
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Glycosylation profiling of intact target proteins by high-resolution mass spectrometry (MS) and glycan analysis using ion mobility-MS/MS†
Alessandro
Quaranta
 *a,
Maya
Spasova
a,
Elena
Passarini
a,
Isabella
Karlsson
a,
Lorena
Ndreu
a,
Gunnar
Thorsén
b and
Leopold L.
Ilag
a
*a,
Maya
Spasova
a,
Elena
Passarini
a,
Isabella
Karlsson
a,
Lorena
Ndreu
a,
Gunnar
Thorsén
b and
Leopold L.
Ilag
a
aDepartment of Environmental Science and Analytical Chemistry, Stockholm University, 10691 Stockholm, Sweden. E-mail: alessandro.quaranta@aces.su.se; alessandro.quaranta@ki.se
bIVL Swedish Environmental Research Institute, 11428 Stockholm, Sweden
First published on 17th December 2019
Abstract
Glycosylation influences the structure and functionality of glycoproteins, and is regulated by genetic and environmental factors. The types and abundance of glycans on glycoproteins can vary due to diseases such as cancer, inflammation, autoimmune and neurodegenerative disorders. Due to the crucial role glycans play in modulating protein function, glycosylation analysis could lead to the discovery of novel biomarkers and is of prime importance in controlling the quality of glycoprotein biopharmaceuticals. Here, we present a method for the identification and quantification of glycoforms directly on intact proteins, after immunoaffinity purification from biological fluids. The method was validated and applied to serum transferrin and the biopharmaceutical trastuzumab. The accuracy of the method, expressed as the relative error (RE), ranged from 2.1 (at high concentrations) to 7.9% (at low concentrations), and intra- and inter-day precision, expressed as relative standard deviation (RSD), was 3.2 and 8.2%, respectively. The sensitivity and linearity of the method were suitable for serum analysis and the LOQ was calculated to be 3.1 and 4.4 μg mL−1 for transferrin (TFN) and trastuzumab (TRA), respectively. Its application to transferrin from five healthy human serum samples yielded concentrations between 1.61 and 3.17 mg mL−1, which are in agreement with blood reference levels. In parallel, the structure of the identified glycans was determined by ion mobility spectrometry coupled with tandem mass spectrometry. No chromatographic separation was required and sample preparation was performed in a semi-automatic manner, facilitating the handling of up to 12 samples at a time. This method should be useful for clinical laboratories and for the quality control of large batches of biopharmaceuticals.
Introduction
Glycosylation is a post-translational modification (PTM) that significantly influences a large number of biological functions of proteins; it ensures the correct folding, regulates the residence time in blood, and modulates a large variety of signaling processes.1 Unlike nucleic acid and protein syntheses, the addition of carbohydrate units to form glycan structures is not template driven, but is carried out in the endoplasmic reticulum and in the Golgi apparatus by several glycosidases and glycosyltransferases.2 These enzymes can be influenced by several factors, both genetic and environmental, leading to a variety of glycosylation patterns that are preserved on proteins as they are released from the cell into the bloodstream.3,4 Since several diseases, such as cancer, neurodegenerative disorders, and autoimmune diseases, are known to affect glycosylation, the identification of associated alterations could lead to the discovery of reliable glycan biomarkers.3,5–7 Additionally, due to the crucial role glycans play in modulating protein function, the analysis of glycosylation is of prime importance in the quality control of glycoprotein biopharmaceuticals.8 It has been reported that alteration in the glycosylation pattern of biopharmaceuticals can impair their function, decrease their serum lifetime, and cause immune reactions.8,9Three major strategies are currently employed for characterization of protein glycosylation, namely: release of glycans, glycopeptide analysis, and intact glycoprotein analysis.10–12 Glycans can be released from proteins both chemically and enzymatically, separated by liquid chromatography (LC) or capillary electrophoresis (CE), and then analyzed by mass spectrometry (MS) or fluorescence detection (FLD). Derivatization is required to incorporate chromophores for spectroscopic detection or to improve separation and MS detection.13–16
Analysis of the released glycans is often performed by ESI-MS and MALDI-ToF MS. MALDI-based methods have advantages in terms of required analysis time, solvent consumption, and simplicity of the generated data.17,18 The limitation of this approach, however, is the loss of information about sialic acids since these sugars are commonly lost under MALDI ionization conditions, unless derivatization to stabilize the labile α-glycosidic bond is performed.19
Glycopeptides are analyzed after enzymatic digestion both by LC-MS and MALDI-ToF MS, providing additional information on the glycosylation sites and their occupancy rate.20,21 This approach requires long chromatographic separations, high energies to fragment glycopeptides, and extremely reproducible protein digestion, which are difficult to achieve for highly glycosylated proteins.20 All these procedures involve the use of enzymes and reagents, and their efficiencies cannot be quantitatively accounted for, thus introducing uncertainty in the evaluation of the results. In recent years, ion mobility spectrometry-mass spectrometry (IMS-MS) has become a valid tool for the analysis of carbohydrates, providing an additional dimension of structural information based on mass, charge, and shape.22,23 IMS, often combined with other separation techniques, has been used for oligosaccharides24 and N-glycans17,25,26 and has proved quite successful in isomer separation; although, mostly for smaller oligosaccharides, given the current resolution achieved by commercially available instruments.27
The characterization of glycosylation directly on intact proteins by high-resolution MS provides an alternative approach, as the different glycoforms will be determined based on the mass difference from the deglycosylated proteins, eliminating potential work-up artifacts. Moreover, other PTMs can be detected simultaneously with glycosylation.28 However, structural information can be limited, unless advanced instrumentation capable of fragmenting the intact proteins is available (so-called top-down analysis), and even then the identification of proteoforms larger than 30 kDa has proven difficult due to the loss of sensitivity for large molecules.29
Here, we have developed a method that combines the benefits of high-resolution MS of intact proteins with the possibility of performing structural elucidation by IMS-MS/MS. This approach was then applied to determine the glycosylation profile of two intact glycoproteins in complex matrices, namely, the iron transporter transferrin and the biopharmaceutical trastuzumab. Transferrin is a negative acute phase protein with a molecular weight of 77 kDa (excluding glycosylation) having two N-glycosylation sites, one of which is buried and inaccessible to enzymatic digestion.30 Alterations in its glycosylation pattern have been found to be related to hepatocellular and stomach cancer,5,31 chronic alcoholism,32 and Alzheimer's disease.6 Trastuzumab, sold under the commercial name of Herceptin, is a recombinant humanized IgG1 with a molecular weight of 147 kDa. It is produced in Chinese hamster ovary (CHO) cells and used in targeted therapy for HER2-positive metastatic breast cancer.33 Glycosylation occurs in both the heavy chains of the fragment crystallizable (Fc) region.34 TFN was extracted from human serum by a magnetic bead-based immunoaffinity purification method and after elution from the beads and desalting, it was directly injected into the high-resolution mass spectrometer without any chromatographic separation except for a C18 guard column used to focus the injection band and to avoid ion suppression. In the case of TRA, being the major component of the cell supernatant in production systems, no immunoaffinity purification was required so the proteins were analyzed after dilution and solvent exchange. The obtained spectra were deconvoluted using maximum entropy-based software, allowing for a quantitative approach, and the method was validated for both proteins. In parallel, to provide structural elucidation of the identified glycoforms, PNGase F-released glycans were analyzed by ESI-IMS-MS/MS. In this case, the guard column used was a porous graphitized carbon (PGC) cartridge.
Methods
Chemicals and reagents
Pierce streptavidin-coated magnetic beads, Zeba Spin Desalting Columns (7K MWCO), HyperSep porous graphitized carbon (Hypercarb) spin tips 10–200 μL, CaptureSelect Biotin anti-transferrin conjugate, and mouse serum were purchased from Thermo Scientific (Waltham, MA, USA). Standard human transferrin, formic acid (98%), and ammonium acetate (purity 98%) were bought from Sigma-Aldrich (St Louis, MO, USA). Trastuzumab, produced in Chinese hamster ovary (CHO) cells, and the non-transfected cell supernatant were obtained from Gyros (Uppsala, Sweden). Acetonitrile (ACN, analytical grade), isopropanol (IPA, analytical grade), and dimethyl sulfoxide (DMSO, analytical grade) were bought from Honeywell (Charlotte, NC, USA). Water was purified using a Millipore water purification system to a resistance >18 MΩ cm−1.Human serum samples
Commercial human blood samples were acquired from healthy donors through Karolinska Institutet, Sweden, compliant with the Swedish Act on Biobanks – SFS 2002:297 and GDPR. Informed consents were obtained from all human participants of this study. Once received, the samples were centrifuged at 1100g for 15 minutes and the clear serum was aliquoted into 500 μL Eppendorf microtubes and stored at −80 °C until further use.Immunoaffinity purification of TFN
The immunoaffinity capture of transferrin from serum was performed by employing and adapting a bead-based method previously described and characterized by the authors.35,36 Five serum samples were analyzed in parallel: for each sample, four aliquots of streptavidin-coated magnetic bead slurry (50 μL, 10 mg mL−1) were loaded into a 96-well plate and the storage solution was discarded while the beads were kept in place by using a magnetic plate holder. The first two aliquots were used for the duplicate analysis of glycosylation on the intact proteins, while the remaining two aliquots were used for the structural analysis of the released glycans. After washing with phosphate buffered saline (PBS, pH 7.4, 3 × 300 μL), each aliquot was coupled with the anti-transferrin biotinylated CaptureSelect™ camelid single domain antibody fragments (VHHs), by incubation with 150 μL of a 0.04 mg mL−1 (0.4 μmol) solution of VHH in PBS. The incubation was carried out for 1 hour at room temperature under shaking (750 rpm on a Thermomixer, Eppendorf, Hamburg, DE). The unbound VHH was washed away with PBS (3 × 300 μL), and each serum sample (diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 in PBS) was incubated with the bead-bound VHH for 1 hour at room temperature under shaking (750 rpm). Finally, the beads were again washed with 4 × 300 μL of PBS to remove all the unbound components. All the solvent transfer procedures were performed using an electronic automatic 12-channel pipette (Finnpipette F2, 20–300 μL, Thermo Scientific).
:15 in PBS) was incubated with the bead-bound VHH for 1 hour at room temperature under shaking (750 rpm). Finally, the beads were again washed with 4 × 300 μL of PBS to remove all the unbound components. All the solvent transfer procedures were performed using an electronic automatic 12-channel pipette (Finnpipette F2, 20–300 μL, Thermo Scientific).
TFN elution from VHHs, solution exchange, and TRA preparation
To recover the purified TFN from the bead-immobilized VHH, elution was performed by incubation with 150 μL of a solution consisting of H2O/IPA (1:1) and formic acid (1% v/v) for 5 minutes (shaking at room temperature, 750 rpm). The eluted protein was subsequently desalted and solvent exchanged to 150 mM ammonium acetate by using size exclusion spin columns (7 kDa molecular cut-off), operated according to the manufacturer's instructions. Before injection, the volume was adjusted to 200 μL with 150 mM ammonium acetate solution.
TRA was directly spiked in the non-transfected cell supernatant at a concentration of 1.0 mg mL−1. After 1:15 dilution in 150 mM ammonium acetate, the protein was solvent exchanged as described for TFN.
MS analysis and data treatment of intact proteins
All analyses of intact proteins were carried out on a Waters SYNAPT G2S Q-ToF mass spectrometer (Milford, MA, USA), equipped with an ACQUITY UPLC system. To focus the injection band and to avoid ion suppression, a C18 guard column (ACQUITY UPLC CSH™, 2.1 × 5 mm, 1.7 μm, Waters) was used in an isocratic elution system, with the mobile phase consisting of H2O/ACN (7:3) modified with 0.1% v/v formic acid. The flow rate was kept at 0.3 mL min−1, the injection volume was set to 3 μL and, to control the carryover, solvent blanks were injected between the runs. For acquisition, the instrument was operated in resolution mode and in positive ionization mode. The charge envelopes of the obtained proteins were subsequently deconvoluted using Waters MaxEnt1™ software to obtain the mass values of the uncharged protein species. The glycoforms were identified according to the mass difference with the deglycosylated proteins by using the bioinformatics software GlycoWorkbench v2.137 and by comparison with the IMS-MS/MS data generated on the enzymatically released glycans. MS and deconvolution parameters are listed in ESI, Table S1.†
Glycan release for IMS analysis
Before enzymatic release could take place, the bead aliquots for glycan analysis were moved to clean wells in the plate, to avoid the contribution of glycans coming from proteins unspecifically adsorbed to the plastic of the used wells. TFN was first denatured for 10 minutes at 90 °C in the presence of a reducing buffer, in order to expose the buried glycosylation site and, after cooling to room temperature, deglycosylated by incubation with 500 U of PNGase F for 3 h at 37 °C under shaking (750 rpm). After incubation, the supernatant containing the released glycans was collected and purified by using porous graphitized carbon (PGC) solid-phase extraction (SPE) spin tips. The tips were first conditioned with 200 μL of ACN and 2 × 150 μL of ACN/H2O (6:4), followed by 2 × 150 μL of H2O. After sample loading, the tips were washed with 150 μL of H2O and the glycans were subsequently eluted in 300 μL of ACN/H2O (6:4).
TRA was directly spiked in the non-transfected cell supernatant at a concentration of 1.0 mg mL−1 and was deglycosylated in solution, after solvent exchange to H2O, under the same conditions as described for TFN.
IMS-MS/MS analysis of the released glycans
Analyses of the released glycans were carried out on the same SYNAPT G2S Q-ToF mass spectrometer used for the intact proteins. Similarly to intact proteins, the injection band was focused by means of a guard column, which also separated the glycans from non-retained components. The guard column was a PGC cartridge (10 × 4 mm, 5 μm, Thermo Scientific), operated in an isocratic elution system. Different mobile phases were used for the glycans coming from the two proteins: TFN glycans (sialylated) were analyzed in negative ionization mode, while TRA glycans (neutral) were analyzed in positive ionization mode. The two mobile phases consisted of ACN/H2O (3:7) and NH4OH (0.1 M) for the negative mode, and ACN/H2O (3:7) and formic acid (0.1% v/v) for the positive mode. The flow rate was kept at 0.3 mL min−1 and the injection volume was set to 5 μL.
The MS spectrometer was operated in a single reflectron “sensitivity mode” for the analysis of glycans in both positive and negative modes. Glycans were separated in the mobility cell and subsequently fragmented by collision-induced dissociation (CID) in the transfer cell, positioned between the IMS cell and the ToF. The detailed parameters for ESI ionization, IMS separation, and MS/MS fragmentation in both positive and negative modes are reported in Tables S2 and S3 in the ESI.† Data acquisition and processing were accomplished by using the Waters DriftScope software (version 2.8) and MassLynx (version 4.1). The fragmentation spectra of glycans were interpreted with the help of the bioinformatics software GlycoWorkbench v2.1.37
Method validation
The use of MaxEnt1™ allowed quantitative analysis directly from the deconvoluted mass spectra, as it provides zero charge spectra where the areas under the peaks reflect the summed intensity of the charge states that generated the peaks.38 Six-point calibration curves (ranging from 0.005 to 0.1 mg mL−1) were prepared for both the target proteins to quantify the different glycoforms and to calculate the recoveries for the elution and solution exchange steps. Quality control (QC) samples at two concentration levels (low 0.008 mg mL−1 and high 0.08 mg mL−1) were also prepared and used to calculate the inter- and intra-day accuracy of the method.The matrix effect was estimated by subjecting a blank mouse serum to the analytical procedure, post-spiking TFN at low (LCME, 0.008 mg mL−1) and high (HCME, 0.08 mg mL−1) concentration levels, and comparing the response factors in the matrix with the ones from the standard solutions of TFN in pure solvent. The matrix effect was estimated in the same way as for TRA, although the non-transfected cell supernatant was used, as it was deemed a more relevant matrix.
Results
Analysis of intact proteins
Direct analysis of the intact target proteins was first performed on standard compounds to test the possibility of discriminating the different glycoforms. Typical spectra with related deconvolution are shown in Fig. 1. The structure, m/z values, nomenclature, and mass accuracy of the identified glycans are reported in Table S4.† In the following text, glycans will be named according to their saccharide composition, with conventional abbreviations: H, hexose; N,N-acetyl hexose; F, fucose; S, sialic acid, followed by the number of respective units.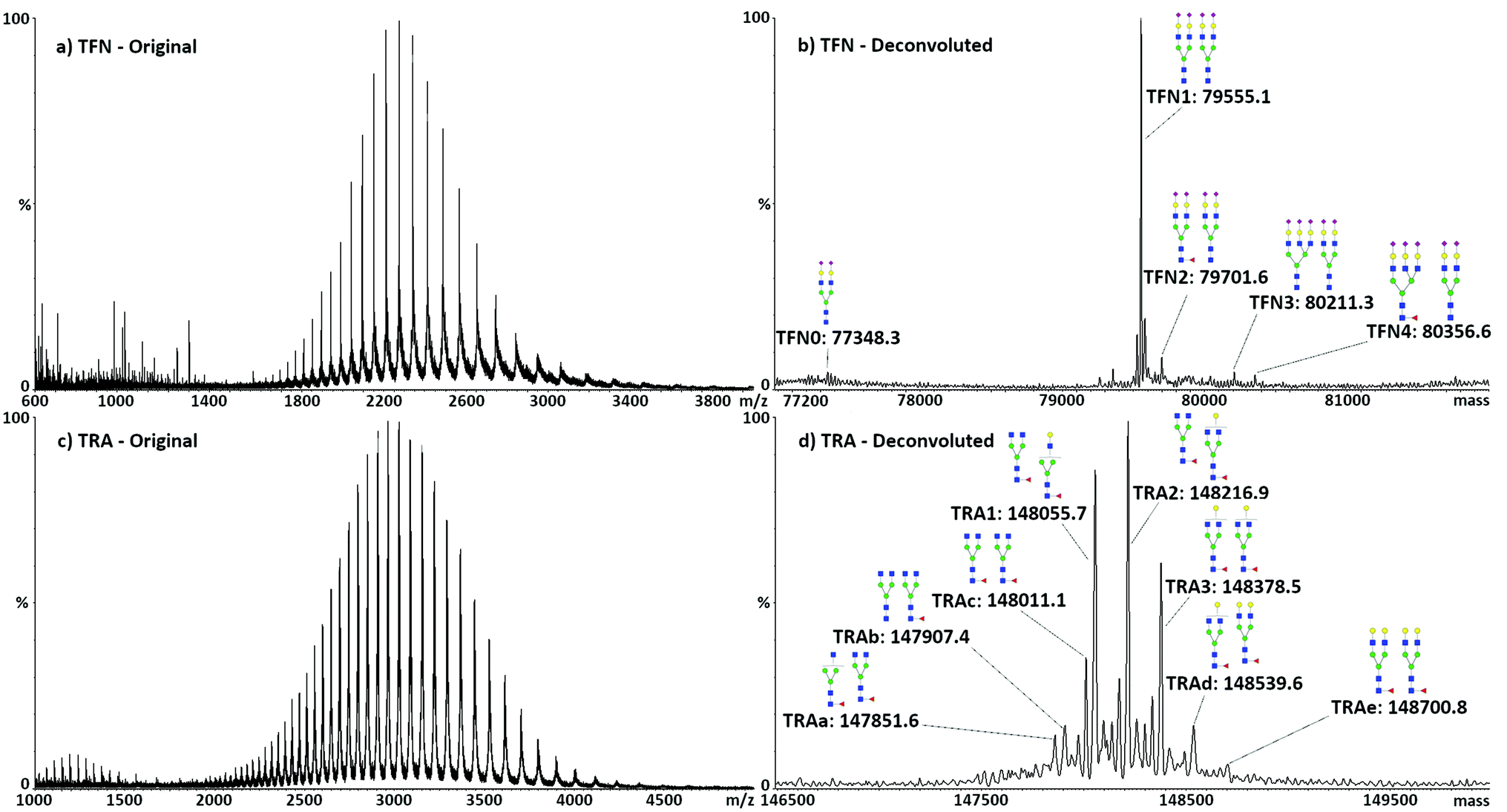 | ||
| Fig. 1 Representative MS spectra of intact TFN and TRA, and subsequent deconvolution in the relevant mass range. (a) MS full scan of TFN standard solution (0.1 mg mL−1) and (b) related deconvolution and identification of the protein glycoforms. (c) MS full scan of TRA standard solution (0.1 mg mL−1) and (d) related deconvolution and identification of the protein glycoforms. Monosaccharide symbols follow the SNFG (symbol nomenclature for glycans): N-acetylglucosamine (GlcNAc): blue squares; mannose (Man): green circles; galactose (Gal): yellow circles; fucose (Fuc): red triangles; sialic acid (Neu5Ac): purple diamonds. | ||
As expected, the major TFN glycoform consisted of two biantennary di-sialylated glycans (H5N4S2), accounting for 80% of the total protein. The remaining glycoforms always expressed H5N4S2 in one of the two glycosylation sites, with the other showing different complex species, namely, H5N4S2F (10%), H6N5S3 (5%), and H6N5S3F (4%). Traces of mono-glycosylated TFN, showing only one H5N4S2 glycan, were also identified, accounting for less than 1% of the total, with a mass of 77330.6 Da. As analysis is performed on the intact proteins, both glycosylation sites were accounted for without the necessity to denature the protein to make the hindered glycosylation site accessible to enzymes for glycan release. The obtained results showed good agreement with the literature30 and previous MALDI-MS analyses of desialylated TFN glycans performed by our group.35 The five identified glycoforms of TFN were named TFN0 (mono-glycosylated TFN, only H5N4S2, mass 79348.3 Da), TFN1 (H5N4S2 and H5N4S2, mass 79555.1 Da), TFN2 (H5N4S2 and H5N4S2F, mass 79701.6 Da), TFN3 (H5N4S2 and H6N5S3, mass 80211.3 Da), and TFN4 (H5N4S2 and H6N5S3F, mass 80356.6 Da). TFN1 was chosen for the following quantitative analysis.
Similar to TFN, TRA has two glycosylation sites on the heavy chains of the FC region. Several combinations of glycans were observed in these two sites, consisting of biantennary species with different levels of galactosylation and possibility of fucosylation. Seven glycoforms could be identified: the three major ones, accounting for almost 70% of the total protein, were named TRA1 (H3N4F and H3N4F, mass 148055.7 Da), TRA2 (H4N4F and H3N4F, mass 148216.9 Da), and TRA3 (H4N4F and H4N4F, mass 148378.5 Da). Quantitative analysis was subsequently performed on the sum of these glycoforms. The other minor forms were named TRAa (H3N3F and H3N4F, mass 147851.6 Da), TRAb (H3N4 and H3N4F, mass 147907.4 Da), TRAc (H4N3F and H3N4F, mass 148011.1 Da), TRAd (H5N4F and H4N4F, mass 148539.6 Da), and TRAe (H5N4F and H5N4F, mass 148700.8 Da).
Method evaluation
The parameters studied to characterize the performance of the method were linearity, limit of detection (LOD), limit of quantification (LOQ), accuracy, precision, and the matrix effect. The linear range was studied between 0.005 and 0.1 mg mL−1 for both the target proteins, with correlation coefficients (R2) higher than 0.99 for both proteins. The calibration curves consisted of 6 concentration levels; two quality control samples, one at a high concentration (HQC: 0.08 mg mL−1) and one at a low concentration (LQC: 0.008 mg mL−1), were used to calculate the accuracy and inter- and intra-day precision. Equations and correlation coefficients are reported in Fig. S2.† Accuracy, expressed as the relative error (RE) between the measured and theoretical concentration values of the QC samples, and precision, expressed as relative standard deviation (RSD) calculated on triplicate samples, are reported in Table 1. The LOD and LOQ were defined as the concentration equivalent to signal/noise = 3 and 10, respectively. All the values, calculated using the aforementioned definitions for the two proteins, are reported in Table 1.| HQC accuracy (RE, %) | LQC accuracy (RE, %) | Inter-day precision (RSD, %) | Intra-day precision (RSD, %) | LOD concentration (μg mL−1) | LOQ concentration (μg mL−1) | |
|---|---|---|---|---|---|---|
| TFN | 2.1 | 7.9 | 8.2 | 3.8 | 0.9 | 3.1 |
| TRA | 3.1 | 5.9 | 4.2 | 3.2 | 1.3 | 4.4 |
Recovery experiments were carried out for the whole immunoaffinity purification procedure on the standard TFN, and the results showed an average value of 37.8 ± 1.7%. Considering the reference concentration values of TFN in human serum, these values would correspond to the final TFN concentrations in solution ranging between 0.02 and 0.03 mg mL−1, which would be in the middle of the calibration range. Characterization of the method showed that elution of the protein from the VHH was the limiting step in the total yield, and several attempts to improve this step were made. The best results were obtained by incubating the VHH-bound protein with HCOOH (0.1% v/v) and isopropanol/H2O (1:1) for 5 minutes, followed by solvent exchange to 150 mM ammonium acetate. The low recovery values can be explained by the very high binding affinity of the VHHs for their target proteins, which resulted in extremely efficient capture and difficult release.
Selectivity of the immunoaffinity purification
To assess the selectivity of the immunoaffinity recovery and to identify eventual contributions from proteins aspecifically captured during the bead-based procedure, negative control experiments were performed. As it is not possible to obtain human serum without TFN, mouse serum was used as a target protein-free matrix, since the anti-TFN VHH does not cross-react with murine TFN. To test the cross-reactivity with human proteins, mouse serum was spiked at relevant blood concentration levels with both human serum albumin and TRA; the latter was used to mimic the presence of IgG. Again, no significant signal could be observed. In both cases, no significant signal could be detected, indicating the absence of relevant cross-reactivity or contaminations and the efficiency of the sample preparation procedure. The potential presence of unspecifically adsorbed proteins was tested by subjecting human serum samples to the whole analytical procedure on non-derivatized beads. The eluates were subsequently analyzed, revealing the absence of detectable protein signals. Representative spectra for the negative control experiments are shown in Fig. S3.† Matrix blanks for TRA were prepared by diluting and solvent exchanging the non-transfected non-spiked cell supernatant, also showing the absence of relevant signals. An example is shown in Fig. S3.†Matrix effect estimation
The matrix effect was estimated for both proteins in their related matrices, both at high and low concentrations. TFN was post-spiked in mouse serum at two different concentration levels, 0.08 mg mL−1 (HC) and 0.008 mg mL−1 (LC), with the response factor compared to the one produced by the same protein concentration in 150 mM ammonium acetate. Similarly, TRA was post-spiked in the non-transfected cell supernatant at the same two concentration levels, and the response factor compared with the ones in solvent. The values for both proteins are reported in Table 2, expressed as the percent difference of the relative error.| HC matrix effect (RE, %) | LC matrix effect (RE, %) | |
|---|---|---|
| TFN | −2.2 | 2.9 |
| TRA | 8.6 | 9.1 |
Structural elucidation by IMS-MS/MS
To structurally identify the glycoforms expressed by the target proteins, the enzymatically released glycans were analyzed by travelling wave ion mobility (TWIMS)39 coupled with tandem MS. Acidic glycans released from TFN were ionized in negative mode. All the four expected species were identified as [M − 2H]2− and, as expected, H5N4S2 produced the most intense peak, while only traces of tri-antennary glycans could be detected, ionizing as well as [M − 2H]2− (Fig. 2a). Furthermore, several minor components were observed, in some cases representing sodium adducts (mostly for doubly charged tri-antennary species) or triply charged species. The signal appearing at m/z 1170.9, which was present when analyzing the glycans released from both the standard and serum TFN, could not be assigned. Signals compatible with this species were not detected when analyzing the same solutions by MALDI-MS, both with and without sialic acids (data not shown).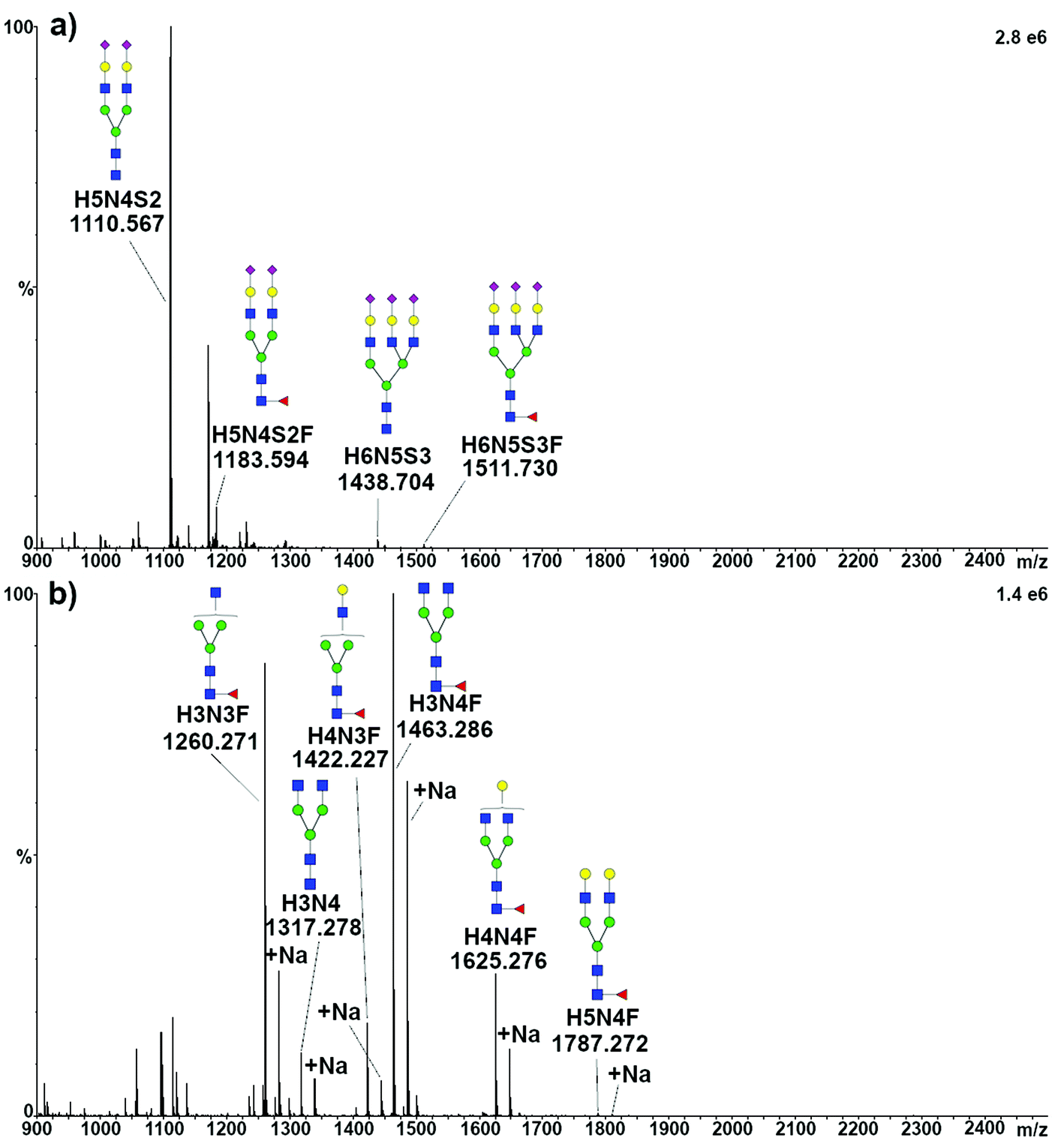 | ||
| Fig. 2 Representative full scan MS spectra of the glycans released from TFN and TRA. (a) Negative ionization mode analysis of sialylated glycans released from TFN. (b) Positive ionization mode analysis of glycans released from TRA. Glycans are named according to their saccharide composition, with conventional abbreviations: H, hexose; N,N-acetyl hexose; F, fucose; and S, sialic acid, followed by the number of respective units. | ||
Neutral glycans released from TRA were ionized in positive mode, resulting in the identification of five species, together with the respective sodium adducts. As expected, the most intense peak was related to the H3N4F glycan, the major component of TRA1 and TRA2, while a very intense signal was generated by the H3N3F glycan, identified only in the minor glycoform TRAa (Fig. 2b).
The SYNAPT G2S instrument used is equipped with a tri-wave cell, which consists of a trap and a transfer cell, where CID can be performed, and a travelling wave drift tube, where the separation takes place. For structural elucidation, glycans were separated in the TWIMS cell and subsequently fragmented in the transfer cell, in order to maintain a relationship between the arrival distribution time of each parent and its fragments.
Analysis of acidic glycans released from TFN
TWIMS of acidic glycans released from TFN allowed the baseline separation of di-antennary and tri-antennary glycans, but only minor separation was achieved between species differing only in terms of the presence of a fucose unit. Mobilograms related to the analysis of acidic glycans released from TFN and related tandem MS spectra acquired in negative ionization mode are shown in Fig. 3.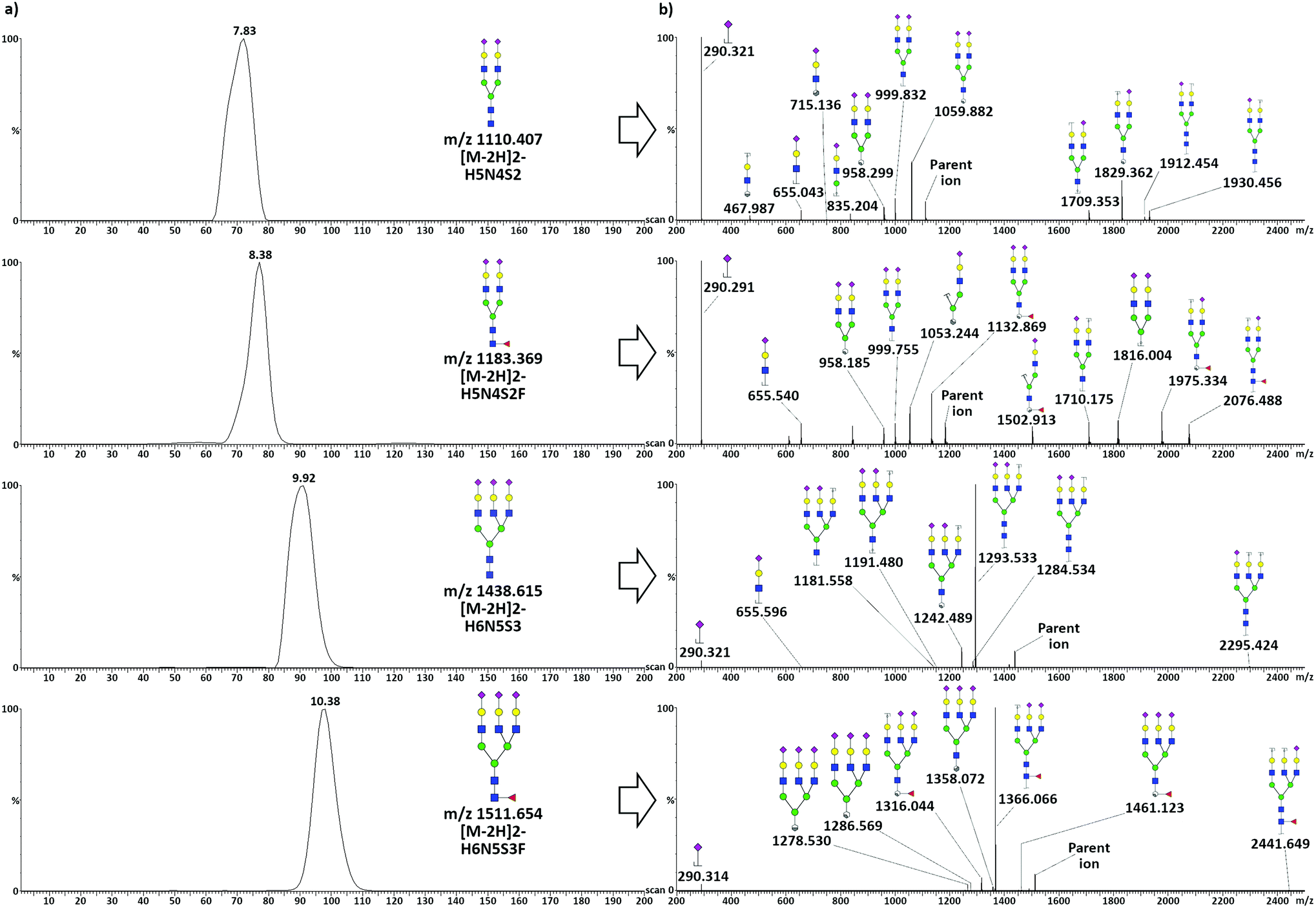 | ||
| Fig. 3 Structural elucidation of acidic glycans after the enzymatic release from TFN and subsequent TWIMS separation. (a) TWIMS mobilograms illustrating the resolved mobilities of the four target glycans and (b) related MS/MS spectra obtained by using the quadrupole as a mass filter and fragment identification. An intensity threshold limit of 3% of the base peak intensity was set to exclude low-intensity fragments. In the case of H6N5S3 and H6N5S3F, due to the high absolute intensity of the main fragment (m/z 1293.533 and 1366.066 respectively), the threshold limit was set on the 3% of the intensity of the peak of the parent ion. | ||
To obtain pure fragmentation spectra for each glycan, the quadrupole was used as a mass filter, allowing the separate transmission of the four precursor glycan ions to the mobility cell. After fragmentation in the transfer tube, only fragments reflecting the arrival time distribution of each parent were considered, and an intensity threshold limit was set to exclude low-intensity (i.e. unreliable) fragments. Three percent of the base peak was set for the signal coming from di-antennary glycans, whereas for the tri-antennary glycans, due to the intensity of the main fragment relative to the minor ones, the value was set to 3% of the intensity of the parent ion. For all the considered species, the loss of a sialic acid was the most common fragmentation: i.e. the released sialic acid was the most intense fragment for the di-antennary glycans, while the tri-antennary glycans showed a very intense signal for [M − S]2−. Additionally, several A-type cross-ring fragments (according to the nomenclature scheme of Domon and Costello40) generated by cleavage at different positions in the chitobiose core, together with a glycosidic cleavage both in the core and in the antennae, could be observed. Core fucosylation was assigned for both fucosylated species due to the presence of diagnostic fragments. In the case of H5N4S2F, the presence of the B6/Y6 (m/z 1710.175) and C5 (m/z 1816.004) ions could not be explained by antennary fucosylation. The core fucosylation of H6N5S3F was confirmed by the presence of two diagnostic cross-ring fragments produced by the cleavage of the terminal GlcNac, the 2,4A7 (m/z 1358.072) and the 0,2A7 (m/z 1461.023). The mass difference between these two doubly charged fragments was calculated to be 206.1 Da, indicating the presence of a fucose bound to the terminal GlcNac. The presence of such fragments is commonly used to indicate core fucosylation, whereas antennary fucosylation is typically highlighted by tri- and tetra-saccharide B ions produced by the fragmentation of the antenna.41
It should be considered that the re-arrangement of labile residues, such as fucose and sialic acids, has been reported for the CID fragmentation of protonated or deprotonated ions of glycans.42 Unambiguous identification would then require an improvement in the TWIMS separation to exclude the contemporary presence of more than one isomer before MS/MS fragmentation. Unfortunately, this could not be achieved given the actual instrument resolution.
Analysis of neutral glycans released from TRA
TWIMS of neutral protonated glycans released from TRA resulted in the separation of two overlapped pairs of species: the heptasaccharides H3N3F–H3N4 and the octasaccharides H4N3F–H3N4F, and in the partial separation of the larger H4N4F and H5N4F. The separation of sodiated adducts was also considered, due to the high intensity of such species, to investigate the effects of cation coordination. Adduct formation with group I and II cations has been used to improve the separation of species up to pentasaccharides, due to different coordination altering the collisional cross-section of various species in a diverse manner.43,44 When considering sodiated species, the three TRA glycans with asymmetry in the antennary portion (H3N3F, H4N3F, and H4N4F) showed a partial separation between two isomeric conformations, which was not observed in the protonated homologues, with the exception of H4N3F (Fig. S7†). Fragmentation spectra resulting from the two species of each glycan showed no difference in terms of the obtained fragments and relative intensities. The effect could thus be related to the position of the larger antenna: extension of the α-3 antenna would result in more elongated structures compared to the same extension on the α-6 antenna. Noticeably, even though the octasaccharide H4N3F had a larger difference in terms of size of the antenna, a better separation was obtained between the two isomers of the smaller heptasaccharide H3N3F.Since sodium coordination stabilizes the structures and prevents re-arrangements of the sugar units during CID,42 sodiated adducts were considered for MS/MS fragmentation. This was not the case for H5N4F, as its mono-sodium adduct had too low intensity. Similarly to TFN, to obtain pure fragmentation spectra from unresolved species, the quadrupole was used as a mass filter, allowing the distinct transmission of the six precursor glycan ions to the mobility cell. Mobilograms of neutral glycans released from TRA and related tandem MS spectra acquired in positive ionization mode are shown in Fig. 4.
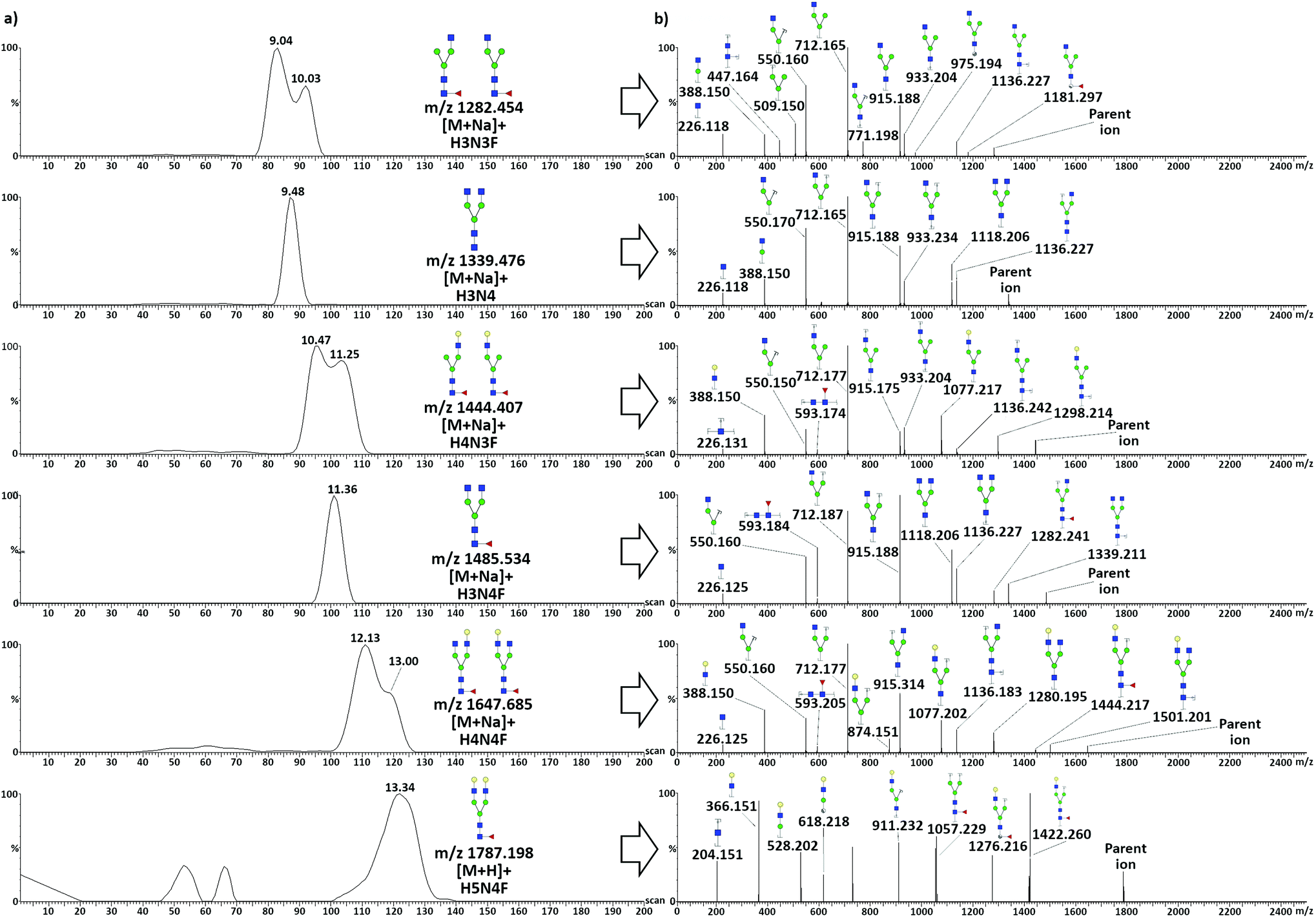 | ||
| Fig. 4 Structural elucidation of neutral glycans after the enzymatic release from TRA and TWIMS separation. (a) TWIMS separation with mobilograms illustrating the resolved mobilities of the six target glycans and (b) related MS/MS spectra and fragment identification. An intensity threshold limit of 3% of the base peak intensity was set to exclude low-intensity fragments. | ||
Positive mode fragmentation took place mostly at the glycosidic bond level, with the production of mostly B and Y ions, with a very limited presence of A-type cross-ring fragments. Fucosylation was assigned to the core of all the fucosylated species due to the presence of the characteristic trisaccharide GlcNac-GlcNac-Fuc Y2 ion (m/z 593.2). This ion was absent in H4N3, H3N3F, and H5N4F, the first being non-fucosylated. Fucosylation of H3N3F was still assigned to the core due to the presence of the diagnostic pair of terminal A-type fragments, the 2,4A5 (m/z 975.194) and the 0,2A5 (m/z 1181.297) ions, with a mass difference compatible with the presence of a fucose unit on the terminal GlcNac. For H5N4F, core fucosylation was confirmed by the presence of the diagnostic Y4,α/Y4,β (m/z 1057.229) and 3,5A6 (m/z 1276.216) ions. As a protonated ion of this glycan was fragmented, the possibility of a certain degree of re-arrangement during CID must be taken into account, as discussed above. H5N4F was anyway present at very low levels, which was expected as it is only present in the minor glycoforms TRAd and TRAe, which together accounted for less than 10% of the total TRA.
Application of the method
Compared to the standard TFN, one additional glycoform variably emerging in the samples could also be identified, having a mass value of 79263.7 Da, and was assigned to the loss of a sialic acid unit from TFN1. The loss of sialic acids naturally takes place over time and leaves exposed galactose units which are recognized by a Gal-binding lectin on liver parenchymal cells, leading to the removal of glycoproteins from the circulation.45 Among other modifications, a proteoform having a mass value of 79863.9 Da was observed in individual S4 and, less pronounced, in individual S2 (Fig. S4b and S4d†). The mass difference between this form and TFN2 was 162.05, indicating the possible addition of a hexose unit, with a mass accuracy of −60 ppm. Since no TFN2 glycan with hexose addition was identified by TWIMS-MS/MS in individuals S2 and S4, it is hypothesized that this modification could correspond to the glycation of a side chain amino group. Finally, individual S3 showed intense +32 Da peaks for every glycoform, accounting for almost 50% of the intensity (Fig. S4c†). Such peaks were also identified in individuals S2 and S4, but did not exceed 5–10% of the related glycoform. The absence of modifications on the mass values of the released glycans, as determined by TWIMS-MS/MS, suggested that this modification could originate from the protein backbone. In particular, the oxidation of some amino acids, such as methionine (to methionine sulfone) and proline (to glutamic acid), would result in a +32 Da shift in the mass of a protein, and could be related to oxidative stress.46
The quantification of the five main glycoforms of TFN in the 5 analyzed samples is reported in Fig. 5a.
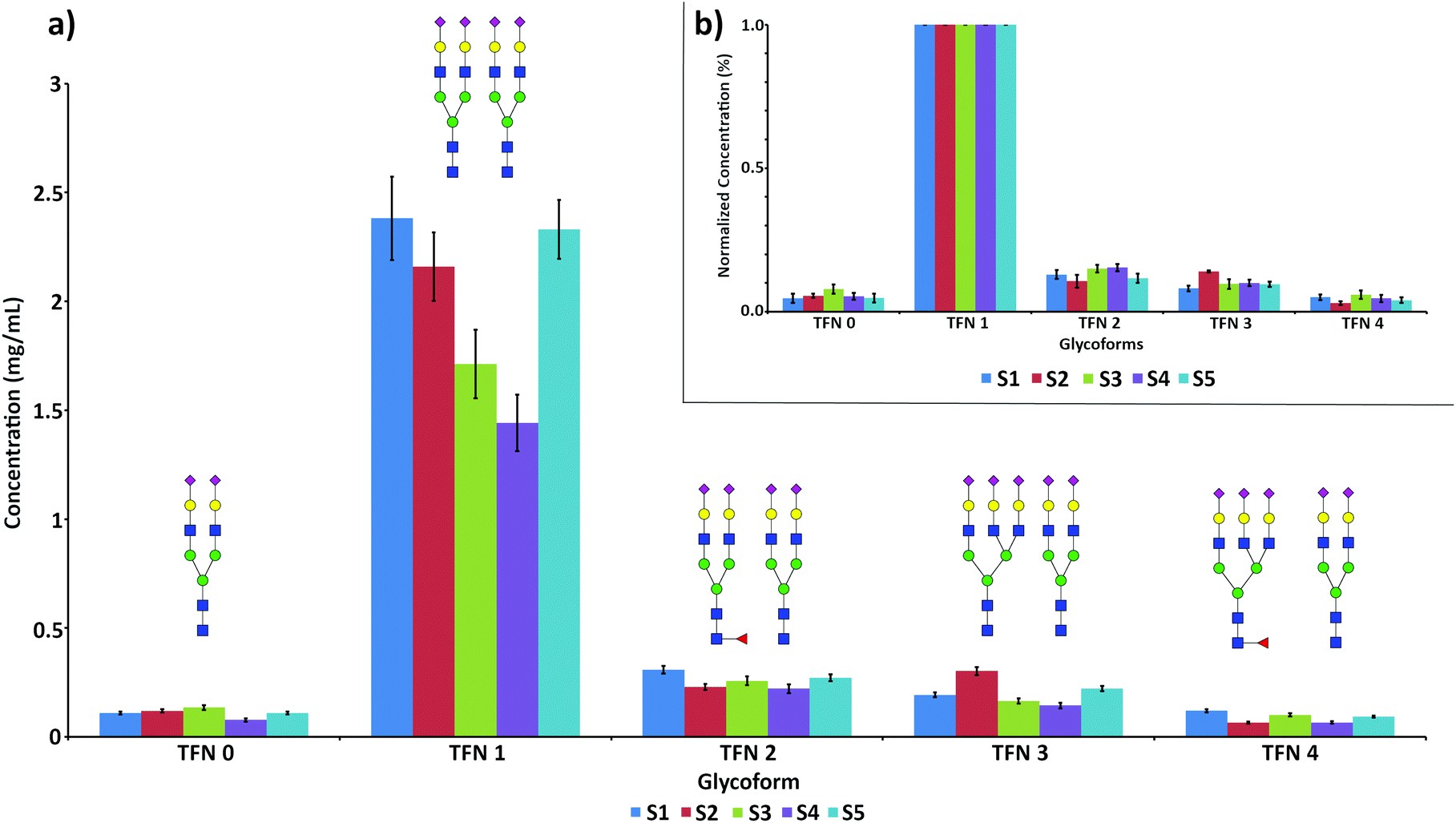 | ||
| Fig. 5 Quantification of the five identified TFN glycoforms in the 5 analyzed serum samples. (a) Concentration values for all the glycoforms, expressed in mg mL−1 as an average of triplicate analysis of duplicate samples (n = 2). Error bars expressed as pooled standard deviation. (b) Normalization on the concentration of TFN1 to highlight relative variations. | ||
Concentration values for total TFN, obtained by the sum of the concentrations of all the glycoforms, ranged between 1.61 and 3.17 mg mL−1, in good agreement with the reference serum values.30 The detailed concentration values for all the glycoforms are reported in Table 3.
| TFN 0 (mg mL−1) | TFN 1 (mg mL−1) | TFN 2 (mg mL−1) | TFN 3 (mg mL−1) | TFN 4 (mg mL−1) | TFN total (mg mL−1) | |
|---|---|---|---|---|---|---|
| Sample S1 | 0.11 ± 0.01 | 2.42 ± 0.25 | 0.32 ± 0.07 | 0.20 ± 0.02 | 0.12 ± 0.01 | 3.17 ± 0.32 |
| Sample S2 | 0.12 ± 0.01 | 2.23 ± 0.26 | 0.22 ± 0.05 | 0.31 ± 0.05 | 0.069 ± 0.017 | 2.95 ± 0.33 |
| Sample S3 | 0.13 ± 0.01 | 1.58 ± 0.20 | 0.23 ± 0.05 | 0.15 ± 0.02 | 0.09 ± 0.01 | 2.18 ± 0.27 |
| Sample S4 | 0.062 ± 0.007 | 1.18 ± 0.15 | 0.20 ± 0.04 | 0.12 ± 0.02 | 0.056 ± 0.014 | 1.61 ± 0.20 |
| Sample S5 | 0.11 ± 0.01 | 2.13 ± 0.26 | 0.26 ± 0.03 | 0.20 ± 0.03 | 0.084 ± 0.014 | 2.78 ± 0.33 |
Sample S4 showed the lowest concentration values for all the glycoforms, followed by sample S3. In the latter case, the low values could be ascribed to the high influence of the oxidized form of TFN, accounting for almost 50% of each glycoform and not considered in the quantification (Fig. S4c†).
As expected, TFN 1 was largely the main glycoform of TFN in all the samples. Minor variations could be observed in the relative abundances of the other glycoforms in the analyzed serum samples, with slightly higher branching (S2) and a lower degree of fucosylation (S2 and S5). These effects are more evident when the concentration values are normalized against the concentration value of TFN 1, thus accounting for the variable levels of TFN in the different serum samples (Fig. 5b). Such individual variations in the glycosylation patterns are expected and are due to both genetic and environmental factors.47
The structure of the identified glycans was subsequently controlled by enzymatic release and TWIMS-MS/MS analysis. The examples of full scan and fragmentation spectra are reported in Fig. S5,† while the identities and the structures of the detected glycans are reported in Table S5.† A procedural blank was also performed, by incubating an aliquot of serum collected from individual S3 with VHH-free beads, and performing the whole experimental procedure. This analysis showed the total absence of glycan-related signals, indicating the specificity of the immunoaffinity purification method. The representative full scan spectrum is reported in Fig. S5d.† All the serum samples showed similar glycan profiles, with H5N4S2 as the main species, followed by H5N4S2F and traces of H6N5S3 and H6N5S3F. The peak related to the monosialylated species H5N4S was not identified in any of the samples, probably due to the fact that, as it would be mono-charged, its ionization would not be favourable compared to doubly charged species. The tandem-MS analysis of H5N4S2F confirmed the structure that is already observed in the standard for all the five analyzed samples due to the presence of the singly charged 2,4A7/Y6 (m/z 1769.5) and 0,2A7/Y6 (m/z 1975.7) ions, with a mass difference of 206.2 Da, compatible with the presence of the core fucose.
Noticeably, many of the fragments identified on the standard H5N4S2F could not be observed in the serum samples. A reason for this could be that the amount of released glycans was limited by the capture of TFN, as opposed to in-solution release from the standard proteins. As a result, minor intensity fragments of low abundance species could be lost as their intensity could easily fall below the technique's limit of detection. Additionally, the sensitivity of the method was sufficient for the analysis of di-antennary species but was not enough for obtaining useful MS–MS spectra of the tri-antennary glycans, which are only present at a trace level.
Structural elucidation by TWIMS-MS/MS analysis showed the same results as obtained in solvent, confirming the structures already shown in Fig. 4.
Discussion
Quantification of TFN glycoforms is commonly carried out at the glycopeptide level by triple quadrupole MRM experiments.48,49 Compared to these methods, our method showed similar performances in terms of detection limits (ranging from 0.5 to 0.9 μg mL−1), precision, accuracy, and linearity. An example of quantification of TFN at the intact protein level was performed by LC-ICP-MS, focusing on the different sialoforms present on the protein.50 Qualitative analysis of TFN glycoforms by high-resolution MS has been performed by Van Scherpenzeel et al. and applied to the diagnosis of congenital disorder of glycosylation (CDG).51 In their work, a similar method employing bead-based immunoaffinity purification of TFN and nano-LC Q-ToF analysis was employed, resulting in the characterization of several glycoforms in CDG samples. Our method showed similar performances in terms of accuracy and precision of the obtained mass values, but allowed for absolute quantification with a significantly shorter analysis time.Quantitative analysis of biopharmaceuticals in their production medium is not often performed, as their determination in biological fluids is deemed to be more interesting to study their behavior after administration. In our procedure, the possibility of obtaining absolute quantitation of all the TRA glycoforms minimizing the sample preparation is beneficial for a quality control setup. Obtaining absolute concentration values would precisely highlight alterations in the glycosylation machinery of the production systems, potentially decreasing the drug efficiency or being harmful to the health of the patient.
The developed IMS method was able to separate glycan species with more than one saccharide unit difference, but failed to discriminate between different sugars with the same number of monosaccharides. Complex glycans released from TFN were fully separated only when they differ in an antenna or more, and fucosylated species showed partial overlap with the respective non-fucosylated glycans. Separation between species differing in the position of fucosylation could not be observed. It cannot be concluded whether this was due to the absence of differential fucosylation or to the inability of resolving such positional isomers due to the lack of IMS resolution. Positional isomers could be partially separated for sodium adducts of smaller species, such as the ones released from TRA, where the change in the position of one unit, the addition of a single unit, or the exchange of a sugar with a different one can have a higher impact on the alteration of the three-dimensional shape of the molecule.24,52 It was concluded that the instrumental resolution was not enough to fully implement the methodology. A new generation IMS instrument, namely cyclic IMS or TIMS, may abate these issues, as the resolution is about 20 and 50 times higher compared to that of the instrument used in this work.53,54 This was recently shown by Boons et al., who demonstrated the possibility of discriminating between differently fucosylated N-glycan isomers on a new generation cyclic IMS instrument.55
Conclusions
Currently, quantitative methods for glycan analysis are not so extensively developed, as most applications rely on relative quantification and focus on providing a relative profile of the glycans present in a sample.56 The possibility of combining a fast and robust quantitative method with the identification and structural elucidation of specific glycan species provides an important tool for the development of glycan-based biomarkers for clinical application. In this work, the focus was on transferrin, based on its involvement in several pathologies and on trastuzumab, to show its applicability in the quality control of biopharmaceuticals. Due to the nature of the method, other proteins can be easily targeted by derivatizing the beads with appropriate immunoaffinity binders, giving the possibility of having several targets for creating biomarker panels.Application of the developed method to TFN extracted from five healthy donor serum samples showed protein concentration values in good agreement with the reference values, and the analysis of the protein glycoforms highlighted the naturally occurring interpersonal variations. The identified glycoforms were subsequently characterized by TWIMS-MS/MS, to structurally elucidate the glycans expressed on the two proteins. TWIMS separation did not provide the resolution required to achieve the separation of glycans with minor structural differences, but positional isomers were partially separated for smaller glycans, for which a difference in the position of a unit or antenna had a more prominent impact on the shape of the molecule.
Overall, the method could allow facile implementation of immunoaffinity extraction for several samples in parallel, with a high degree of automation if a pipetting robot is used for the solvent transfer steps. This, together with the short required analyses times, makes the approach suitable for clinical laboratories and quality control of large batches of biopharmaceuticals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Kurt Benkestock and Peter Teiwik from Waters, Sweden, for the help and support while setting up the IMS method. Niclas Karlsson is gratefully acknowledged for his fundamental contribution to the interpretation of the MS/MS data.Notes and references
- P. Stanley, H. Schachter and N. Taniguchi, in Essential of glycobiology, ed. A. Varki, J. D. Esko, et al., C. S. H. L. Press, New York, 2009, ch. 8, pp. 101–114 Search PubMed.
- M. Yamamoto-Hino, Y. Kanie, W. Awano, K. F. Aoki-Kinoshita, H. Yano, S. Nishihara, H. Okano, R. Ueda, O. Kanie and S. Goto, PLoS Genet., 2010, 6, 1–11 CrossRef PubMed.
- H. J. An, S. R. Kronewitter, M. L. de Leoz and C. B. Lebrilla, Curr. Opin. Chem. Biol., 2009, 13, 601–607 CrossRef CAS PubMed.
- V. Zoldos, M. Novokmet, I. Beceheli and G. Lauc, Glycoconjugate J., 2013, 30, 41–50 CrossRef CAS PubMed.
- B. Adamczyk, T. Tharmalingam and P. M. Rudd, Biochim. Biophys. Acta, 2012, 1820, 1347–1353 CrossRef CAS PubMed.
- R. Barone, L. Sturiale, A. Palmigiano, M. Zappia and D. Garozzo, J. Proteomics, 2012, 75, 5123–5139 CrossRef CAS PubMed.
- S. S. Pinho and C. A. Reis, Nat. Rev. Cancer, 2015, 15, 540–555 CrossRef CAS PubMed.
- E. Higgins, Glycoconjugate J., 2010, 27, 211–225 CrossRef CAS PubMed.
- A. Beck, mAbs, 2014, 3, 107–110 CrossRef PubMed.
- K. Marino, J. Bones, J. J. Kattla and P. M. Rudd, Nat. Chem. Biol., 2010, 6, 713–723 CrossRef CAS PubMed.
- M. Baerenfaenger and B. Meyer, J. Proteome Res., 2018, 17, 3693–3703 CrossRef CAS PubMed.
- I. M. Lazar, J. Deng, F. Ikenishi and A. C. Lazar, Electrophoresis, 2015, 36, 225–237 CrossRef CAS PubMed.
- S. Houel, M. Hilliard, Y. Q. Yu, N. McLoughlin, S. M. Martin, P. M. Rudd, J. P. Williams and W. Chen, Anal. Chem., 2014, 86, 576–584 CrossRef CAS PubMed.
- G. S. M. Kammeijer, B. C. Jansen, I. Kohler, A. A. M. Heemskerk, O. A. Mayboroda, P. J. Hensbergen, J. Schappler and M. Wuhrer, Sci. Rep., 2017, 7, 3733 CrossRef PubMed.
- O. Trenchevska, R. W. Nelson and D. Nedelkov, Proteomes, 2016, 4, 13 CrossRef PubMed.
- O. Aizpurua-Olaizola, J. Sastre Toraño, J. M. Falcon-Perez, C. Williams, N. Reichardt and G. J. Boons, TrAC, Trends Anal. Chem., 2018, 100, 7–14 CrossRef CAS.
- D. J. Harvey, Mass Spectrom. Rev., 2017, 36, 255–422 CrossRef CAS PubMed.
- Y. Hu and Y. Mechref, Electrophoresis, 2012, 33, 1768–1777 CrossRef CAS PubMed.
- K. R. Reiding, D. Blank, D. M. Kuijper, A. M. Deelder and M. Wuhrer, Anal. Chem., 2014, 86, 5784–5793 CrossRef CAS PubMed.
- D. Falck, B. C. Jansen, R. Plomp, D. Reusch, M. Haberger and M. Wuhrer, J. Proteome Res., 2015, 14, 4019–4028 CrossRef CAS PubMed.
- G. Zauner, A. M. Deelder and M. Wuhrer, Electrophoresis, 2011, 32, 3456–3466 CrossRef CAS PubMed.
- Z. Chen, M. S. Glover and L. Li, Curr. Opin. Chem. Biol., 2018, 42, 1–8 CrossRef CAS PubMed.
- J. Hofmann and K. Pagel, Angew. Chem., Int. Ed., 2017, 56, 8342–8349 CrossRef CAS PubMed.
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Nature, 2015, 526, 241–244 CrossRef CAS PubMed.
- D. J. Harvey, C. A. Scarff, M. Edgeworth, M. Crispin, C. N. Scanlan, F. Sobott, S. Allman, K. Baruah, L. Pritchard and J. H. Scrivens, Electrophoresis, 2013, 34, 2368–2378 CrossRef CAS PubMed.
- K. Pagel and D. J. Harvey, Anal. Chem., 2013, 85, 5138–5145 CrossRef CAS PubMed.
- J. P. Williams, M. Grabenauer, R. J. Holland, C. J. Carpenter, M. R. Wormald, K. Giles, D. J. Harvey, R. H. Bateman, J. H. Scrivens and M. T. Bowers, Int. J. Mass Spectrom., 2010, 298, 119–127 CrossRef CAS.
- T. Nagel and B. Meyer, Biochim. Biophys. Acta, 2014, 1844, 2284–2289 CrossRef CAS PubMed.
- L. V. Schaffer, R. J. Millikin, R. M. Miller, L. C. Anderson, R. T. Fellers, Y. Ge, N. L. Kelleher, R. D. LeDuc, X. Liu, S. H. Payne, L. Sun, P. M. Thomas, T. Tucholski, Z. Wang, S. Wu, Z. Wu, D. Yu, M. R. Shortreed and L. M. Smith, Proteomics, 2019, 19, e1800361 CrossRef PubMed.
- F. Clerc, K. R. Reiding, B. C. Jansen, G. S. Kammeijer, A. Bondt and M. Wuhrer, Glycoconjugate J., 2016, 33, 309–343 CrossRef CAS PubMed.
- K. Yamashita, N. Koide, T. Endo, Y. Iwaki and A. Kobata, J. Biol. Chem., 1989, 264, 2415–2423 CAS.
- C. Flahaut, J. C. Michalski, T. Danel, M. H. Humbert and A. Klein, Glycobiology, 2003, 13, 191–198 CrossRef CAS PubMed.
- P. Zhang, S. Woen, T. Wang, B. Liau, S. Zhao, C. Chen, Y. Yang, Z. Song, M. R. Wormald, C. Yu and P. M. Rudd, Drug Discovery Today, 2016, 21, 740–765 CrossRef CAS PubMed.
- C. W. Damen, W. Chen, A. B. Chakraborty, M. van Oosterhout, J. R. Mazzeo, J. C. Gebler, J. H. Schellens, H. Rosing and J. H. Beijnen, J. Am. Soc. Mass Spectrom., 2009, 20, 2021–2033 CrossRef CAS PubMed.
- I. Karlsson, L. Ndreu, A. Quaranta and G. Thorsen, J. Pharm. Biomed. Anal., 2017, 145, 431–439 CrossRef CAS PubMed.
- A. Sroka-Bartnicka, I. Karlsson, L. Ndreu, A. Quaranta, M. Pijnappel and G. Thorsen, J. Pharm. Biomed. Anal., 2017, 132, 125–132 CrossRef CAS PubMed.
- A. Ceroni, K. Maas, H. Geyer, R. Geyer, A. Dell and S. Haslam, J. Proteome Res., 2008, 7, 1650–1659 CrossRef CAS PubMed.
- J. C. Cottrell and B. N. Green, Application note 212.
- R. Cumeras, E. Figueras, C. E. Davis, J. I. Baumbach and I. Gracia, Analyst, 2015, 140, 1376–1390 RSC.
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS.
- D. Sagi, J. Peter-Katalinic, H. S. Conradt and M. Nimtz, J. Am. Soc. Mass Spectrom., 2002, 13, 1138–1148 CrossRef CAS PubMed.
- M. Wuhrer, C. A. M. Koeleman and A. M. Deelder, Anal. Chem., 2009, 81, 4422–4432 CrossRef CAS PubMed.
- Y. Huang and E. D. Dodds, Anal. Chem., 2013, 85, 9728–9735 CrossRef CAS PubMed.
- Y. Huang and E. D. Dodds, Anal. Chem., 2015, 87, 5664–5668 CrossRef CAS PubMed.
- P. J. Coombs, M. E. Taylor and K. Drickamer, Glycobiology, 2006, 16, 1C–7C CrossRef CAS PubMed.
- B. S. Berlett and E. R. Stadtman, J. Biol. Chem., 1997, 272, 20313–20316 CrossRef CAS PubMed.
- A. Knezevic, O. Polasec, O. Gornik, I. Rudan, H. Campbell, C. Hayward, A. Wright, I. Kolcic, N. O'Donoghue, J. Bones, P. M. Rudd and G. Lauc, J. Proteome Res., 2009, 8, 694–701 CrossRef CAS PubMed.
- S. Miyamoto, C. D. Stroble, S. Taylor, Q. Hong, C. B. Lebrilla, G. S. Leiserowitz, K. Kim and L. R. Ruhaak, J. Proteome Res., 2018, 17, 222–233 CrossRef CAS PubMed.
- Y. Yu, J. Xu, Y. Liu and Y. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 902, 10–15 CrossRef CAS PubMed.
- Y. N. Ordonez, R. F. Anton and W. C. Davis, Anal. Methods, 2014, 6, 3967–3974 RSC.
- M. van Scherpenzeel, G. Steenbergen, E. Morava, R. A. Wevers and D. J. Lefeber, Transl. Res., 2015, 166, 639–649 CrossRef CAS PubMed.
- J. Hofmann, A. Stuckmann, M. Crispin, D. J. Harvey, K. Pagel and W. B. Struwe, Anal. Chem., 2017, 89, 2318–2325 CrossRef CAS PubMed.
- K. Giles, J. Ujma, J. Wildgoose, S. Pringle, K. Richardson, D. Langridge and M. Green, Anal. Chem., 2019, 91(13), 8564–8573 CrossRef CAS PubMed.
- M. E. Ridgeway, M. Lubeck, J. Jordens, M. Mann and M. A. Park, Int. J. Mass Spectrom., 2018, 425, 22–35 CrossRef CAS.
- G. J. Boons, J. Sastre Toraño, I. A. Gagarinov, G. M. Vos, F. Broszeit, A. D. Srivastava, M. Palmer, J. I. Langridge, O. Aizpurua-Olaizola and V. J. Somovilla, Angew. Chem., 2019, 58(49), 17616–17620 CrossRef PubMed.
- J. Etxebarria and N. C. Reichardt, Biochim. Biophys. Acta, 2016, 1860, 1676–1687 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an02081k |
| This journal is © The Royal Society of Chemistry 2020 |