to acknowledge the (CSC) for China Scholarship Council (CSC) program for a full scholarship. EA is grateful for the support of the “Programme national d'aide à l'Accueil en Urgence des Scientifiques en Exil” (PAUSE) from the Collège de France.References
- C. D. Chouinard, M. S. Wei, C. R. Beekman, R. H. J. Kemperman and R. A. Yost, Clin. Chem., 2016, 62, 124–133 CrossRef CAS PubMed.
- I. Kohler, A. Verhoeven, R. J. E. Derks and M. Giera, Bioanalysis, 2016, 8, 1509–1532 CrossRef CAS PubMed.
- E. Theodorsson, Bioanalysis, 2012, 4, 305–320 CrossRef CAS PubMed.
- A. Sreekumar, L. M. Poisson, T. M. Rajendiran, A. P. Khan, Q. Cao, J. D. Yu, B. Laxman, R. Mehra, R. J. Lonigro, Y. Li, M. K. Nyati, A. Ahsan, S. Kalyana-Sundaram, B. Han, X. H. Cao, J. Byun, G. S. Omenn, D. Ghosh, S. Pennathur, D. C. Alexander, A. Berger, J. R. Shuster, J. T. Wei, S. Varambally, C. Beecher and A. M. Chinnaiyan, Nature, 2009, 457, 910–914 CrossRef CAS PubMed.
- D. H. Spackman, W. H. Stein and S. Moore, Anal. Chem., 1958, 30, 1190–1206 CrossRef CAS.
- M. M. Khamis, D. J. Adamko and A. El-Aneed, Mass Spectrom. Rev., 2017, 36, 115–134 CrossRef CAS PubMed.
- G. J. Patti, O. Yanes and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2012, 13, 263–269 CrossRef CAS PubMed.
- H. Prinsen, B. G. M. Schiebergen-Bronkhorst, M. W. Roeleveld, J. J. M. Jans, M. G. M. de Sain-van der Velden, G. Visser, P. M. van Hasselt and N. M. Verhoeven-Duif, J. Inherited Metab. Dis., 2016, 39, 651–660 CrossRef CAS PubMed.
- A. Desmons, E. Thioulouse, J.-Y. Hautem, A. Saintier, B. Baudin, A. Lamazière, C. Netter and F. Moussa, J. Chromatogr. A, 2020, 461135, DOI:10.1016/j.chroma.2020.461135.
- H. S. Jonasdottir, C. Papan, S. Fabritz, L. Balas, T. Durand, I. Hardardottir, J. Freysdottir and M. Giera, Anal. Chem., 2015, 87, 5036–5040 CrossRef CAS PubMed.
- P. R. S. Baker, A. M. Armando, J. L. Campbell, O. Quehenberger and E. A. Dennis, J. Lipid Res., 2014, 55, 2432–2442 CrossRef CAS PubMed.
- T. P. I. Lintonen, P. R. S. Baker, M. Suoniemi, B. K. Ubhi, K. M. Koistinen, E. Duchoslav, J. L. Campbell and K. Ekroos, Anal. Chem., 2014, 86, 9662–9669 CrossRef CAS PubMed.
- A. Basit, S. Pontis, D. Piomelli and A. Armirotti, Metabolomics, 2016, 12, 10 CrossRef PubMed.
- C. Lapthorn, F. Pullen and B. Z. Chowdhry, Mass Spectrom. Rev., 2013, 32, 43–71 CrossRef CAS PubMed.
- B. B. Schneider, E. G. Nazarov, F. Londry, P. Vouros and T. R. Covey, Mass Spectrom. Rev., 2016, 35, 687–737 CrossRef CAS PubMed.
- P. Dwivedi, A. J. Schultz and H. H. Hill, Int. J. Mass spectrom., 2010, 298, 78–90 CrossRef CAS PubMed.
- G. Paglia and G. Astarita, Nat. Protoc., 2017, 12, 797–813 CrossRef CAS PubMed.
- F. A. Fernandez-Lima, D. A. Kaplan and M. A. Park, Rev. Sci. Instrum., 2011, 82, 126106 CrossRef CAS PubMed.
- I. K. Webb, S. V. B. Garimella, A. V. Tolmachev, T. C. Chen, X. Y. Zhang, R. V. Norheim, S. A. Prost, B. LaMarche, G. A. Anderson, Y. M. Ibrahim and R. D. Smith, Anal. Chem., 2014, 86, 9169–9176 CrossRef CAS PubMed.
- M. A. Baird, P. V. Shliaha, G. A. Anderson, E. Moskovets, V. Laiko, A. A. Makarov, O. N. Jensen and A. A. Shvartsburg, Anal. Chem., 2019, 91, 6918–6925 CAS.
- M. A. Baird, G. A. Anderson, P. V. Shliaha, O. N. Jensen and A. A. Shvartsburg, Anal. Chem., 2019, 91, 1479–1485 CrossRef CAS PubMed.
- A. A. Shvartsburg, W. F. Danielson and R. D. Smith, Anal. Chem., 2010, 82, 2456–2462 CrossRef CAS PubMed.
- R. Fernandez-Maestre, C. Wu and H. H. Hill, Rapid Commun. Mass Spectrom., 2012, 26, 2211–2223 CrossRef CAS PubMed.
- D. G. Beach, E. S. Kerrin, S. D. Giddings, M. A. Quilliam and P. McCarron, Sci. Rep., 2018, 8, 11 CrossRef PubMed.
- M. McCooeye and Z. Mester, Rapid Commun. Mass Spectrom., 2006, 20, 1801–1808 CrossRef CAS PubMed.
- R. Perez-Miguez, B. Bruyneel, M. Castro-Puyana, M. L. Marina, G. W. Somsen and E. Dominguez-Vega, Anal. Chem., 2019, 91, 3277–3285 CrossRef CAS PubMed.
- J. D. Zhang, K. M. M. Kabir, H. E. Lee and W. A. Donald, Int. J. Mass Spectrom., 2018, 428, 1–7 CrossRef CAS.
- K. J. D. Fouque, A. Garabedian, J. Porter, M. Baird, X. Q. Pang, T. D. Williams, L. J. Li, A. Shvartsburg and F. Fernandez-Lima, Anal. Chem., 2017, 89, 11787–11794 CrossRef PubMed.
- D. L. Winter, J. Mastellone, K. M. M. Kabir, M. R. Wilkins and W. A. Donald, Anal. Chem., 2019, 91, 11827–11833 CrossRef CAS PubMed.
- M. Hernandez-Mesa, A. Escourrou, F. Monteau, B. Le Bizec and G. Dervilly-Pinel, TrAC, Trends Anal. Chem., 2017, 94, 39–53 CrossRef CAS.
- R. Guevremont, D. A. Barnett, R. W. Purves and J. Vandermey, Anal. Chem., 2000, 72, 4577–4584 CrossRef CAS PubMed.
- S. Wernisch, F. Afshinnia, T. Rajendiran and S. Pennathur, Anal. Bioanal. Chem., 2018, 410, 2865–2877 CrossRef CAS PubMed.
- E. Sinclair, K. A. Hollywood, C. Y. Yan, R. Blankley, R. Breitling and P. Barran, Analyst, 2018, 143, 4783–4788 RSC.
- S. Goscinny, L. Joly, E. De Pauw, V. Hanot and G. Eppe, J. Chromatogr. A, 2015, 1405, 85–93 CrossRef CAS PubMed.
- J. Regueiro, N. Negreira, R. Hannisdal and M. H. G. Berntssen, Food Control, 2017, 78, 116–125 CrossRef CAS.
- J. Regueiro, N. Negreira and M. H. G. Berntssen, Anal. Chem., 2016, 88, 11169–11177 CrossRef CAS PubMed.
- Z. W. Zhou, X. T. Shen, J. Tu and Z. J. Zhu, Anal. Chem., 2016, 88, 11084–11091 CrossRef CAS PubMed.
- X. Y. Zheng, N. A. Aly, Y. X. Zhou, K. T. Dupuis, A. Bilbao, V. L. Paurus, D. J. Orton, R. Wilson, S. H. Payne, R. D. Smith and E. S. Baker, Chem. Sci., 2017, 8, 7724–7736 RSC.
- C. M. Nichols, J. N. Dodds, B. S. Rose, J. A. Picache, C. B. Morris, S. G. Codreanu, J. C. May, S. D. Sherrod and J. A. McLean, Anal. Chem., 2018, 90, 14484–14492 CrossRef CAS PubMed.
- L. C. Nye, J. P. Williams, N. C. Munjoma, M. P. M. Letertre, M. Coen, R. Bouwmeester, L. Martens, J. R. Swann, J. K. Nicholson, R. S. Plumb, M. McCullagh, L. A. Gethings, S. Lai, J. I. Langridge, J. P. C. Vissers and I. D. Wilson, J. Chromatogr. A, 2019, 1602, 386–396 CrossRef CAS PubMed.
- L. D. Roberts, A. L. Souza, R. E. Gerszten and C. B. Clish, Curr. Protoc. Mol. Biol., 2012, 98, 30 Search PubMed.
- O. Hernandez, S. Isenberg, V. Steinmetz, G. L. Glish and P. Maitre, J. Phys. Chem. A, 2015, 119, 6057–6064 CrossRef CAS PubMed.
- S. L. Isenberg, P. M. Armistead and G. L. Glish, J. Am. Soc. Mass Spectrom., 2014, 25, 1592–1599 CrossRef CAS PubMed.
- F. Berthias, B. Maatoug, G. Glish, F. Moussa and P. Maitre, J. Am. Soc. Mass Spectrom., 2018, 29, 752–760 CrossRef CAS PubMed.
- Y. Wang, E. Alhajji, B. Rieul, F. Berthias and P. Maitre, Int. J. Mass Spectrom. Ion Processes, 2019, 443, 16–21 CrossRef CAS.
- A. A. Shvartsburg, Differential Mobility Spectrometry: Nonlinear Ion Transport and Fundamentals of FAIMS, CRC Press, 2008 Search PubMed.
- I. A. Buryakov, E. V. Krylov, E. G. Nazarov and U. K. Rasulev, Int. J. Mass Spectrom. Ion Processes, 1993, 128, 143–148 CrossRef CAS.
- J. L. Campbell, M. Zhu and W. S. Hopkins, J. Am. Soc. Mass Spectrom., 2014, 25, 1583–1591 CrossRef CAS PubMed.
- A. A. Shvartsburg, K. Q. Tang, R. D. Smith, M. Holden, M. Rush, A. Thompson and D. Toutoungi, Anal. Chem., 2009, 81, 8048–8053 CrossRef CAS PubMed.
- E. V. Krylov, S. L. Coy, J. Vandermey, B. B. Schneider, T. R. Covey and E. G. Nazarov, Rev. Sci. Instrum., 2010, 81, 024101 CrossRef PubMed.
- K. L. Davidson and M. F. Bush, Anal. Chem., 2017, 89, 2017–2023 CrossRef CAS PubMed.
- L. W. Beegle, I. Kanik, L. Matz and H. H. Hill, Anal. Chem., 2001, 73, 3028–3034 CrossRef CAS PubMed.
- G. R. Asbury and H. H. Hill, J. Chromatogr. A, 2000, 902, 433–437 CrossRef CAS PubMed.
- M. R. Menlyadiev, A. Tarassov, A. M. Kielnecker and G. A. Eiceman, Analyst, 2015, 140, 2995–3002 RSC.
- M. T. Campbell and G. L. Glish, Int. J. Mass Spectrom., 2018, 425, 47–54 CrossRef CAS.
- Z. D. Chen, S. L. Coy, E. L. Pannkuk, E. C. Laiakis, A. B. Hall, A. J. Fornace and P. Vouros, J. Am. Soc. Mass Spectrom., 2016, 27, 1626–1636 CrossRef CAS PubMed.
- M. M. Sanagi, S. L. Ling, Z. Nasir, D. Hermawan, W. A. W. Ibrahim and A. Abu Naim, J. AOAC Int., 2009, 92, 1833–1838 CrossRef CAS PubMed.
- D. A. Barnett, R. W. Purves and R. Guevremont, Nucl. Instrum. Methods Phys. Res., Sect. A, 2000, 450, 179–185 CrossRef CAS.
- P. Pathak, M. A. Baird and A. A. Shvartsburg, Anal. Chem., 2018, 90, 9410–9417 CrossRef CAS PubMed.
- P. Pathak, M. A. Baird and A. A. Shvartsburg, J. Am. Soc. Mass Spectrom., 2020, 31, 137–145 CrossRef CAS.
- D. G. Beach, J. Am. Soc. Mass Spectrom., 2017, 28, 1518–1530 CrossRef CAS PubMed.
- E. Varesio, J. C. Y. Le Blanc and G. Hopfgartner, Anal. Bioanal. Chem., 2012, 402, 2555–2564 CrossRef CAS PubMed.
- H. B. Xu, F. R. Boucher, T. T. Nguyen, G. P. Taylor, J. J. Tomlinson, R. A. Ortega, B. Simons, M. G. Schlossmacher, R. Saunders-Pullman, W. Shaw and S. A. L. Bennett, J. Lipid Res., 2019, 60, 200–211 CrossRef CAS PubMed.
- Y. L. Fu, Y. Q. Xia, J. Flarakos, F. L. S. Tse, J. D. Miller, E. B. Jones and W. K. Li, Anal. Chem., 2016, 88, 3655–3661 CrossRef CAS PubMed.
- G. A. Eiceman, E. V. Krylov, E. G. Nazarov and R. A. Miller, Anal. Chem., 2004, 76, 4937–4944 CrossRef CAS.
- N. Krylova, E. Krylov, G. A. Eiceman and J. A. Stone, J. Phys. Chem. A, 2003, 107, 3648–3654 CrossRef CAS PubMed.
- D. Ruskic and G. Hopfgartner, Anal. Chem., 2019, 91, 11670–11677 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Composition of standard mixture; concentrations of AAs and related compounds in plasmas; composition and concentrations of calibration solutions; compensation voltage and collision cross section values for a set of AAs and related compounds; limit of detection values; day-to-day confidence interval on CV values. See DOI: 10.1039/d0an00377h |
| This journal is © The Royal Society of Chemistry 2020 |