
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sustainable amperometric biosensor for the analysis of ascorbic, benzoic, gallic and kojic acids through catechol detection. Innovation and signal processing
Ana
Casanova†
a,
María
Cuartero
 *b,
Yolanda
Alacid
a,
Carmen M.
Almagro
a,
Francisco
García-Cánovas
c,
María S.
García
a and
Joaquín A.
Ortuño
*a
*b,
Yolanda
Alacid
a,
Carmen M.
Almagro
a,
Francisco
García-Cánovas
c,
María S.
García
a and
Joaquín A.
Ortuño
*a
aDepartment of Analytical Chemistry, Faculty of Chemistry, University of Murcia, Murcia E-30100, Spain. E-mail: jortuno@um.es
bDepartment of Chemistry, School of Engineering Science in Chemistry, Biotechnology and Healthcare, KTH Royal Institute of Technology, 10044, Stockholm, Sweden. E-mail: mariacb@kth.se
cDepartment of Biochemistry and Molecular Biology-A, Faculty of Biology, University of Murcia, Murcia E-30100, Spain
First published on 11th March 2020
Abstract
In this work, we present a new catechol amperometric biosensor fabricated on the basis of naturally available enzymes in common mushrooms. The biosensor response mechanism comprises the reduction of the quinone exclusively produced in the oxidation of the catechol present in the sample, which is catalyzed by tyrosinase enzyme. The new catechol biosensor has demonstrated excellent analytical performance at increasing catechol concentrations in the sample solution, which includes superior reproducibility for several electrodes and long-term stability. On top of that, the biosensing element used in the fabrication is a sustainable material, of low-cost and presents an excellent lifetime of years. Whether the catechol biosensor is operating in the presence of a compound influencing the reactions underlying the amperometric response (such as ascorbic, benzoic, gallic and kojic acids), this serves as an analytical platform to detect these compounds in real samples. Particularly, we introduce herein for the first time different treatments to process the current signal of the biosensor pursuing the linearity needed for the analytical application in real samples. In this sense, the catechol biosensor has been successfully applied to the detection of benzoic, gallic and kojic acids in juices, teas and cosmetic products, respectively.
Introduction
Enzymatic biosensors have largely demonstrated excellent analytical applicability for the detection of targets that are involved either as substrates or inhibitors in the enzymatic reaction in which the sensor is based. Specifically, biosensors based on polyphenol oxidase (also called tyrosinase) allow for the determination of phenolic compounds as substrates of this enzyme (such as catechol, phenol, tyrosine and dopamine),1–9 as well as a number of inhibitors (such as benzoic acid among others).10–13Different strategies have been reported for the construction of this sort of biosensors, mainly involving the immobilization of the enzyme by distinct approaches. The enzyme immobilization may in turn comprise different grades of complexity. Furthermore, commercially available enzymes are traditionally used despite being rather expensive. As a result, the entire processing and immobilization of the enzyme cause the preparation of the biosensor to be often considered as complex, expensive, time-consuming and risky to present a shortcoming that impedes/retrogresses the optimal analytical functioning.
Aiming at providing simplicity and more affordability to the general process for the construction of enzymatic biosensors, a wave of platforms operating on the basis of bio-tissues that are rich in the required enzymes were introduced some years ago.14–21 Particularly, it is well known that common mushrooms have very rich polyphenol oxidase activity. However, the fast degradation of the raw bio-tissues limits the lifetime of the manufactured biosensor, being therefore necessary to further preserve the enzymatic activity of the vegetal material.
The freeze–drying process, also known as lyophilization in the jargon, is currently conceived as a smooth methodology for the suppression of water content in vegetal materials and biologically active molecules. Hereby, lyophilization is very effective for the preservation of the inherent features of these natural materials. Indeed, the lyophilization process is well-entrenched and worldwide utilized, especially in the enzymological and food fields.
In recent years, it has been observed that stability and final activity of freeze-dried materials critically depend on how the entire approach is performed.22–25 Essentially, this is based on different freezing and drying stresses, such as solute concentration, formation of ice crystals, pH changes and others, that are prone to denature proteins in the material to various degrees.22 Thus, stabilizers are often required in a protein formulation to protect its stability in the final processed material.22,24
As an alternative to the use of commercially available (purified) enzymes and to fresh bio-tissues, we report herein a catechol biosensor based on mushroom lyophilized powder as the enzymatically active core of the biosensor. Despite being promising, this type of material has been scarcely used in biosensor development and mainly restricted to oxygen observation.26 Our studies are based on the monitoring of the reduction of the quinone formed in the oxidation of catechol catalysed by the tyrosinase enzyme present in the mushroom powder. In addition, the analytical determination of the response of certain inhibitors of the biosensor to catechol (i.e. ascorbic, benzoic, gallic and kojic acids) is explored, this being important in the analysis of real samples such as beverages and cosmetic products among others.
It is crucial to consider here that the mechanism in which each of the selected compounds influences the biosensor response to catechol varies. While ascorbic acid reacts with the quinone formed, benzoic, gallic and kojic acids inhibit the enzymatic reaction. Particularly, gallic acid is in real an alternative substrate to the tyrosinase enzyme but shows a low catalytic constant27,28 and therefore generally considered as an inhibitor.29 Indeed, there are many substrates and inhibitors of the tyrosinase enzyme and therefore, the applicability possibilities for the developed catechol biosensor are really wide.30,31 One of us has indeed been involved in numerous investigations concerning tyrosinase enzyme and notably participated in the development and tuning of a test for the discrimination between alternative substrates (i.e. compounds different from catechol that are also substrates of the tyrosinase) and inhibitors.32
Tyrosinase-based biosensors have been extensively reported for the detection of benzoic acid,10–13 whereas we have found only one paper related to the detection of kojic acid.33 While both (benzoic and kojic acids) are competitive and reversible inhibitors, it is known that the kinetics involved in kojic acid-based inhibition is slower. Accordingly, the data treatment developed by us in the present paper is different for these two inhibitors in order to optimize the required analysis time. Then, to date and as far as we know, all the biosensors reported for the detection of ascorbic acid are based on the ascorbate oxidase enzyme34 and none has been reported for the determination of gallic acid. Therefore, the studies in the present paper are of high interest from an analytical point of view.
Experimental
Reagents, materials and instrumentation
Aqueous solutions were prepared by dissolving the appropriate salts in deionized water (18.2 MΩ cm). Catechol (≥99.5%), benzoic acid (≥99.5%), L-ascorbic acid (≥99.7%), gallic acid monohydrate (≥98%), kojic acid (≥99.5%), synthetic graphite powder (<20 μm), potassium dihydrogen phosphate (≥99%) and sodium hydroxide (≥98%) were purchased from Panreac. Mineral oil and Folin-Ciocalteu reagent were acquired from Sigma-Aldrich. Mushrooms (Agaricus bisporus) already laminated and all the analyzed beverages and products were provided by a local supplier. Phosphate buffer solution (PBS) at pH 6.5 was prepared at 0.05 M concentration. Standard solutions of catechol (0.1 M), L-ascorbic acid (0.01 M), benzoic acid (0.01 M) and gallic acid (0.01 M) were prepared in PBS background and stored in a fridge at 4 °C.For the amperometric measurements, a potentiostat that was in-house fabricated at the University of Murcia, a CHI220 Cell Stand (IJ Cambria Scientific) coupled to a regular beaker, a CHI111 Ag/AgCl reference electrode (3 M KCl inner solution, IJ Cambria Scientific) and a CHI115 platinum wire counter electrode (IJ Cambria Scientific) were used. A MF-2010 Carbon Paste Electrode Body (2.87 mm ID) (BASi, West Lafayette) was used to accommodate the enzymatic-active paste to prepare the catechol biosensor, which was the working electrode of the electrochemical cell. A freeze dryer (ALPHA 1-2 LDplus, Christ, Germany), a manual grinder (A505 2HF, Moulinex, France), a vacuum packing machine (Boxer 35, Henkelman, the Netherlands), a microwave oven (LG), a spectrophotometer (Thermo Spectronic Unicam UV-510 UV–Visible), a stirrer (F203A0161, Velp Scientifica, Italy), a pH-meter (pH & ION-Meter GLP + 22, Crison, Spain), an agate mortar (01442-AB, Spi Supplies, China) and a sieve (Filtra vibración S.L., Spain) were also utilized.
Preparation of the lyophilized mushroom powder
Mushrooms were acquired in a local shop in the laminated format and the lyophilization process was carried out at the ‘Sección de Investigación Agroforestal laboratory’ (SEAF) at the University of Murcia. Briefly, the laminated mushrooms were stored in lyophilization bags. Each bag was treated at −60 °C under 0.021 mbar for 48 h. This process allowed the water evaporation at low temperature. The lyophilized mushrooms were vacuum-packed and stored in the fridge at 4 °C. Before use, the mushrooms were passed through an electric grinder and subsequently screened to obtain particle sizes between 60–250 μm and 250–420 μm. These mushroom powders were stored in closed recipients in the fridge at 4 °C.Preparation of the biosensor
For the preparation of the electrode pastes comprising 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 and 1:5 wt. vegetal tissue/graphite, 0.11 g or 0.22 g of the corresponding mushroom powder 1.10 g of graphite, and 0.9 g of mineral oil were manually mixed in an agate mortar and pestle until the obtention of a homogeneous paste. Either of these two pastes was placed in the electrode body generating a compact block whose external surface was slightly polished using glossy paper. Although electrode conditioning was not necessary, it was stored in PBS at pH 6.5 over its use to maximize its lifetime.
:10 and 1:5 wt. vegetal tissue/graphite, 0.11 g or 0.22 g of the corresponding mushroom powder 1.10 g of graphite, and 0.9 g of mineral oil were manually mixed in an agate mortar and pestle until the obtention of a homogeneous paste. Either of these two pastes was placed in the electrode body generating a compact block whose external surface was slightly polished using glossy paper. Although electrode conditioning was not necessary, it was stored in PBS at pH 6.5 over its use to maximize its lifetime.
Procedure for the biosensor calibration towards catechol
A volume of 25 mL of PBS at pH 6.5 was transferred to the electrochemical cell. The biosensor was then activated at a constant potential of 0.1 V and at a constant moderate stirring of the solution (approx. 200 rpm). When the measured current reached a constant value, the calibration graph for catechol started by the addition of increasing concentrations to the PBS background by adding small volumes of the 0.1 M catechol standard solution. Subsequently, the calibration graph was constructed using the current value reached after exactly 30 s of each catechol addition, i.e., coinciding with the response time of the biosensor.Procedure for the obtention of the calibration graph for ascorbic, benzoic, gallic and kojic acids
For experiments based on benzoic, gallic and kojic acids, the base line of the biosensor was first registered in 4 × 10−4 M catechol in PBS at pH 6.5 until a constant current value was reached. In the case of ascorbic acid, two different concentrations were used: 4 × 10−4 M (for comparison purposes with the rest of the inhibitors) and 4 × 10−5 M as the optimized concentration for the calibration graph.Subsequently, increasing concentrations of each acid were added by small volumes of the 0.01 M standard solution of the corresponding acid. A time of 30 s for ascorbic and benzoic acids, 90 s for gallic acid and no time for kojic acid were fixed between each volume addition. The observed current value at the corresponding time was then plotted against the inhibitor concentration and a linear calibration graph was obtained according to the different data treatments developed for each acid (see below in the Results and Discussion section).
Detection of inhibitory capacity of fruit-based juices and teas
Commercial fruit-based juices were analyzed as purchased without any pre-treatment. The standard addition method was applied as follows. After the catechol biosensor reached a constant current signal in 25 mL of 4 × 10−5 M catechol in PBS at pH 6.5, a volume of 25 μl of the corresponding juice was added and then 25 μl of the 0.01 M ascorbic acid solution by 4 times, waiting a fixed time of 30 s between each addition. The inhibitory capacity of each juice expressed as ascorbic acid concentration was calculated from the fitting of the logΔI (logarithmic current increment, I − I0) versus logCT, where CT is the total ascorbic acid concentration (considering additions) in the solution.
In the case of the tea samples, 1.5 g of the corresponding leaf powder was used for the extraction procedure in 50 mL of deionized water. This was accomplished in a microwave oven at 800 W for 1 min. Subsequently, the tea was left in a closed beaker for 5 min to avoid any type of evaporation and/or contamination and then diluted to a final volume of 50 mL with deionized water. The standard addition method was applied as follows. After the catechol biosensor reached a constant current signal in 25 mL of 4 × 10−4 M catechol in PBS at pH 6.5, a volume of 100 μl of the corresponding tea was added and then 50 μl of the 0.01 M gallic acid solution by 4 times, waiting a fixed time of 90 s between each addition. The inhibitory capacity of each tea expressed as gallic acid concentration was calculated from the plot of the accumulative signal (A.S.) against the added gallic acid concentration. All the tea samples were additionally analyzed by the Folin-Ciocalteu method in order to calculate the total amount of polyphenols, according to the procedure reported elsewhere.35
Detection of the ‘total depigmenting efficacy’ in cosmetic products
Ampoules for the treatment of skin blemishes without any kind of pre-treatment were analyzed by the standard addition method described as follows. After the catechol biosensor reached a constant current signal in 25 mL of 4 × 10−4 M catechol in PBS at pH 6.5, a volume of 200 μl of the ampoule was added and then 10, 10, 10 and 15 μl of the 0.01 M kojic acid solution, waiting a fixed time of 60 s between each addition. The ‘total depigmenting efficacy’ in the ampoule expressed as kojic acid concentration was calculated from the plot of the A.S. versus the added kojic acid concentration.Results and discussion
Evaluation of the response of the biosensor towards catechol
The influence of the applied potential on the biosensor response was studied by using a potential window from 0.2 V to −0.5 V every 0.1 V. For this purpose, the biosensor was based on a paste comprising lyophilized vegetal tissue (60–250 μm of particle size) and vegetal tissue/graphite in 1:5 weight ratio. The experiment consisted of registering the current in both PBS at pH 6.5 and then 4 × 10−5 M catechol solution during the application of different potentials. Fig. 1a shows the values for the steady-state current in these two solutions, together with the corresponding difference. As observed, the current in PBS displays low values in the potential window from 0.2 V to −0.2 V, from which the current increases up to a relatively high value at −0.5 V.
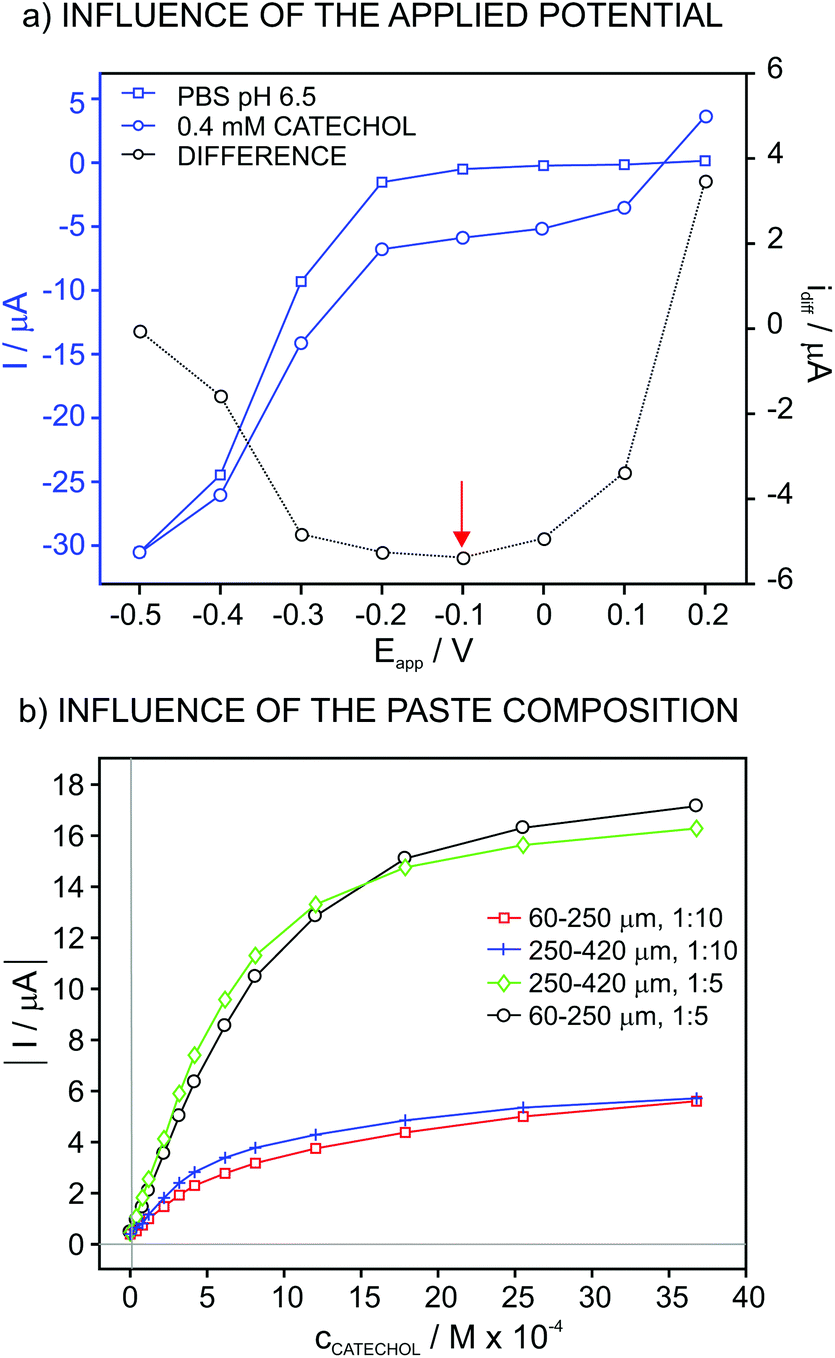 | ||
| Fig. 1 Influence of (a) the applied potential and (b) paste composition and particle size on the recorded current. | ||
The difference of the current acquired in PBS and the catechol solution is purely associated with the reduction of the quinone generated in the catechol oxidation by the dissolved O2 in the solution, which is in turn catalyzed by the enzyme that is immobilized on the electrode surface (i.e. tyrosinase). In this case, the potential window at which the acquired current does not experience any change, considering the curve with the difference between the PBS and the catechol solution, is from 0.0 V to −0.3 V. Thus, a potential of −0.1 V, which is in within this range, was selected for further studies.
Aiming at confirming that the observed current is related to the presence of quinone uniquely formed in the enzymatic catechol oxidation at the biosensor surface, therefore discarding the influence of any side reaction (such as the non-catalyzed quinone formation) as well as any contamination (e.g. quinone traces already present in the catechol solution), the same experiment was accomplished using an electrode comprising only graphite paste and no vegetal tissue. Thus, the same curves (current versus potential) were obtained in the absence and presence of catechol (data not shown). It is hence concluded that, in the presence of the vegetal tissue, the observed current originates from the reduction of the quinone that is only produced in the catechol oxidation process catalyzed by the lyophilized tissue enzyme in the biosensor, as depicted in Fig. 2.
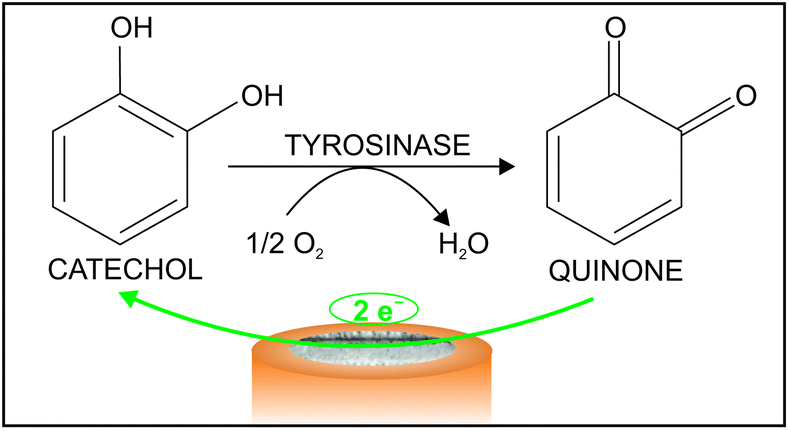 | ||
| Fig. 2 Illustration of the response mechanism of the biosensor. | ||
Fig. 1b presents the plots for the current at increasing catechol concentrations with an applied potential of −0.1 V for biosensors based on different compositions of the vegetal tissue/graphite (1:10 and 1:5) paste as well as the particle size of the tissue (60–250 and 250–420 μm). In addition, Table 1 collects the tested compositions with the corresponding fitting parameters along the linear range of response (LRR). The sensitivity (i.e. slope) improved for the mixtures based on a higher content in the vegetal tissue. However, when the tissue amount was increased even more (up to 1:2), no response was observed at increasing catechol concentrations, likely due to the low content in the electrical conductor as a result of reducing the graphite amount. Therefore, a ratio of 1:5 was selected for further studies. On the other hand, the particle size did not really influence the response of the biosensors based on this ratio and therefore, a size of 60–250 μm was used further on.
| Particle size (μm) | Tissue/graphite (wt. ratio) | Slope (μA M−1) × 104 | LRR (M) | r 2 |
|---|---|---|---|---|
| 60–250 | 1:5 |
−1.26 | 4 × 10−5–8 × 10−4 | 0.993 |
| 60–250 | 1:10 |
−0.47 | 4 × 10−5–4 × 10−4 | 0.995 |
| 250–420 | 1:5 |
−1.51 | 4 × 10−5–6 × 10−4 | 0.991 |
| 250–420 | 1:10 |
−0.60 | 4 × 10−5–4 × 10−4 | 0.993 |
The response of the biosensor for increasing concentrations of catechol at an applied potential of −0.1 V is shown in Fig. 3a. In this experiment, the concentration of catechol was changed in PBS background (pH of 6.5) from 4 × 10−5 to 4 × 10−3 M concentrations. The absolute value of the recorded current increases after each catechol addition and reaches a near steady-state value after 30 s. For the construction of the calibration graph (catechol concentration versus current), final values for the current achieved after having increased the catechol concentration were plotted against the total concentration of catechol. This was selected as the criteria for the analytical signal rather than waiting for the steady-state of the signal in order to reduce the analysis time.
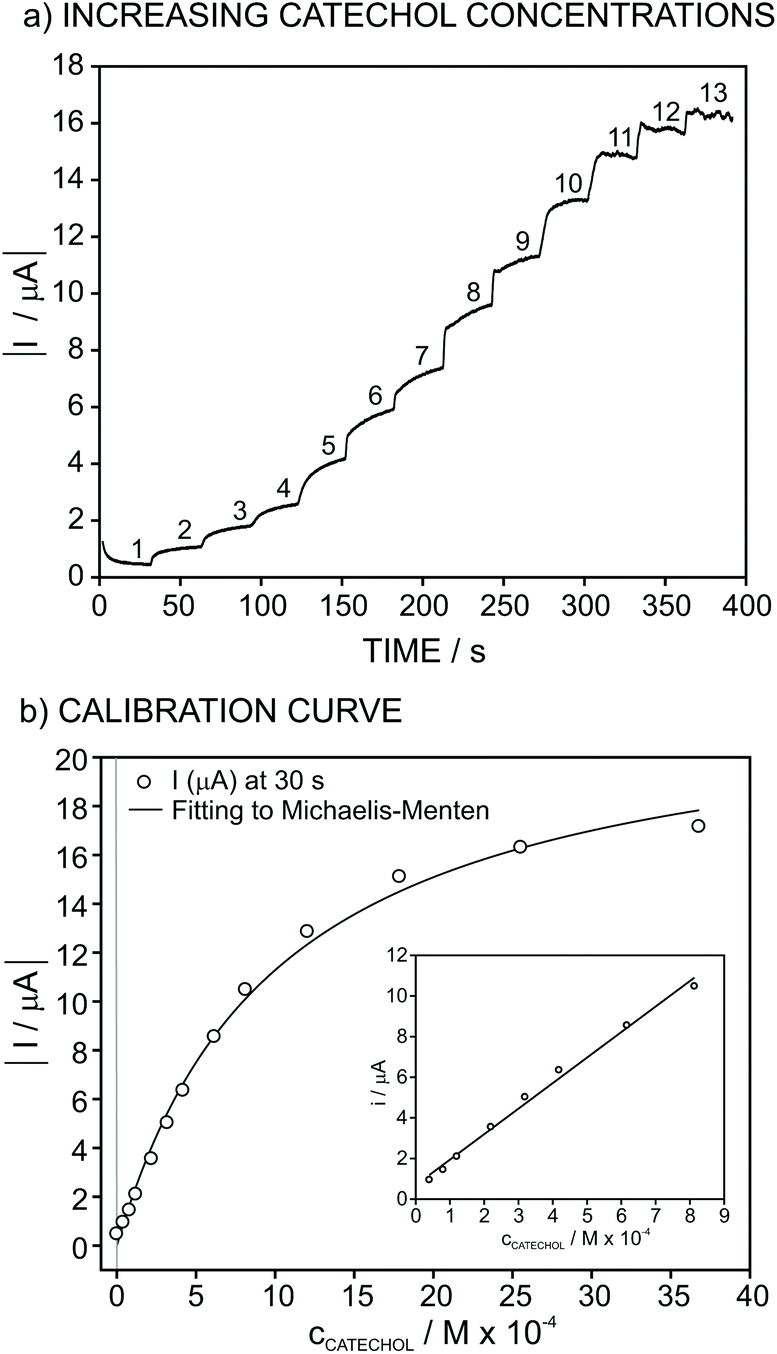 | ||
| Fig. 3 Calibration graph under the optimized conditions (applied potential of −0.1 V and 1:5 tissue/graphite wt. composition with the particle size of the tissue of 60–250 μm for the composition of the electrode material): (a) Recorded dynamic current and (b) corresponding calibration curve. The entire calibration graph was fitted to the Michaelis–Menten equation while the concentration range between 4 × 10−5 and 8 × 10−4 M comprises the linear range of response of the biosensor (inset plot). | ||
As observed in Fig. 3b (inset), the recorded current obeys linearity from 4 × 10−5 to 8 × 10−4 M catechol concentration, and the fitted curve is as follows:
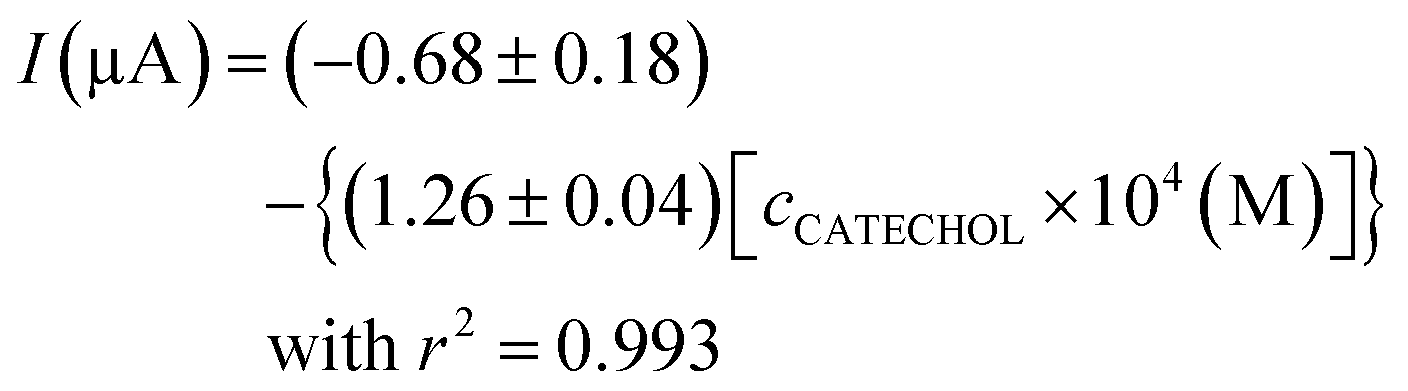 | (1) |
In contrast, from 8 × 10−4 M catechol concentration, the recorded current gradually tends to reach a constant value, with the entire current versus concentration curve approaching a definition by the Michaelis–Menten equation as follows:
| I = ImaxcCATECHOL/KM + cCATECHOL | (2) |
Concerning the range in which the current response is linear for increasing catechol concentrations (Fig. 3b, inset), this may be used as a calibration graph towards the analytical application of the developed biosensor in real samples. The limit of detection of the biosensor was calculated to be 7 × 10−6 M catechol concentration, which was obtained as the concentration for which the response is equal to three times the standard deviation of the response of the biosensor in PBS (pH = 6.5).
The lifetime of the paste as well as the repeatability and reproducibility of the calibration parameters was subsequently evaluated. Thus, the calibration graph of the biosensor at increasing catechol concentrations was recorded under different conditions: (i) when the paste used for the preparation of the sensor was stored at different temperatures (room temperature of 22 °C, in the fridge at 4 °C and in the freezer at −18 °C), (ii) three consecutive calibration graphs using the same electrode, (iii) reproducibility of the electrode response using the same paste batch over 15 days from its preparation and (iv) two sensors prepared with different batches of vegetal tissue.
With the paste stored for 1 day at 22 °C, 4 °C and −18 °C, three electrodes were prepared according to the procedure described in the Experimental section and the calibration graphs were obtained for comparison. The slopes observed within the linear range of response were −1.10, −1.11 and −0.78 × 104 μA mol−1 L−1 for stored pastes at 22 °C, 4 °C and −18 °C, respectively. While the paste stored in a protected environment at room temperature and in the fridge responds in a similar way, the electrode based on the paste stored in the freezer displayed a significantly lower sensitivity. This is likely due to the fact that the freezing/thawing process affects the entire structure of the vegetal tissue and therefore, the enzymatic activity. On the other hand, when the paste was stored in the fridge for more than 3 months, this still presented the same response to catechol (data not shown).
Regarding the response repeatability (3 consecutive calibrations using the same electrode), the initial baseline of the biosensor experienced a slight decrease with consecutive uses. If this shift is corrected, and therefore an increment in current with respect to the baseline is considered, the three curves obtained at increasing catechol concentrations are overlapped and the portions for the linear response are very similar. Thus, the variation coefficient for the slopes was ca. 3.5%, with exactly the same linear range of response (4 × 10−5–8 × 10−4 M) and limit of detection (7 × 10−6 M), displaying excellent repeatability. In addition, the variation for the 3 replicate measurements of 5 × 10–5 M catechol solution was ± 2.9%.
Despite the repeatability of the biosensor response to catechol being excellent, this slightly deteriorated in subsequent days although different storages were tried (i.e. protected environment at room temperature or in the fridge, as the storage used for the paste). Accordingly, the use of freshly prepared electrodes is convenient. However, the same active paste can be used over time after its preparation. The variation coefficient of the slope in the linear range of response of different biosensors prepared with the same paste over 15 days after its preparation was ca. 5.5%, while rather maintaining the linear range of response. This manifests the disposable nature of the sensor, which is not a big drawback at all owing to its easy and fast preparation as well as the use of sustainable materials (i.e. the mushroom tissue). On the other hand, as the paste can be stored for long time, the same batch for the vegetal tissue is available for the preparation of many twin electrodes (in the scale of hundreds of electrodes, depending on the processed initial amount). Notably, the loss of the biosensor response seems to be associated with its consecutive use, i.e. with the gradual depletion of the enzyme in the tissue, rather than its deterioration.
Two different batches of vegetal tissue (i.e. they were purchased in a local supermarket two different days) were also evaluated. Interestingly, a variation of 12% between the slopes of the linear range of response displayed for each one and practically the same linear range was found. This points out that the tyrosinase activity in each batch is slightly different, affecting more the sensitivity than the linear range of response and limit of detection of the biosensor. For all the experiments accomplished in the present paper, the vegetal tissue showing a higher sensitivity was exclusively used.
Overall, the analytical features provided for the developed biosensor are comparable to those reported for other biosensors based on tyrosinase, with the advantage of presenting herein a wide range of applications (see next Sections). Seemingly, after inspecting several papers reported in the last few years, different methods for the fabrication of the biosensor provided very similar analytical performances (Table 2). Thus, the limit of detection and linear range of responses were reported in the range of micromolar catechol concentration, with the exception of some biosensors prepared on the basis of nanoparticles. In particular, the group of Pingarron reported on two different biosensors prepared with polyfunctionalized gold nanoparticles with crosslinked tyrosinase as well as Fe3O4 nanoparticles modified with cyclodextrins and with the tyrosinase immobilized by supramolecular interaction; both presented an impressive limit of detection of 20 nM.7,8 However, no analytical applications were reported for such sensors.
| Description | Analytical Performances | Applications | Ref. |
|---|---|---|---|
| a Prepared with sinusoidal voltages. b For dopamine/catechol mixtures. c Biosynthesized polyphenol oxidase extract. d 3-n-Propylpyridinium silsesquioxane chloride, thiolactic acid self-assembled monolayer, horseradish peroxidase. e Supramolecular immobilization. Tyr = tyrosinase; PEDOT = poly(3,4-ethylenedioxythiophene); LRR = linear range of response; LOD = limit of detection; S = sensitivity; tresponse = response time; PPy = polypyrrole; NPs = nanoparticles; CD = cyclodextrin. | |||
| MWCNTs/Nafion/Tyr nanobiocomposite | S = 346 A μM−1 | — | 1 |
| LRR = 1–23 μM | |||
| LOD = 0.22 μM | |||
| t response < 8 s | |||
| K m,app = 26 μM | |||
| Tyr/PEDOT compositea | S = 0.27 nA μM−1 | — | 2 |
| LRRb = 10–60 μM | |||
| Graphite/Epoxy/Tyr biocomposite | — | ANN for phenolic compounds | 9 |
| S = 37.45 μA (mMcm−1)−1 | Catechol in fruit wines | 5 | |
| LRR = 1–60 μM | |||
| PPy encapsulationc | LOD = 1.8 μM | ||
| I max = 0.223 μA | |||
| K m = 0.070 mM | |||
| AuNPs-SiPy+Cl−/TLA-SAM/HRPd | S = 0.026 μA μM−1 | — | 6 |
| LRR = 6.0–46.0 μM | |||
| LOD = 0.852 μM | |||
| Lifetime = 25 days | |||
| Polyfuntionalized Au-NPs/crosslinked Tyr | S = 1.94 A M−1 cm2 | — | 8 |
| LRR = 50 nM–10 μM | |||
| LOD = 20 nM | |||
| Lifetime = 16 days | |||
| K m = 21.9 μM | |||
| I max = 505 nA | |||
| Fe3O4 NPs/CD/Tyre | S = 12 mA M−1 | — | 7 |
| LRR = 100 nM–12 μM | |||
| LOD = 22 nM | |||
| K m = 74 μM | |||
| I max = 1.1 μA | |||
Evaluation of the response of the catechol biosensor in the presence of benzoic acid, ascorbic acid, gallic acid and kojic acid
Once the analytical performance of the developed biosensor was characterized, we subsequently studied its response towards catechol in the presence of four compounds that modify the biosensor response by influencing any of the reactions there involved: benzoic acid, gallic acid and kojic acid as inhibitors of the enzymatic reaction and ascorbic acid reducing the formed quinone (see Fig. 2). It is expected that, in the presence of higher amounts of the inhibitor (or compound) under study, the enzymatic reaction occurs in a lesser extent and, as a result, the current provided by the biosensor gradually decreases.In this sense, Fig. 4 depicts the dynamic current displayed by the biosensor in PBS, then for 4 × 10−4 M catechol and with the subsequent addition of increasing inhibitor concentration. As observed, the magnitude of the absolute dynamic current gradually decreased with the concentration of the inhibitor for all those tested. Then, the profiles for the current change in the form of I − I0 (i.e. absolute current change with respect to the current registered in the absence of any inhibitor of the biosensor response, i.e. base line) versus the added inhibitor concentration are shown in Fig. 5.
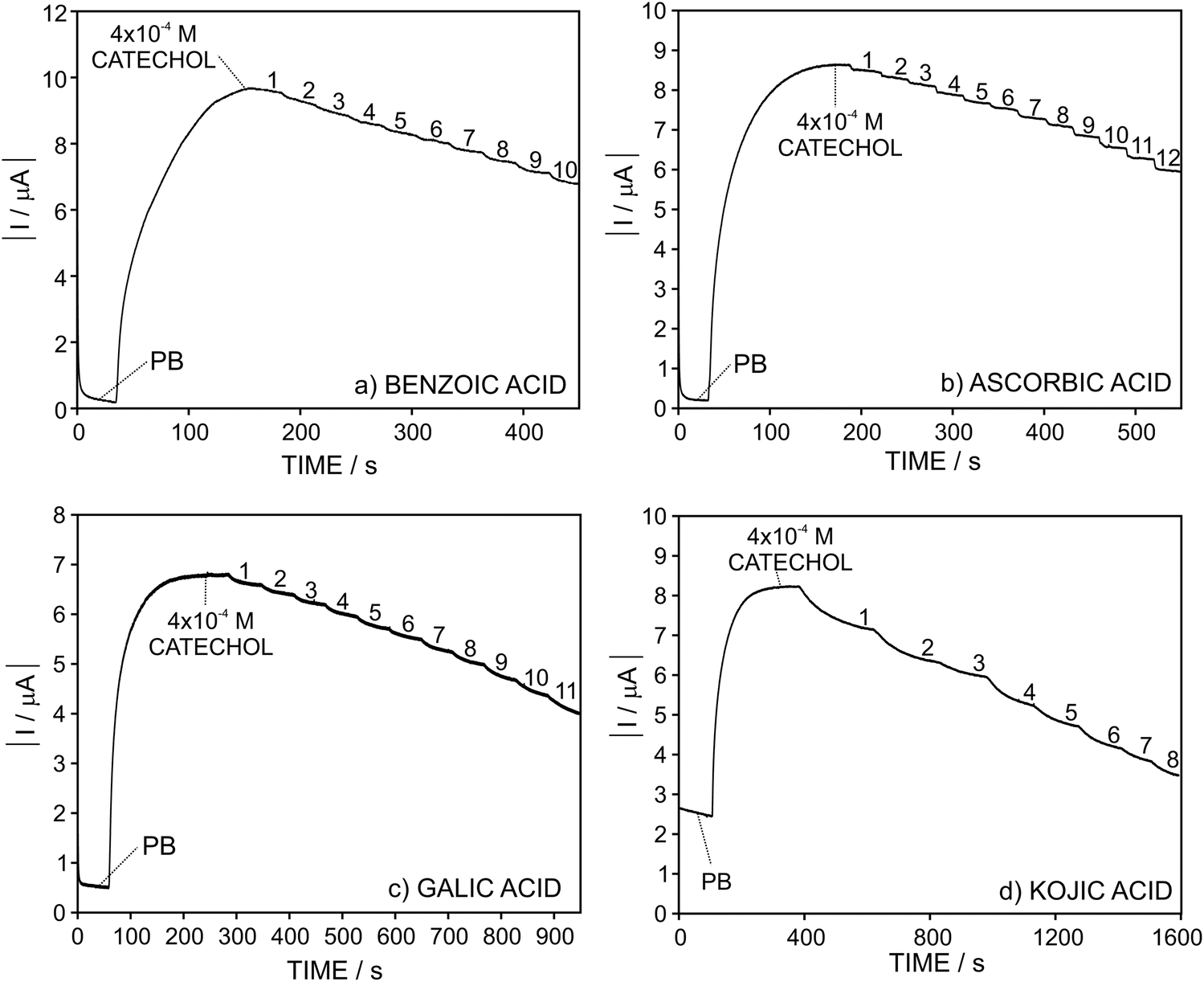 | ||
| Fig. 4 Dynamic response of the sensors at a fixed concentration of catechol (4 × 10−4 M) and gradually increasing the inhibitor concentration in the solution: (a) benzoic acid, (b) ascorbic acid, (c) gallic acid, and (d) kojic acid. | ||
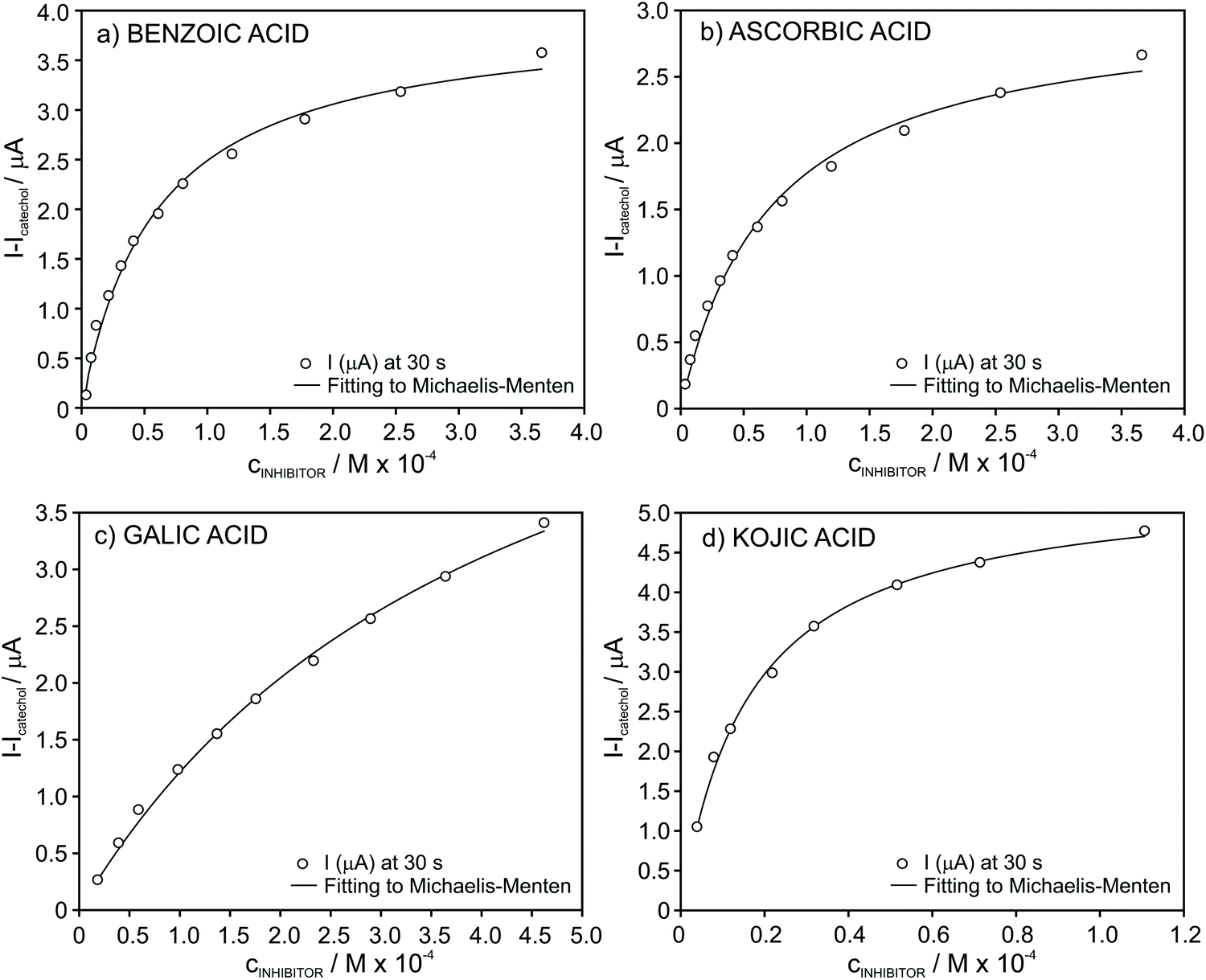 | ||
| Fig. 5 Plot of the absolute current change for increasing concentrations of the inhibitors in the presence of 4 × 10−4 M catechol concentration in PB: (a) benzoic acid, (b) ascorbic acid, (c) gallic acid, and (d) kojic acid. | ||
Importantly, in the case of ascorbic acid, the current displayed by the biosensor evolved to a constant value after every increase of the inhibitor concentration in the solution in a faster way than the rest of the inhibitors. This is likely explained by the fact that ascorbic acid is not a true inhibitor of the enzymatic reaction. Instead, it acts by re-reducing the quinone formed in the enzymatic reaction.
For all the tested inhibitors, the absolute current differently increased at increasing concentrations follows the Michaelis–Menten equation in the well-known form for inhibited (reversible) reactions.39 The fitting parameters KappI and Imax were (5.9 ± 0.5) × 10−5 and 4.0 ± 0.1 for benzoic acid, (7.1 ± 0.6) × 10−5 and 3.0 ± 0.1 for ascorbic acid, (4.3 ± 0.4) × 10−4 and 6.5 ± 0.3 for gallic acid and finally, (1.6 ± 0.1) × 10−5 and 5.4 ± 0.1 for kojic acid. From an analytical point of view considering the developed biosensor for the determination of these inhibitors in real samples, the fitting of the data to the Michaelis–Menten equation is evidently not appropriate and therefore, we further searched for different data processing to reach linearity as the basis of the analytical calibration graph.
Protocols for the linearization of the biosensor response in the presence of ascorbic, gallic and kojic acids
In the case of ascorbic acid, we found an excellent agreement of the raw data with the empirical function of the type y = axb (Fig. 6a). As a result, rather good linearity along the entire tested inhibitor concentration range (4.0 × 10−6–4.0 × 10−4 M, r2 = 0.996) was obtained by considering logarithmic I − I0 (ΔI) versus logarithmic inhibitor concentration, as presented in Fig. 6b. This straight line may be considered as the calibration graph for the analytical detection of ascorbic acid in real samples within this wide concentration range. Indeed, the calibration parameters presented excellent repeatability logΔI=(1.76 ± 0.07) + [(0.62 ± 0.01)·logcAA] (n = 3 biosensors) and reproducibility logΔI=(1.75 ± 0.04) + [(0.61 ± 0.01)·logcAA] (n = 3 calibration graphs over a week of operation).
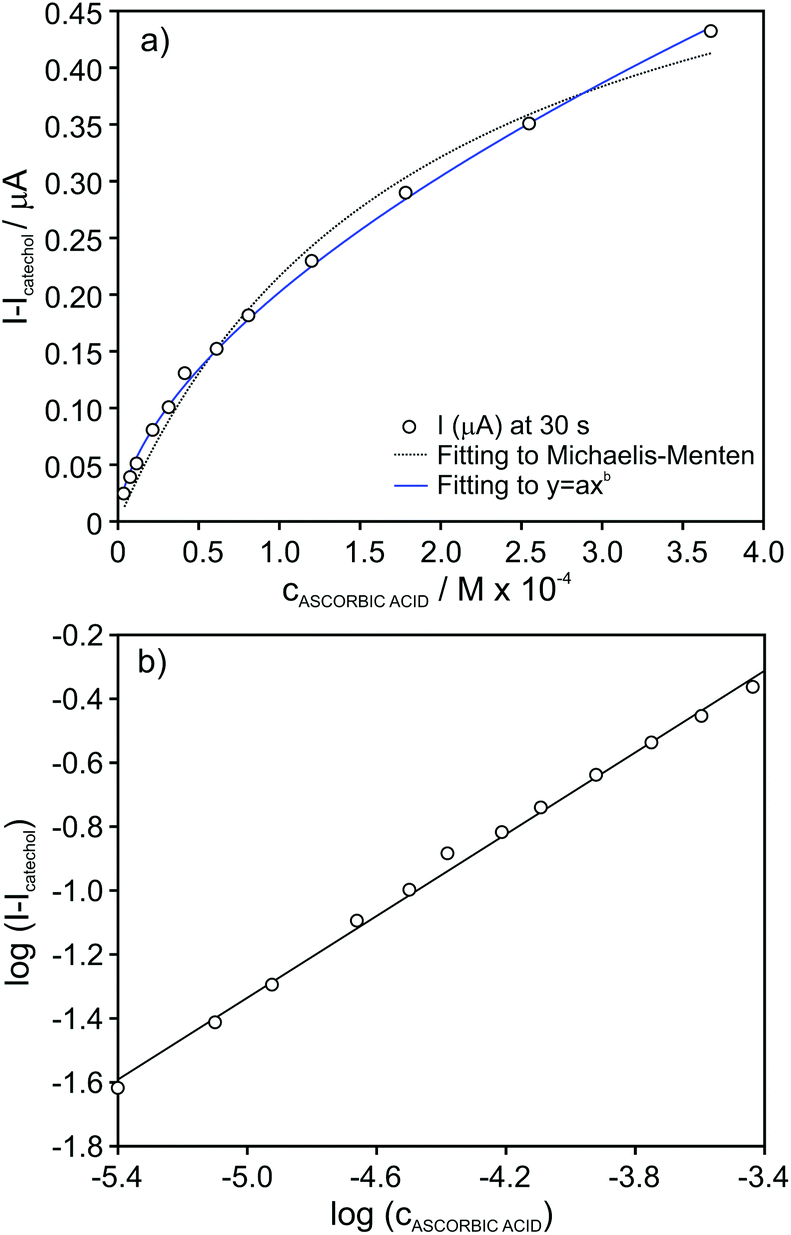 | ||
| Fig. 6 (a) Plot of the absolute current change for increasing concentration of ascorbic acid in the presence of 4 × 10−5 M catechol concentration. (b) Calibration graph after data linearization. | ||
For gallic and kojic acids, the treatment proposed for the observed current signal was different. Essentially, this considers that the maximum change possible for the signal gradually decreases with the consecutive additions of inhibitor, as illustrated in Fig. 7a for the case of gallic acid. This maximum change was calculated from the current corresponding to the baseline of the biosensor in PBS (IBL) and the current displayed as a consequence of the presence of catechol in the sample, which was gradually decreasing from I0 to Ii (with i from 1 to n) after each inhibitor addition. Because the biosensor signal changes after each inhibitor addition (in a magnitude equal to ΔIn), the maximum ΔI reached after each inhibition addition gradually decreases, as illustrated in Fig. 7a. This allows one to calculate a correction for the biosensor signal, called ‘accumulative signal’ (A.S.), as follows:
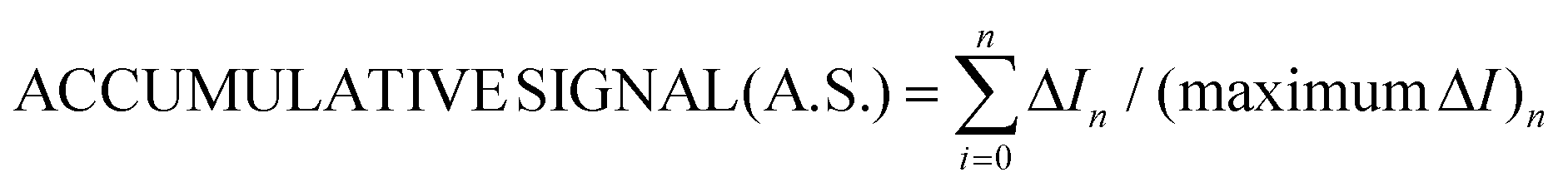 | (3) |
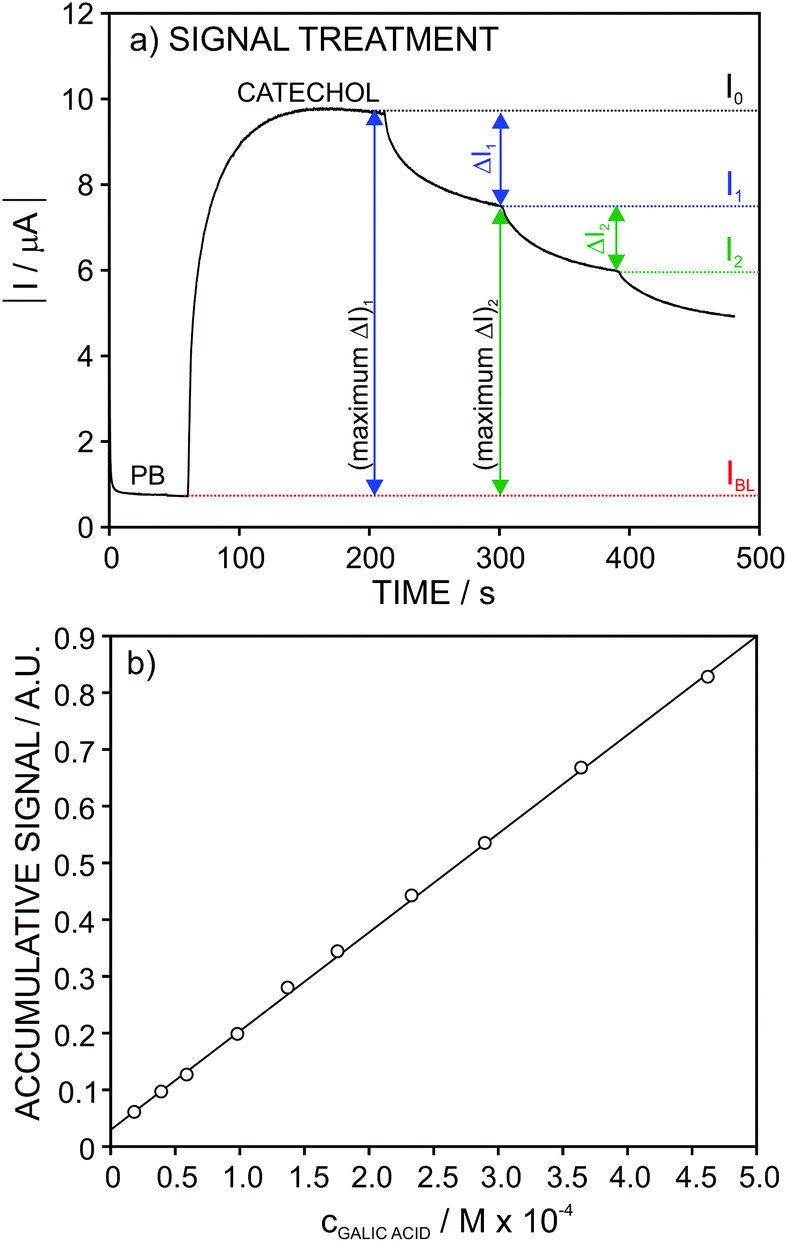 | ||
| Fig. 7 (a) Recorded current response for increasing gallic acid concentration and the description of the parameters used for calculation of the accumulative signal. (c) Calibration graph obtained after linearization. | ||
This new parameter leads to the calibration graph shown in Fig. 7b for gallic acid. An excellent linear fitting was observed (r2 = 0.9996), and it was kept along the entire concentration range (from 2.0 × 10−5 to 4.6 × 10−4 M) tested for the inhibitor. The repeatability and between-electrode reproducibility (n = 3 over a 3-month period) were also evaluated, and the observed calibration was A.S. = (−0.9 ± 2.3) + (25.5 ± 1.6)·cgallic(M × 104) and A.S. = (0.1 ± 1.1) + (23.6 ± 1.6) ·cgallic(M × 104), respectively, confirming excellent performance.
Considering now the data acquired for the kojic acid, one extra step was introduced in the signal treatment seeking for the best linear range of response. While for the ascorbic and gallic acids the analytical signal was considered at a fixed time of 30 and 90 s, respectively, for kojic acid (slow inhibitor) we decided to evaluate a different data treatment in which the experimental dynamic response is fitted to an appropriate equation and then, the extrapolated currents at t = ∞ are used for the construction of the calibration graph. This treatment serves for a double purpose. First, it avoids the need for using a fixed recording time for the current after each analyte addition. And second, for slow inhibitors, it permits considerably reducing the recording time by using recorded signals very far from steady-state currents.
The I–t signals observed at increasing concentrations of kojic acid were then fitted using the following equation:
| I = I∞ + ae−bt | (4) |
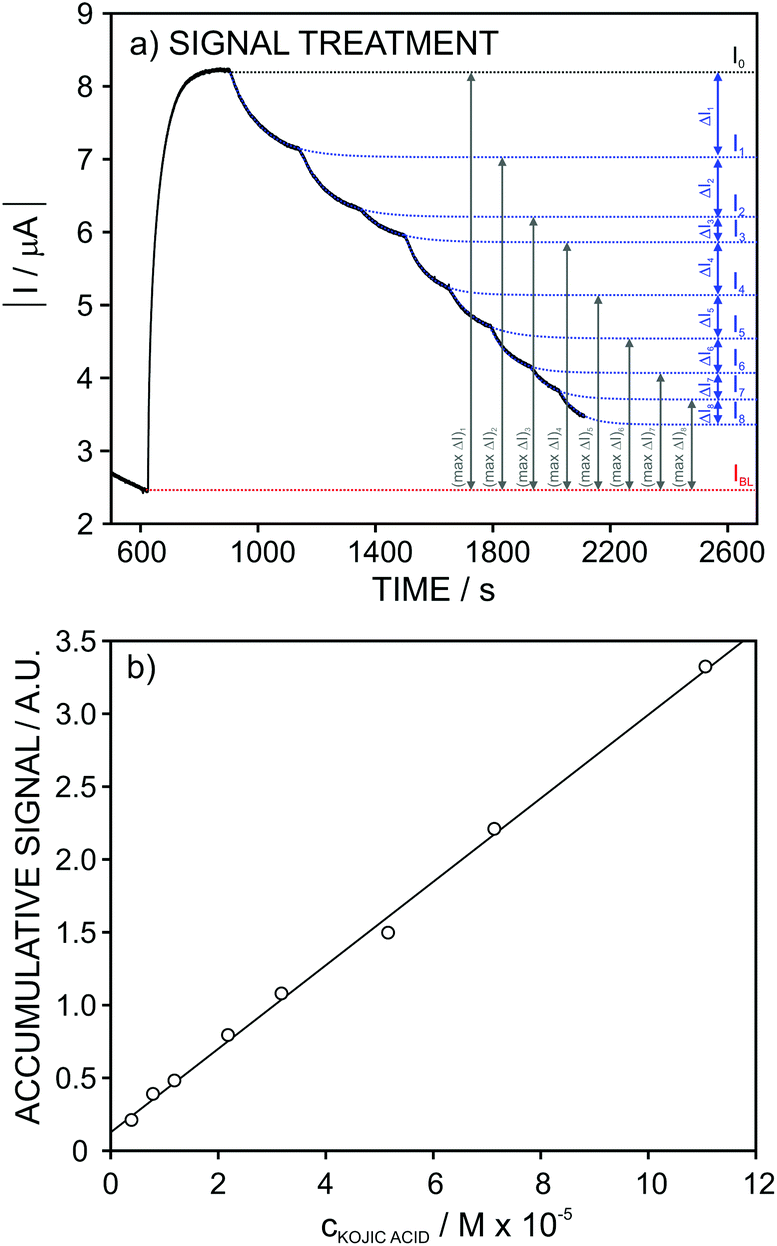 | ||
| Fig. 8 (a) Recorded current response for increasing kojic acid concentration (black) and the corresponding extrapolated fitted curves (dashed blue line). (b) Calibration graph observed after linearization. | ||
Once the current signal was extrapolated to t = ∞, the treatment was exactly the same as for the gallic acid, thus obtaining the accumulative signal (A.S.) at each inhibitor concentration (Fig. 8a).
Fig. 8b depicts the A.S. against the kojic acid concentration, displaying a good linearity (r2 = 0.994) in the range from 4 × 10−6 to 7 × 10−5 M. The repeatability (n = 3) of the calibration parameters was found to be excellent: A.S. = (0.14 ± 0.03) + (0.28 ± 0.01)·ckojic(M × 105). This pointed out that the same electrode may be suitable to be consecutively used when dealing with the analysis of real samples. Furthermore, rather good results were obtained in reproducibility for several electrodes studies over a period of 3 months: A.S. = (0.12 ± 0.03) + (0.29 ± 0.04)·ckojic(M × 105).
Analytical applications
The catechol biosensor was firstly applied to the determination of the total inhibitory capacity expressed as ascorbic acid concentration (g L−1) in different commercial juices. This capacity is due to all the compounds present in the sample capable of influencing the reactions underlying the biosensor amperometric response. For this purpose, the standard addition method was applied as described in the Experimental section.The obtained values are collected in Table 3. In all the samples, the values gathered by the developed biosensor were much higher than the ascorbic acid content labeled by the manufacturer. This may be due to the presence of other compounds in the juices that contribute to the total inhibitory capacity. Also, since the commercial juices are enriched in ascorbic acid, the labeled value in the beverage could mean the added amount instead of the total.
| Juice | Inhibitory capacitya | Labelled (g L−1 ascorbic acid) |
|---|---|---|
| a Expressed as g L−1 ascorbic acid. Mean ± standard deviation (n = 3). | ||
| Orange | 0.82 ± 0.04 | 0.44 |
| Pineapple | 0.76 ± 0.04 | 0.4 |
| Peach | 0.44 ± 0.04 | 0.2 |
| Apple | 0.19 ± 0.04 | — |
| Red grape, redcurrant and pomegranate | 1.25 ± 0.09 | — |
| Peach and grape | 0.47 ± 0.09 | 0.24 |
| Carrot and orange | 0.30 ± 0.03 | 0.32 |
| Apple, kiwi and lime | 0.24 ± 0.01 | — |
| Pear and apple | 0.48 ± 0.05 | 0.2 |
| Apple and mango | 0.61 ± 0.02 | 0.4 |
| Red fruits and acerola | 1.58 ± 0.11 | 0.12 |
The total inhibitory capacity expressed as gallic acid in different kinds of teas was also obtained. The standard addition method was applied according to the protocol described in the Experimental section. This time, the linearity of the current signal recorded at the increasing additions of gallic acid to the corresponding sample was reached by plotting the accumulative signal versus the added concentration.
Table 4 presents the total biosensor inhibitory capacity of the different tea samples expressed as gallic acid together with the total amount of polyphenol content obtained by the well-established Folin-Ciocalteu method also expressed as gallic acid. Interestingly, the inhibitory capacity followed this order: green > white > black > red. Similar inhibitory capacity and total polyphenol content were found for green tea, while lower inhibitory capacity than total polyphenols was found for white and black teas. This may be because not all polyphenols present in the teas contribute to the inhibitory capacity to the same extent.
Finally, we obtained the ‘total depigmenting efficacy’ in one cosmetic product. The application of the biosensor for this purpose is very interesting due to the high demand of this type of cosmetics in the market, as depigmenting cosmetics help in the treatment of skin hyperpigmentation. Notably, its composition traditionally includes species such as arbutin, ferulic acid, ascorbic acid and kojic acid among others.40
The ‘total depigmenting efficacy’ is herein presented as the kojic acid concentration expressed in mg L−1 for a commercial ampoule with the mentioned characteristics. Thus, the ampoule was analysed following the procedure described in the Experimental section, providing a value of 24.5 ± 1.11 mg L−1 (n = 3) for the ‘total depigmenting efficacy’.
Conclusions
Powder lyophilised mushrooms are an excellent and low-cost material for the preparation of polyphenol oxidase-based amperometric electrodes. Importantly, no lyophilisation protector is necessary because this sustainable material keeps its enzymatic activity over years just by storing it in a refrigerator. The carbon paste prepared with this enzymatically active vegetal tissue is also rather stable and provides reproducible electrodes for the determination of the substrate catechol and of other compounds influencing the reactions underlying the amperometric response: reversible competitive inhibitors, rapid (benzoic acid) and slow (kojic acid), alternative substrates as inhibitors (gallic acid) and quinone reductants (ascorbic acid). Signal processing innovations presented expand the concentration range of linear calibration, avoid the need for using a fixed recording time and permit reducing it considerably. The biosensors are suitable for the analysis of juices, wines and cosmetics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under Marie Skłodowska-Curie grant agreement no. 792824.References
- Y. C. Tsai and C. C. Chiu, Sens. Actuator, B, 2007, 125, 10–16 CrossRef CAS.
- S. Lupu, C. Lete, P. C. Balaure, D. I. Caval, C. Mihailciuc, B. Lakard, J. Y. Hihn and F. J. del Campo, Sensors, 2013, 13, 6759–6774 CrossRef CAS PubMed.
- F. A. Abd Manan, W. W. Hong, J. Abdullah, N. A. Yusof and I. Ahmad, Mater. Sci. Eng., C, 2019, 99, 37–46 CrossRef PubMed.
- I. M. Apetrei and C. Apetrei, Materials, 2019, 12, 1009 CrossRef CAS PubMed.
- R. M. Apetrei, G. Carac, G. Bahrim and P. Camurlu, Enzyme Microb. Technol., 2019, 128, 34–39 CrossRef CAS PubMed.
- R. Mossanha, C. A. Erdmann, C. S. Santos, K. Wohnrath, S. T. Fujiwara and C. A. Pessoa, Sens. Actuator, B, 2017, 252, 747–756 CrossRef CAS.
- P. Diez, R. Villalonga, M. L. Villalonga and J. M. Pingarron, J. Colloid Interface Sci., 2012, 386, 181–188 CrossRef CAS PubMed.
- R. Villalonga, P. Diez, S. Casado, M. Eguilaz, P. Yanez-Sedeno and J. M. Pingarron, Analyst, 2012, 137, 342–348 RSC.
- A. Gutes, F. Cespedes, S. Alegret and M. del Valle, Biosens. Bioelectron., 2005, 20, 1668–1673 CrossRef CAS PubMed.
- J. Kochana, J. Kozak, A. Skrobisz and M. Wozniakiewicz, Talanta, 2012, 96, 147–152 CrossRef CAS PubMed.
- D. Shan, Q. B. Li, H. G. Xue and S. Cosnier, Sens. Actuator, B, 2008, 134, 1016–1021 CrossRef CAS.
- S. Q. Li, Y. Y. Tan, P. Wang and J. Q. Kan, Sens. Actuator, B, 2010, 144, 18–22 CrossRef CAS.
- S. E. Stanca and I. C. Popescu, J. Mol. Catal. B: Enzym., 2004, 27, 221–225 CrossRef CAS.
- J. Wang, S. A. Kane, J. Liu, M. R. Smyth and K. Rogers, Food Technol. Biotechnol., 1996, 34, 51–55 CAS.
- J. Wang and M. S. Lin, Anal. Chem., 1988, 60, 1545–1548 CrossRef CAS PubMed.
- A. W. O. Lima, V. B. Nascimento, J. J. Pedrotti and L. Angnes, Anal. Chim. Acta, 1997, 354, 325–331 CrossRef CAS.
- D. C. Wijesuriya and G. A. Rechnitz, Biosens. Bioelectron., 1993, 8, 155–160 CrossRef CAS PubMed.
- S. Topcu, M. K. Sezginturk and E. Dinckaya, Biosens. Bioelectron., 2004, 20, 592–597 CrossRef CAS PubMed.
- M. Rahimi-Mohseni, J. B. Raoof, R. Ojani, T. A. Aghajanzadeh and A. B. Hashkavayi, Int. J. Biol. Macromol., 2018, 113, 648–654 CrossRef CAS PubMed.
- A. Erdem, N. Altinigne, E. Kilinç, L. Gokgunneç, T. Dalbasti and M. Ozsoz, Collect. Czech. Chem. Commun., 1991, 56, 1427–1433 CrossRef.
- P. Skladal, Collect. Czech. Chem. Commun., 1991, 56, 1427–1433 CrossRef CAS.
- W. Wang, Int J Pharm., 2000, 203, 1–60 CrossRef CAS PubMed.
- R. Pisano, V. Rasetto, A. A. Barresi, F. Kuntz, D. Aoude-Werner and L. Rey, Eur. J. Pharm. Biopharm., 2013, 85, 974–983 CrossRef CAS PubMed.
- I. Roy and M. N. Gupta, Biotechnol. Appl. Biochem., 2004, 39, 165–177 CrossRef CAS PubMed.
- J. Frank, M. Richter, C. de Rossi, C. M. Lehr, K. Fuhrmann and G. Fuhrmann, Sci. Rep., 2018, 8, 112377 Search PubMed.
- L. M. C. Silva, A. C. C. de Mello and A. M. Salgado, Environ. Technol., 2014, 35, 1012–1017 CrossRef CAS PubMed.
- J. L. Munoz-Munoz, F. Garcia-Molina, P. A. Garcia-Ruiz, M. Molina-Alarcon, J. Tudela, F. Garcia-Canovas and J. N. Rodriguez-Lopez, Biochem. J., 2008, 416, 431–440 CrossRef CAS PubMed.
- Y. J. Kim, Biol. Pharm. Bull., 2007, 30, 1052–1055 CrossRef CAS PubMed.
- N. Alam, K. N. Yoon, J. S. Lee, H. J. Cho and T. S. Lee, Saudi J. Biol. Sci., 2012, 19, 111–118 CrossRef CAS PubMed.
- T. S. Chang, Int. J. Mol. Sci., 2009, 10, 2440–2475 CrossRef CAS PubMed.
- S. Zolghadri, A. Bahrami, M. T. H. Khan, J. Munoz-Munoz, F. Garcia-Molina, F. Garcia-Canovas and A. A. Saboury, J. Enzyme Inhib. Med. Chem., 2019, 34, 279–309 CrossRef CAS PubMed.
- C. V. Ortiz-Ruiz, M. D. Garcia-Molina, J. T. Serrano, V. Tomas-Martinez and F. Garcia-Canovas, J. Agric. Food Chem., 2015, 63, 2162–2171 CrossRef CAS PubMed.
- Y. Hasebe, K. Oshima, O. Takise and S. Uchiyama, Talanta, 1995, 42, 2079–2085 CrossRef CAS PubMed.
- S. Skrovankova, J. Mlcek, J. Sochor, M. Baron, J. Kynicky and T. Jurikova, Int. J. Electrochem. Sci., 2015, 10, 2421–2431 CAS.
- U. J. Unachukwu, S. Ahmed, A. Kavalier, J. T. Lyles and E. J. Kennelly, J. Food Sci., 2010, 75, C541–C548 CrossRef CAS PubMed.
- J. N. Rodriguezlopez, J. R. Ros, R. Varon and F. Garciacanovas, Biochem. J., 1993, 293, 859–866 CrossRef CAS PubMed.
- L. G. Fenoll, J. N. Rodriguez-Lopez, F. Garcia-Molina, F. Garcia-Canovas and J. Tudela, Int. J. Biochem. Cell Biol., 2002, 34, 332–336 CrossRef CAS PubMed.
- J. C. Espin, R. Varon, L. G. Fenoll, M. A. Gilabert, P. A. Garcia-Ruiz, J. Tudela and F. Garcia-Canovas, Eur. J. Biochem., 2000, 267, 1270–1279 CrossRef CAS PubMed.
- M. Cuartero, M. S. Garcia, F. Garcia-Canovas and J. A. Ortuno, Talanta, 2013, 110, 8–14 CrossRef CAS PubMed.
- Y. Funasaka, A. K. Chakraborty, M. Komoto, A. Ohashi, K. Inoue, K. P. Wittern and M. Ichihashi, J. Invest. Dermatol., 1997, 108, 542–542 Search PubMed.
Footnote |
| † Current address: CEMHTI-CNRS, 45071 Orléans Cedex, France. |
| This journal is © The Royal Society of Chemistry 2020 |