
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVirus-based SELEX (viro-SELEX) allows development of aptamers targeting knotty proteins†
Chandan
Narayan
 a,
Junyoung
Kwon
a,
Chonsaeng
Kim
b,
Seong-Jun
Kim
b and
Sung Key
Jang
*a
a,
Junyoung
Kwon
a,
Chonsaeng
Kim
b,
Seong-Jun
Kim
b and
Sung Key
Jang
*a
aMolecular virology Laboratory, Department of Life Sciences, Postech Biotech Center, Pohang University of Science and Technology, Cheongam-ro 77, Nam-gu, Pohang-si, Gyeongsangbuk-do 37673, Republic of Korea. E-mail: sungkey@postech.ac.kr; Fax: +82-54-279-8009; Tel: +82-54-279-2298
bCenter for Convergent Research of Emerging Virus Infection, Korea Research Institute of Chemical Technology, Daejeon, 34114, Republic of Korea
First published on 23rd December 2019
Abstract
It has been 100 years since the worst flu (Spanish flu) mankind has ever experienced. Rapid, accurate diagnosis and subtyping of flu are still an urgent unmet medical need. By using surrogate virus-based SELEX (viro-SELEX), we report here multiple advances incorporated into the field of flu diagnostics: (i) aptamers that can bind to the native virus well even though they cannot bind strongly to a recombinant protein (hemagglutinin); (ii) a couple of aptamers that can target a broad range of strains belonging to the H1N1 subtype and detect only the H1N1 subtype and nothing else; (iii) a highly sensitive lateral flow assay system (limit of detection is 0.08 HAU) using fluorescence-tagged aptamers. The viro-SELEX method of aptamer selection in conjunction with a fluorescent tag on aptamers is a very useful approach to develop highly sensitive, specific, portable, rapid, and quantitative point-of-care testing diagnostic tools for the future.
Introduction
Influenza A viruses, which are the causative agents of flu, have been grouped into subtypes on the basis of their two surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA).2 18 HAs and 11 NAs of influenza A viruses have been reported.3 According to WHO and CDC reports, H3N2, pandemic 2009 H1N1 (H1N1pdm09) influenza A virus, and influenza B viruses are the most prevailing strains at present (https://www.who.int/influenza/surveillance_monitoring/updates/latest_update_GIP_surveillance/en/).4For the prevention and treatment of influenza virus, inhibitors for the M2 ion channel, neuraminidase, and RNA-dependent RNA polymerase have been approved for treating the flu.5–8 A WHO report says virtually all influenza A viruses circulating in humans have been deemed resistant to one category of antiviral drugs, namely, M2 inhibitors (amantadine and rimantadine). Therefore, M2 inhibitors are not recommended to use for treating the flu.9,10 On the other hand, as of October 2017, the frequency of resistance to the NA inhibitor oseltamivir remains low (1–2%) and this drug is used to treat influenza (https://www.cdc.gov/flu/;11http://www.who.int/mediacentre/factsheets/fs194/en/).12 Oseltamivir is recommended to be used within 48 h after the onset of influenza.6,7,13,14 Thus, early diagnosis of influenza plays a pivotal role in order to minimize hospitalization cases and a rampant unnecessary usage of antibiotics.15–17
Broadly neutralizing antibodies (bnAbs) have been investigated to develop anti-influenza drugs.18 The bnAbs reported so far against the influenza viruses bind to HAs of either group I (including H1, H2, and H5) or group II (including H3 and H4); only a few can bind to both. Along the same lines, a small molecule inhibitor targeting HA and blocking the fusion of the viral envelope binds selectively to group I viruses but not group II viruses.18 Thus, subtyping of viruses in patient samples would be of importance not only for epidemiological studies but also for the treatment of a patient with this kind of potential future drug.
Aptamers are single-stranded nucleic acids (RNA or ssDNA) able to specifically recognize a target molecule with high affinity.19 They combine the advantages of both typical low-molecular-weight small molecules and high-molecular-weight antibodies: in vitro chemical synthesis like small molecules and high specificity like antibodies.20
Previously, SELEX for membrane proteins has been performed by using purified proteins. Purifying full length membrane proteins to-date has been a very cumbersome job. Later, expression of extra-cellular domains from the bacteria21 and baculovirus expression system which demonstrated better post-translational modifications was developed. It should be noted, however, that extra-cellular domain expression alone may not mimic the native conformation of the proteins. For instance, the oligomeric form of a protein may not be formed by expressing only the extracellular region of a protein (ectodomain; e.g. HA trimer).22,23 Moreover, an artificial face generated by deletion of the transmembrane domain of a protein is unavoidable when an unnatural polypeptide containing only an ectodomain is produced. Therefore, many aptamers selected using the ectodomain polypeptide would recognize the artificial face and would not recognize the natural structure of the target protein.21 To avoid this kind of membrane protein-related complexity, scientists developed cell-based SELEX (cell-SELEX) which utilizes cells instead of purified proteins in SELEX.24–26 Theoretically, purification of a target protein or prior knowledge of the target is unnecessary for cell-SELEX, and aptamers can be generated for many targets in their native state.27,28 However, damage of cells during the SELEX process and cleavages of target molecules on the cell surface during trypsinisation used in detaching cells from culture plates are unavoidable. More importantly, the target molecule on the surface of a cell is likely to be in minor population among thousands of cellular proteins on the surface of cells. Due to these issues, only a few aptamers have been developed by using cell-SELEX.29–31 To circumvent these issues our group previously reported a novel SELEX method designated as ‘viro-SELEX’ recently.1 The viro-SELEX utilizes surrogate baculoviruses instead of cells. Baculoviruses are a rod-shaped, enveloped virus pathogenic to insect cells, not to mammalian and plant cells.32–34 Membrane proteins of interest (HA) were incorporated into the surrogate viral envelope using a baculovirus expression system, and purified baculoviruses were stable to perform SELEX processes.1 We also performed consecutive SELEX steps with two different surrogate viruses containing HAs from two strains of H1N1 similarly to toggle SELEX35 to enrich aptamers binding to a range of strains belonging to the H1N1 subtype of influenza virus. Moreover, we performed several rounds of SELEX with purified proteins corresponding to the outer part of the viral envelope of HA (ectodomain) in the viro-SELEX steps to remove aptamers targeting macromolecules of the surface of surrogate virus other than HA. We also used the “advanced SELEX technology” with a modified deoxyribonucleotide instead of “conventional SELEX” with unmodified deoxyribonucleotides in order to acquire aptamers with high affinity to target proteins.36 In other words, we used unmodified deoxyribonucleotide triphosphates (dATP, dCTP, and dGTP) and a modified deoxyribonucleotide benzyl-dUTP (instead of dTTP) in our SELEX process. Gold et al.37 reported that the usage of 5-benzylaminocarbonyl-dU increased the success rate of their SELEX for human proteins from <30% to >50%. Few other articles reported that in addition the modified bases induced hydrophobic interactions, which lead to increased affinity and thus interaction between the targets and the aptamers.36,38,39
Through these optimized SELEX processes, we could generate an aptamer pair strongly binding to a native form of HAs with single digit nanomolar Kds.
Finally, we devised a lateral flow assay (LFA) system using the aptamer pair as a capture probe and a detection probe. The detection probe was conjugated with either a gold nanoparticle or a fluorescent dye. The LFA system had very good selectivity to detect H1N1 influenza virus and a limit of detection (LOD) lower than 0.1 hemagglutination unit (HAU) when the detection aptamer with a fluorescence tag was used in the LFA system. Importantly, the LFA system was more sensitive to virus samples than to protein samples. The result suggests that the aptamers bind better to the native form of HA than to the purified ectodomain of HA. This indicates that the viro-SELEX method is a good approach to develop aptamers that can strongly and specifically interact with the native form of membrane proteins which are usually difficult to purify in the intact forms.
Materials and methods
Cell culture
The insect cell line (Sf9 – Spodoptera frugiperda) was used in this study. Briefly, SF900SFM (Gibco) with 1% penicillin/streptomycin mix and 0.2% Amphotericin B (Sigma) was used for culturing the Sf9 cells under suspension culture on a shaking incubator (at 27 °C & 150 rpm).Influenza virus strains
cDNA clones of HAs from influenza viruses [A/Puerto Rico/8/1934 (H1N1), A/Korea pandemic/2009 (H1N1), A/Hong Kong/1968 (H3N2), A/Thailand/2004 (H5N1), and A/Anhui/1/2013 (H7N9) designated as PR, CJ, HK, Kan, and Anh, respectively, henceforth] were used for the generation of recombinant baculoviruses.Virus preparation
Virus preparation was performed as described in a previous report (Kwon et al. 2019).1 Briefly, two influenza virus strains, PR and HK, were obtained from the ATCC and amplified in chicken embryos at 37 °C. B/Lee/1940 virus was obtained from the ATCC and propagated in MDCK cells at 33 °C. Egg allantoic fluids and cell culture media from infected MDCK cells (B/Lee) were cleared by centrifugation at 2500 rpm for 10 min to remove debris. The viruses were purified by sucrose gradient ultracentrifugation as previously described in ref. 40.Generation of surrogate baculoviruses and protein purification
Recombinant Vector p42 (derived from pFastBac) containing hexa-histidine and Flag tag was prepared containing the gene of our interest using a variety of enzymes for the different constructs. This vector was introduced into target bacmid by homologous recombination. Sf9 cells were transfected with the recombinant bacmid DNA for 72 h. Following infection of baculoviruses, the supernatant which contained the hexa-his and flag-tagged recombinant virus underwent the Ni-NTA method of purification which is routinely used for purifying proteins. Purified proteins were analyzed on a 10% SDS-PAGE gel with Coomassie staining. Immunoblotting confirmed that all the proteins corresponded to their appropriate sizes.The Baculovirus expression vector system designed by Invitrogen was used with minor modifications to purify the baculoviruses. At 3 days after baculovirus infection, the culture media were centrifuged at 500g at 4 °C for 5 min. The supernatant was aliquoted into a new 50 ml centrifuge tube, and again centrifuged at 5000g at 4 °C for 15 min. The supernatant was again spun in an ultracentrifuge at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C for 2 h over 18 ml of sucrose gradient buffer [5 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA and 20% sucrose]. Following the centrifugation, the supernatant was discarded and the translucent pellet was washed delicately 2 times and resuspended in cold PBS (500 μl). The presence of HA proteins on the virion particles was confirmed by western blotting.
000g at 4 °C for 2 h over 18 ml of sucrose gradient buffer [5 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA and 20% sucrose]. Following the centrifugation, the supernatant was discarded and the translucent pellet was washed delicately 2 times and resuspended in cold PBS (500 μl). The presence of HA proteins on the virion particles was confirmed by western blotting.
In vitro evolution of DNA aptamers for HA of H1N1 influenza virus using swap-SELEX and viro-SELEX
We followed the procedures as described by Gold et al. (2010)37 and Kwon et al. (2019)1 with minor modifications. Dextran sulfate (DxSO4) was added in most of the rounds as an anionic competitor. Prothrombin, casein, and human serum albumin were used for negative selection in the competition buffer. Also negative selection was performed with other purified proteins and surrogate viruses of influenza origin (HAs of H3N2, H5N1, and H7N9).In the present selection strategy, we have included HA proteins from 2 different strains of H1N1 (PR and CJ) and also we have included surrogate baculoviruses that contain the HA proteins on the virion particles. This approach would result in selecting aptamers that can bind to native forms of HAs of both PR and CJ. Thus, these aptamers could be very useful in covering the broad range of H1N1 strains of influenza A. We performed 10 rounds of viro-SELEX as described by Kwon (Kwon et al., 2019)1 and analyzed rounds 8–10 for binding capabilities.
Cloning and sequencing of oligonucleotides enriched by viro-SELEX
The eDNA pool of the 9th round of viro-SELEX showed better binding affinities and specificity to the proteins of our interest than the 10th round and thus was amplified by PCR with 5′ and 3′ primers, and the amplified DNAs were purified using a PCR purification kit (Qiagen, Germany). Cloning and sequencing processes were performed by Solgent (Solgent, Daejeon, South Korea). Sequences were analyzed using an in-house program “Aptamer motif searcher”.The equilibrium dissociation constant (Kd) between the aptamer and protein was determined by the nitrocellulose-filter binding method.37
Modifications of aptamers and construction of a lateral flow strip
Lateral flow strips were prepared as described by Kwon et al. (2019).1For subtype specificity tests, 20 pmol of different proteins in 200 μl of sample buffer (10 mM sodium phosphate buffer and 0.1 μM DxSO4) were applied to the sample pad and checked for the appearance of the control and test lines.
For the sensitivity test, different dilutions of either recombinant target proteins (20 pmol–1.25 pmol) or the viruses (64 HAU–0.25 HAU) in 200 μl of sample buffer were applied to the sample pad and checked for the appearance of the control and test lines.
Conjugation of fluorescent tags
To further improve the sensitivity of the LFA system, we conjugated the biotinylated detection aptamers with a fluorescent tag Alexa FluorTM 488 by using Streptavidin-conjugated Alexa FluorTM 488 (Thermo Fisher Scientific). 20 μl of aptamers conjugated with Alexa FluorTM 488 (5 pmol) was applied to the conjugate pad and dried overnight in a desiccator. 20 μl of 10 mM sodium phosphate buffer containing 0.5 mg ml−1 biotin was applied to the conjugate pad and dried overnight in a desiccator. The biotin treatment decreased the background by binding to the aptamer-unoccupied streptavidin-containing dye molecules.Results and discussion
There are a variety of molecular assay-based diagnostic kits for influenza viruses which are FDA approved and can distinguish between A/H1, A/H3, and A/2009 H1, but it takes hours for detection with a sensitivity rate of 70–90%. Some antigen based immunochromatogenic assays are available which are quick (30 min), but their sensitivity ranges from 59–93% and most of them are not for subtype-specific diagnosis.2 Also it is important to note that these methods need a lab set up to perform diagnoses and are thus inconvenient in the field during times like a pandemic outbreak, especially seen in the past with H1N1.Development of aptamers specific for HA proteins of H1N1 influenza viruses
Previously, there have been reports of DNA aptamers being selected against H1N1 influenza viruses using a variety of raw materials like HA proteins and whole viruses. Kd values of these aptamers ranged from 10–100 nM.41 Here we report a subtype-specific influenza virus diagnostic system with high specificity and a good LOD in the laboratory settings. In order to develop this new diagnostic system, we utilized aptamers exhibiting single nanomolar binding affinities which were generated by recently developed viro-SELEX. By using surrogate viruses containing HA proteins on the virion particles, we were able to remove oligonucleotides binding to the artificial faces generated by producing the truncated version of the HA protein containing only the ectodomain of HA (Fig. 1). Moreover, we could remove oligonucleotides binding to the surface HA monomer which is hidden in trimeric HA proteins in influenza virus (Fig. 1).22,23 It should be noted that the hidden surface of HA proteins would be exposed in the truncated HA protein since the truncated HA proteins exist in a monomeric form due to the lack of the region required for trimerization of HA proteins.22,23 In other words, oligonucleotides binding to the artificial parts of the truncated proteins and the unnatural faces of the purified proteins can be removed by viro-SELEX. Detailed procedures are as follows: once all the viruses and proteins were purified in sufficient amounts, we performed positive selections with ectodomain H1 proteins (HA proteins of subtype H1N1) and H1 surrogate viruses (baculoviruses containing HA proteins of subtype H1N1) for enriching the aptamers that can bind to the proteins of our interest. We also performed negative selections with ectodomain of H3 (HA proteins of subtype H3N2), H5 (HA proteins of subtype H5N1), and H7 (HA proteins of subtype H7N9) proteins and control baculoviruses to remove oligonucleotides interacting with HA proteins of other subtype influenza viruses and with envelope proteins of baculoviruses other than H1 proteins of influenza virus, respectively. We included selections with surrogate viruses in at least 2 rounds of our SELEX cycle. | ||
| Fig. 1 Principle of viro-SELEX. Conventional SELEX methods, which utilize the ectodomain of a target protein, may enrich aptamers binding to some portions on the protein which are hidden or inaccessible in the native conformation of the same protein expressed on the influenza virus. But viro-SELEX uses surrogate baculoviruses expressing the protein on their surface, which can mimic the native conformation and thus helps in obtaining aptamers that can bind to the influenza virus in its native state. | ||
Determination of sensitivity and specificity of aptamers using the nitrocellulose filter-binding method
We performed protein-binding affinity tests with DNA pools from the rounds 8, 9, and 10 of SELEX executed as mentioned above. We found that the pool obtained from the 9th round had good protein-binding affinity as analyzed from the filter-binding assay for HA proteins of both strains CJ01 (Kd = 2.58 ± 0.82 nM) and PR08 (Kd = 6.41 ± 1.27 nM). The 9th round pool was investigated for any cross binding activity with HA proteins of subtypes H3N2, H5N1, and H7N9. No cross binding was detected (data not shown). Therefore, the DNA pool of the 9th round of SELEX was cloned and sequenced. Out of a variety of families of sequences, aptamers #1 (A1) and #14 (A14) were chosen since they have high affinities to HA proteins of strain CJ01 (Kd values were 3.36 ± 0.65 nM and 2.98 ± 1.35 nM, respectively; Fig. 2, Table 1), and they did not compete each other for binding to HA proteins (ESI Fig. S1;† see below). In order to investigate the specificity of aptamers A1 and A14, we performed filter binding assays with HA and NA proteins from various subtypes of influenza viruses. HA and NA proteins of H1N1, H3N2, H5N1, and H7N9 were used in the filter binding assays at a fixed concentration of 20 nM. The A1 aptamer showed strong binding to HA proteins of CJ01 and PR08 strains of H1N1, but this aptamer showed only weak binding (less than 20%) to all other proteins tested (Fig. 3A). The A14 aptamer also showed clear specific binding to HA proteins of CJ01 and PR08 strains of H1N1 (Fig. 3A) even though this aptamer showed only weak affinity to the purified HA protein of strain PR08 (Kd > 100 nM) (Table 1).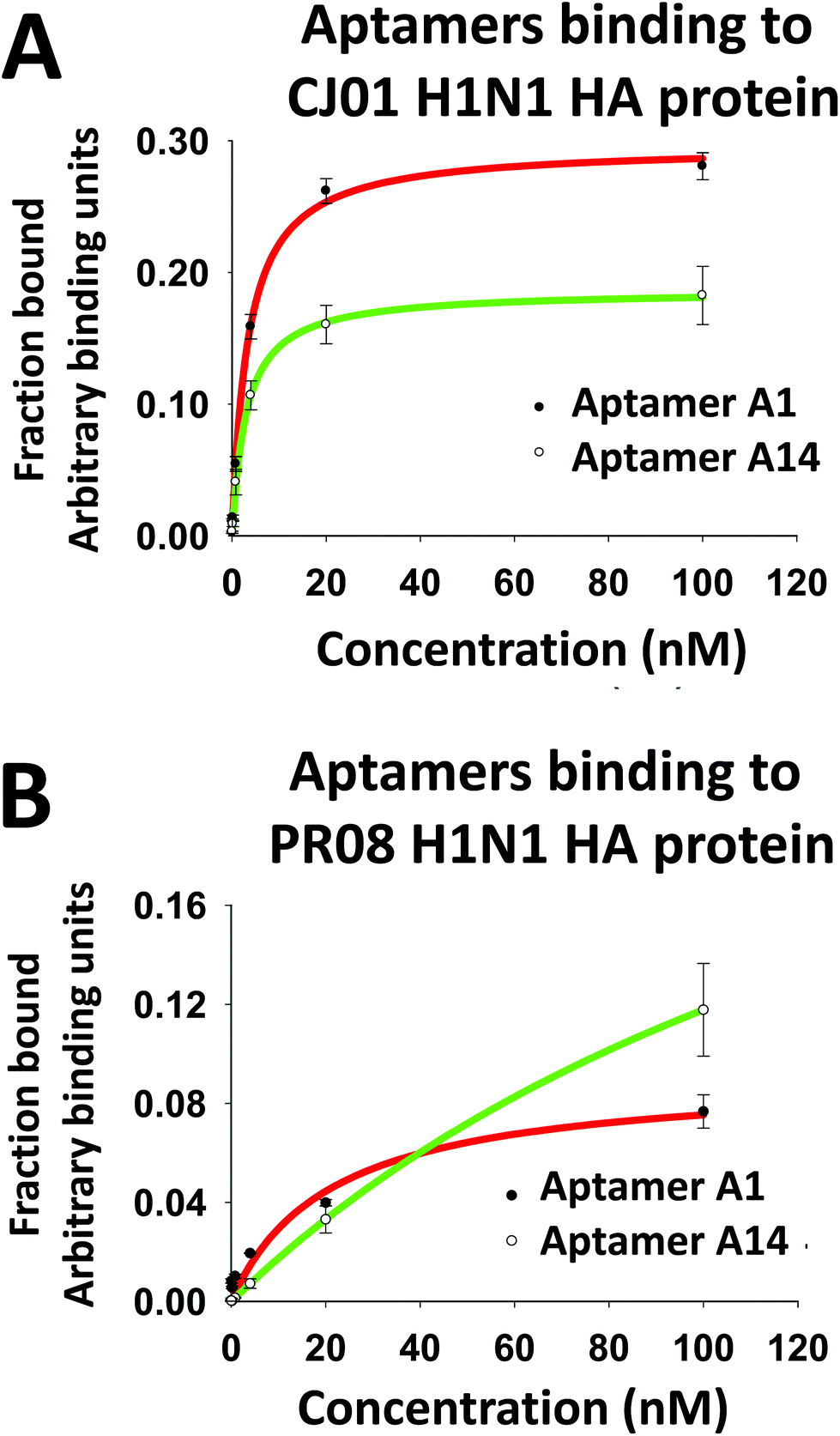 | ||
| Fig. 2 Determination of the binding affinity of aptamers to recombinant proteins CJ01 H1N1 HA (A) and PR08 H1N1 HA (B) using the nitrocellulose-filter binding method. For the binding assays, 5′-ends of the aptamers were labeled with [32P]-ATP using T4 PNK (T4-polynucleotide kinase), and then bound with a range of recombinant HA protein concentrations ranging from 100 nM with five-fold dilution, and then passed through the filter. The fraction of the protein-bound aptamer was quantified with a PhosphorImager. [32P]-labeled library DNAs (Library) were used as a negative control. The radioactivity obtained on each slot was normalized to that of the negative control slot. 0.1 μM of DxSO4 was used as a polyanionic competitor. The binding affinities were calculated using the sigma plot 12.0 version software (Systat Software Inc.). | ||
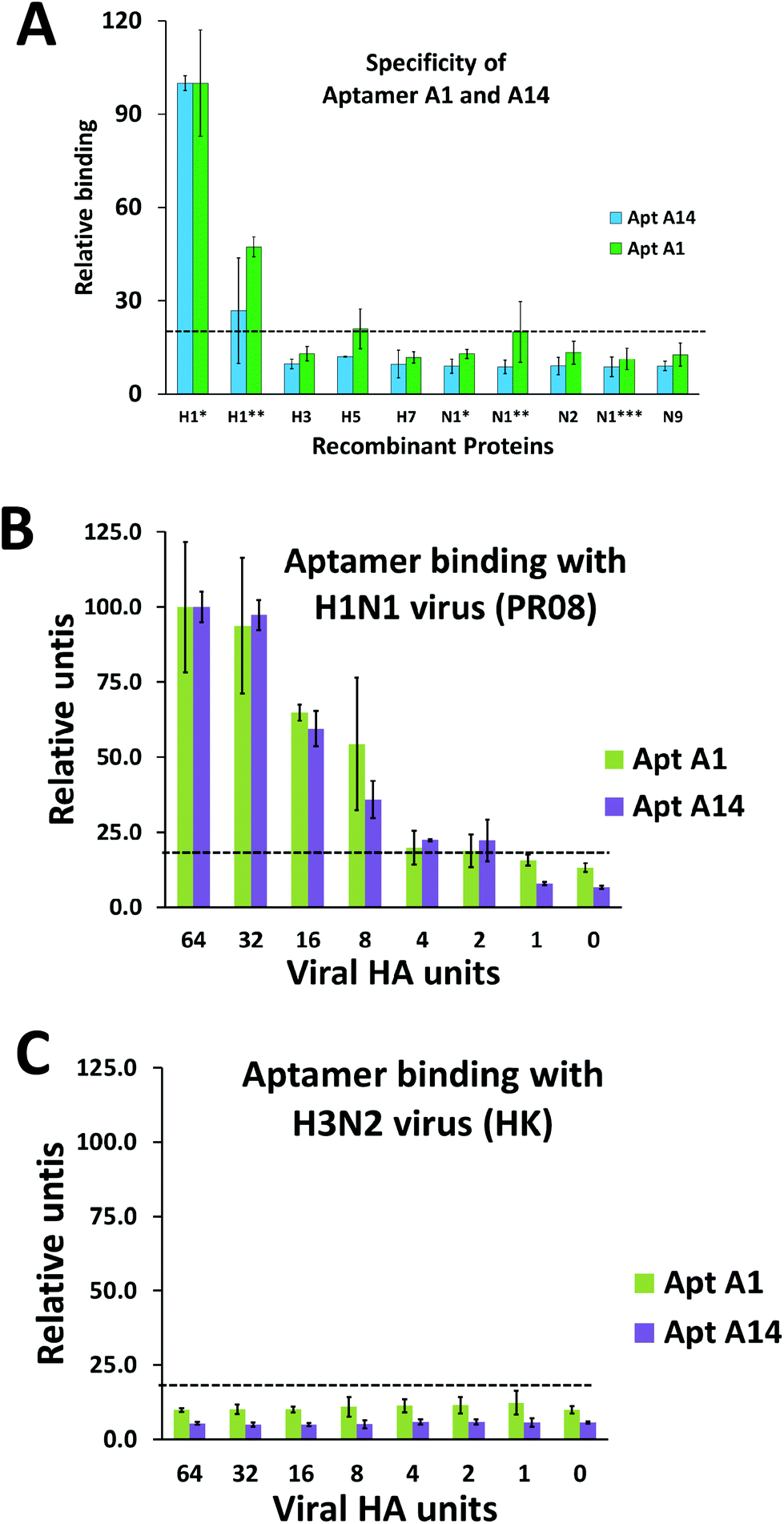 | ||
| Fig. 3 Determination of the specificity and viral binding capacity of the aptamers using the nitrocellulose-filter binding method. The aptamers A1 and A14 (A) were incubated with HA and NA proteins from a variety of strains of influenza A virus to check their specificity. The proteins included HA and NA proteins from H1N1, H3N2, H5N1, and H7N9 influenza viruses. Protein concentrations included in the experiment were a single point concentration at 20 nM, with a negative control with no protein. The abbreviations are as follows: H1*-CJ01 H1N1 HA; H1**-PR08 H1N1 HA; H3-H3N2 HA; H5-H5N1 HA; H7-H7N9 HA; N1*-CJ01 H1N1 NA; N1**-PR08 H1N1 NA; N2-H3N2 NA; N1***-H5N1 NA; N9-H7N9 NA. Binding affinity of aptamers A1 and A14 to influenza A virus PR08 H1N1 (B) and HK H3N2 (C) was analyzed. The aptamers A1 and A14 were incubated with a range of UV-inactivated H1N1 influenza A virion particles (64 – 1 HAU). No virus lane was used as the negative control. 1% Triton-X was used to lyse the virus just before using in the filter binding assay. The relative spot intensities were plotted on the Y axis and the viral HA units on the X axis. | ||
| K d (nM) | B max | K d (nM) | B max | |
|---|---|---|---|---|
| CJ01 HA protein | PR08 HA protein | |||
| Representative Kd and Bmax values of aptamers that specifically bound to HA1. “Kd” stands for “equilibrium dissociation constant” and “Bmax” represents maximum specific binding. Values shown are the mean ± SD of three independent experiments. | ||||
| Aptamer A1 | 3.36 ± 0.65 | 0.29 | 4.20 ± 2.59 | 0.10 |
| Aptamer A14 | 2.98 ± 1.35 | 0.18 | >100 | 0.32 |
| Modified aptamers | |
|---|---|
| “5” represents “benzyl-dU”. | |
| Apt H1 60mer capture | 5′-Thiol HEG*3 CCTGCCTTTG G555AACC5555GACCGC555C55AGGCCG55AGGAAAGG CACCGACAGC-invert dT-3′ |
| Apt H14 60mer detection | 5′-Biotin HEG*3 CCTGCCTTTG 55G5C55AG5CCG55G5G5CG5AA5GGC55AACCCG5A55 CACCGACAGC-invert dT-3′ |
| Control oligo –74mer | 5′-NH2-C6 linker-CTGGGTGGCTGTCGGTGAATACGGGTTAAGCCATTACGACACAACGGACTAAGACAACAAAGGCAGGACGCTCG-3 |
We further performed filter binding assays with two purified viruses (PR08 strain of H1N1 and Hong Kong strain of H3N2 virus). To our surprise, not only the A1 aptamer but also A14 aptamer strongly interacts with the PR08 strain of H1N1 virus (Fig. 3B). And not much of difference in binding affinity to the H3N2 virus was observed between A1 and A14 aptamers (Fig. 3C). It should be noted that these two aptamers showed a big difference in binding affinity to a purified recombinant PR08 HA protein (Table 1). We also confirmed the binding of these aptamers with the surrogate baculoviruses (Fig. S2B and S2C†). We speculate that the viro-SELEX process using a surrogate virus containing intact HA of PR08 virus allowed the selection of A14 which can strongly bind to the native form of the HA protein but only weakly to purified proteins which is not in native configuration.
It is also of interest to notice that our initial full length (74mer) clones bound to A/California/07/2009 (H1N1) virus as well (Fig. S2D†). When we compared the HA protein of the three H1N1 strains, we found that they share around 80% identity in their native state. This might mean that by using the surrogate viruses we may also be able to develop aptamers that can cover a broad range of the H1 family of viruses and at the same time can distinguish them from H3, H5 and H7 which share around 40–65% identity as analyzed (Table 3). This re-iterates the fact that even though the extracellular domain proteins show a low binding affinity, usage of surrogate viruses can come to rescue in finding good binders to the native state of the virus especially in cases of membrane proteins like hemagglutinin.
| A | |||||
|---|---|---|---|---|---|
| PR08 HA | CJ01 HA | H5N1 HA | H3N2 HA | H7N9 HA | |
| PR08 HA | 100 | 81.59 | 65.48 | 42.19 | 42.34 |
| CJ01 HA | 81.59 | 100 | 63.41 | 44.34 | 41.8 |
| H5N1 HA | 65.48 | 63.41 | 100 | 42.34 | 42.88 |
| H3N2 HA | 42.19 | 44.34 | 42.34 | 100 | 48.11 |
| H7N9 HA | 42.34 | 41.8 | 42.88 | 48.11 | 100 |
| B | |||
|---|---|---|---|
| CJ01 HA | CAL09 HA | PR08 HA | |
| CJ01 HA | 100 | 79.12 | 81.59 |
| CAL09 HA | 79.12 | 100 | 86.55 |
| PR08 HA | 81.59 | 86.55 | 100 |
Synthesis, truncation, and modifications of the candidate aptamers
The candidates of varying lengths were tested for their binding capabilities. Initial full length (74mer) aptamers containing a central variable site (40mer) along with primer binding sites (17mer) on both sides were truncated to a smaller length to make it more economical for diagnostic applications. A 50mer retaining only 5mer of the primer binding regions along with the central variable region (40mer) drastically decreased the binding affinity.Next we tried with the 60mer length retaining 10mer of the primer binding regions along with the central variable region (40mer) and checked its binding (Table 2). The 60mer aptamers A1 and A14 retained their binding capabilities and, therefore, we used 60mer aptamers for the development of a diagnostic system.
The aptamers A1 and A14 were used as capture and detection aptamers, respectively. The 5′ and 3′ ends of aptamer A1 were conjugated with the SH-group and idT, respectively, for immobilization of the aptamer to the nitrocellulose membrane filter and stabilization of the aptamer. The 5′ and 3′ ends of aptamer A14 were conjugated with biotin and idT, respectively. The biotin was further associated with either streptavidin-conjugated gold nanoparticles or streptavidin-conjugated fluorescence dyes for visualization of detection aptamers.
Identification of a pair of aptamers that do not compete with each other for binding to H1 proteins through binding competition assay
Lateral flow assay (LFA) needs two probes, in this case, two aptamers binding to different sites on the target protein: one for capturing the target of interest, and the other acting as a detector, for producing a visual read out. We performed binding competition assays in order to find an aptamer pair that does not compete each other for binding to the target protein as described previously in ref. 1. Among the aptamers tested, aptamers A1 and A14 did not compete for binding to CJ01 HA protein, indicating that they do not share a common binding region (Fig. S1†).Determination of the detection limit of gold nanoparticle conjugated aptamer-based LFA
An LFA system was constructed as described by Kwon1 with the aptamers A1 and A14 as capture and detection probes. At first, we conjugated gold nanoparticles to the detection aptamer A14 and measured the detection limits of the assay system with purified ectodomain of H1 protein and H1N1 influenza viruses (Fig. 6). Different dilutions of either purified recombinant HA proteins (ranging from 20 pmol to 1.25 pmol) or PR08 H1N1 influenza viruses (64 HAU–0.25 HAU) in 200 μl of sample buffer [10 mM sodium phosphate buffer (pH 7.0) and 0.1 μM DxSO4] were applied to the sample pad and analyzed the appearance of the test and the control lines.The detection limits of the LFA system for proteins were 0.32 pmol and 3.25 pmol for CH01 HA and PR08 HA proteins, respectively (Fig. 4A and B). The limit of detection (LOD) for CH01 HA was 10-fold lower than that of PR08 HA most likely due to the weak binding affinity of A14 to purified PR08 HA protein (Table 1). Interestingly, the detection limit of the LFA system for PR08 H1N1 virion particles reached 1 HAU roughly equivalent to 0.1 pmol of HA protein (Fig. 4C). This suggests that not only the aptamer A1 but also aptamer A14 binds well to the native HA protein of PR08 H1N1 virus, which was also observed in the filter binding assay (Fig. 3B).
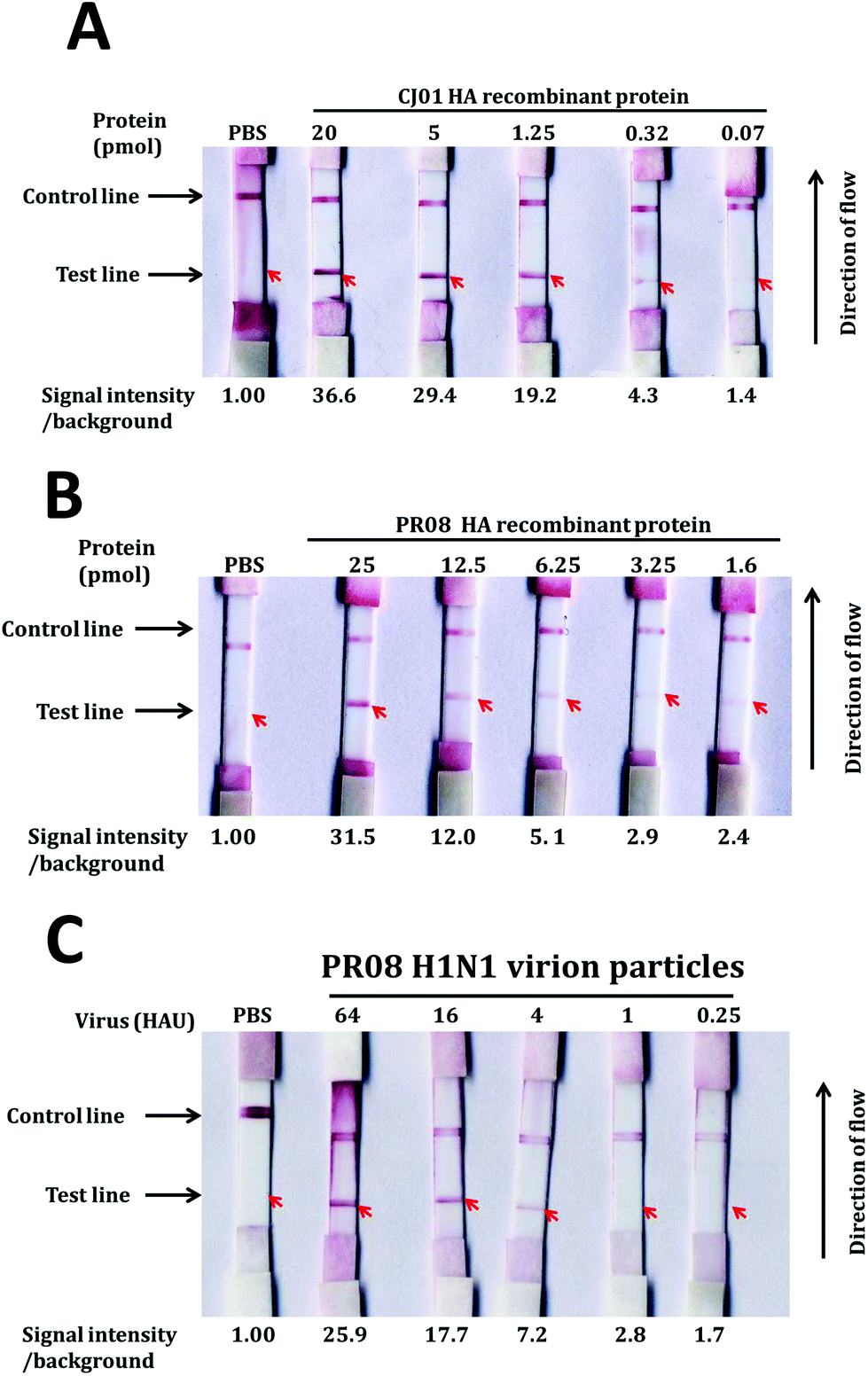 | ||
| Fig. 4 LOD determination of the gold nanoparticle-based LFA system. The LOD was determined by using a gold nanoparticle-based LFA system. Capture aptamers (20 pmol) and oligonucleotides complementary to the detection aptamer (20 pmol) were applied to the test and control lines, respectively, on the membrane. Detection aptamers (20 pmol) were loaded on the conjugation pad. Serially diluted HA proteins of CJ01 (A) and PR08 (B) or PR08 H1N1 virion particles (C) were loaded on the sample pad. The band intensities of the test lines were quantified by using Multi-Gauge (Fuji-film), and the relative band intensities of the test lines are depicted on the bottom of each strip. | ||
We also investigated the specificity of the LFA system using large quantities of purified proteins (20 pmol) and viruses (64 HAU). Positive signals were observed from HA proteins of both CJ01 and PR08 strains of H1N1 influenza virus (Fig. 5A). In contrast, no positive signal was observed from HA proteins of H3N2, H5N1 and H7N9 influenza viruses (Fig. 5A). The specificity of the LFA system was also observed from the tests using virion particles (Fig. 5B). A strong positive signal was observed from the test using PR08 strain of H1N1, but no positive signal was observed from the tests using the Hong Kong strain of H3N2 type A influenza virus and the Lee strain of type B influenza virus. The results indicate that the aptamer-based LFA system is a sensitive and specific diagnostic system for detecting H1N1 influenza virus.
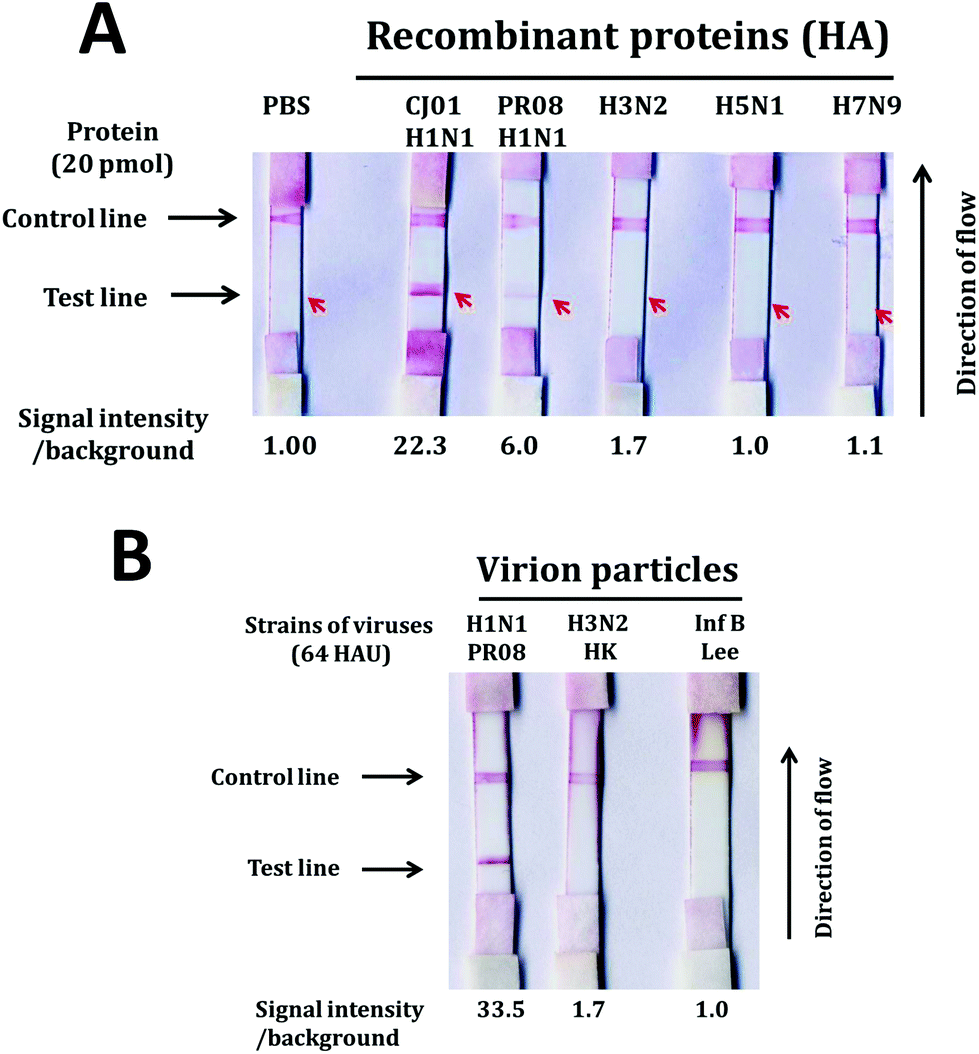 | ||
| Fig. 5 Specificity of the gold nanoparticle-based LFA system. Lateral flow strips were made in a similar fashion as reported in a previous section. HA proteins (20 pmol each) of the CJ01 strain of H1N1, PR08 strain of H1N1, H3N2, H5N1, and H7N9 were applied to the sample pad (A). Virion particles (64 HAU each) of the PR08 strain of H1N1, Hong Kong strain of H3N2, and Lee strain of type B influenza viruses were loaded on the sample pad (B). The band intensities of the test lines were quantified by Multi-Gauge (Fuji-film), and the relative band intensities of the test lines are depicted on the bottom of each strip. | ||
Increasing sensitivity of the LFA system using a fluorescent tag
We noted that patient samples in a hospital setting contained over a wide range of virus particles with a mean range of 6–8 log10 copies per swab sample,42–47 and that 106.8 particles of influenza virus correspond to approximately 1 HAU.48 In order to achieve a LOD of lower than 106 copies, we developed a LFA system using a fluorescent dye (Alexa Fluor 488)-conjugated detection aptamer A14. Alexa Fluor 488 associated with streptavidin (5 pmol) conjugated with biotinylated aptamer (20 pmol) at 1:4 ratio was used in the LFA system.
To demonstrate the performance of the system, the range of detection was determined by measuring the fluorescence signal of samples with different recombinant protein concentrations. Procedures for the LFA were similar to that for the gold nanoparticle-based LFA. The fluorescence signal at the test line was determined by the Cy2 filter of the Amersham™ Imager after 30 min of sample application on the cassette and then subsequently quantified using the Multigauge (Fujifilm) software. The LODs of this LFA system for CJ01 HA and PR08 HA proteins were 0.04 pmol (Fig. 6A and B) and 0.32 pmol, respectively. The enhanced sensitivity of this system is likely attributed to the brightness of the fluorescent Alexa Fluor 488 dyes. And the LOD of this LFA system for the PR08 H1N1 virus stock spiked in with a throat swab sample of a healthy individual was 0.08 HAU (Fig. 6C). These results indicate that this LFA system detects PR08 virus about 40-fold more sensitively that PR08 HA proteins (0.32 pmol is roughly equivalent to 3.2 HAU).48,49
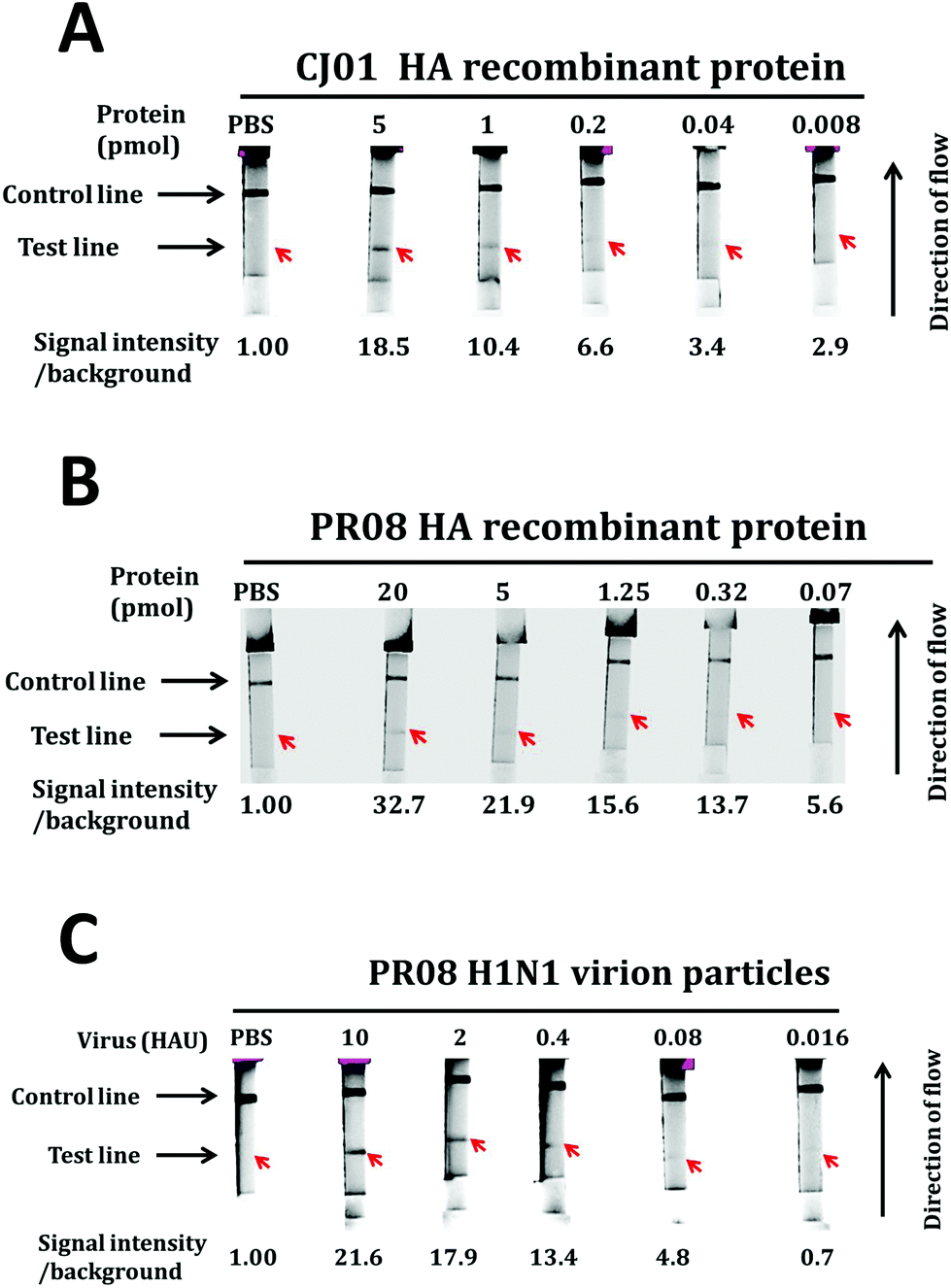 | ||
| Fig. 6 LOD determination of the fluorescence-based LFA system. The LOD was determined by using the fluorescence-based LFA system. Capture aptamers (20 pmol) and oligonucleotides complementary to the detection aptamer (20 pmol) were applied to the test and control lines, respectively, on the membrane. Biotinylated detection aptamers (20 pmol) associated with streptavidin-conjugated Alexa Fluor-488 were loaded on the conjugation pad. Serially diluted HA proteins of CJ01 (A) and PR08 (B) or PR08 H1N1 virion particles (C) were loaded on the sample pad. The band intensities of the test lines were quantified by using the Amersham™ Imager 680 (GE), and the relative band intensities of the test lines are depicted on the bottom of each strip. | ||
In order to evaluate the detection specificity of this LFA system, we performed LFAs with large quantities (20 pmol which is roughly equivalent to 200 HAU)49 of purified HA proteins of CJ01 H1N1, PR08 H1N1, H3N2, H5N1, and H7N9 (Fig. 7A). Strong positive signals were observed from HAs of CJ01 H1N1 and PR08 H1N1, but no signal was observed from HAs of H3N2, H5N1, and H7N9 (Fig. 7A). Moreover, we performed LFAs with purified viruses (64 HAU) of the PR08 strain of H1N1, Hong Kong strain of H3N2, and Lee strain of type B influenza virus (Fig. 7B). A strong positive signal was observed from the PR08 H1N1 virus, but no signal was observed from the H3N2 and type B influenza viruses (Fig. 7B). These results indicate that the LFA system using the aptamers A1 and A14 is highly specific for the detection of the H1N1 subtype of influenza virus. It is worth noting that the aptamers A1 and A14 also bind to the Cal pdm2009 strain of H1N1 virus (Fig. S2D†), which was not used in the SELEX processes of this study. This suggests that the LFA system we describe here is likely to detect a broad spectrum of influenza viruses belonging to subtype H1N1 considering the similarities of CJ01, CAL09 and PR08 strains of H1N1 influenza viruses (Table 3).
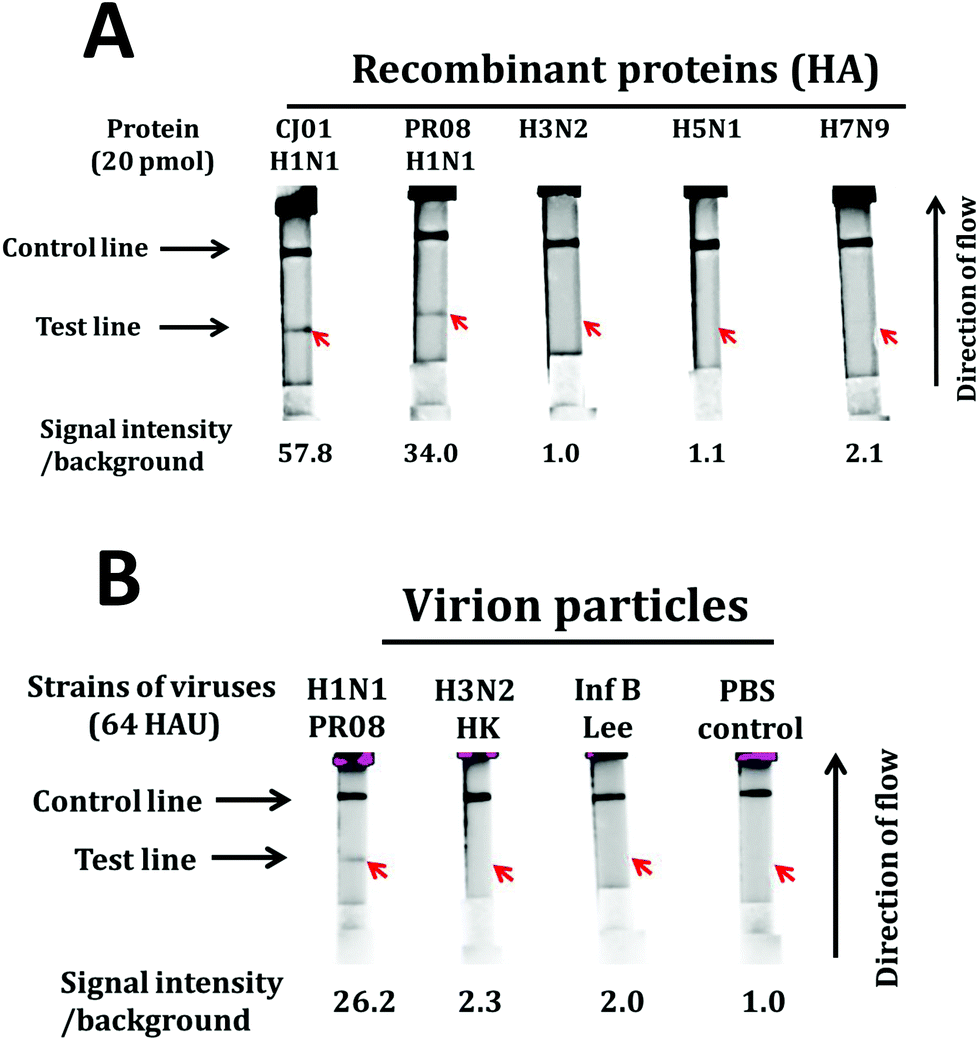 | ||
| Fig. 7 Specificity of the fluorescence-based LFA system. Lateral flow strips were made in a similar fashion as reported in the previous section. HA proteins (20 pmol each) of CJ01 strain of H1N1, PR08 strain of H1N1, H3N2, H5N1, and H7N9 were applied to the sample pad (A). Virion particles (64 HAU each) of PR08 strain of H1N1, Hong Kong strain of H3N2, and Lee strain of type B influenza viruses were loaded on the sample pad (B). The band intensities of the test lines were quantified by using the Amersham™ Imager 680 (GE), and the relative band intensities of the test lines are depicted on the bottom of each strip. | ||
As compared with the conventional aptamer-gold nanoparticle-based LFA system, the aptamer-fluorescence-based LFA system increased sensitivity about 12-fold or more (LOD of 1 HA vs. LOD of 0.08; compare Fig. 6C with Fig. 4C). Considering the virus number from patient samples (106–8 copies) and the detection limit of this system (0.08 HAU or lower), this aptamer-based LFA system is sensitive enough to detect influenza viruses from most of the patient samples.
In our study, we made use of an imager for detecting the fluorescent signals. However, portable lateral flow fluorescence readers such as ESEquant LR3 (Qiagen) are likely to be used for convenient diagnoses of patients in hospitals. By using the viro-SELEX and fluorescence-based LFA system described here, we consider developing multiplex LFAs for H3N2, H1N1 and influenza B viruses which are the most rampant flu viruses circulating at this point of time in the whole world as per CDC and WHO indications.
Conclusion
A highly sensitive LFA system for detecting subtype H1N1 influenza viruses was developed using a novel viro-SELEX and a fluorescence tagging method. The LOD of this system was 0.08 HAU or lower for H1N1 influenza viruses. In order to generate aptamers binding strongly and specifically to the native form of HA proteins of subtype H1N1 of influenza viruses, we used special approaches in SELEX processes. Advanced SELEX using modified nucleosides allowed finding aptamers strongly binding to target proteins; positive selection with HA proteins of H1N1 followed by negative selection with HA proteins of H3N2, H5N1 and H7N9 allowed enrichment of aptamers specifically interacting with H1N1 but not with other subtype influenza viruses; Consecutive positive selections with HA proteins of two different strains of H1N1 allowed enrichment of aptamers binding to a broad spectrum of strains of H1N1 viruses; Viro-SELEX allowed enrichment of aptamers to the native form of HA proteins on the virion particles; finally, the sensitivity of the LFA system was improved by using fluorescence-dye conjugated detection aptamers. These approaches can be used for developing diagnostic systems for other infectious agents and other diseases such as cancer probing membrane proteins.Author contributions
C.N. conceived and performed the experiments and wrote the manuscript. J.K. provided feedback. C.K. and S.J.K. provided materials. S.K.J. designed the project, provided feedback, and contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science and ICT (No. NRF-2017M3A9F6029755) and by the Korean government (MSIT) (No. NRF-2019M3E5D6063871).We would like to thank Dr Hahm (School of Medicine, University of Missouri-Columbia)/Dr Shan-Lu Liu (University of Missouri)/Dr Gary Nabel (NIH), Dr YK Choi (Chungbuk National University), Dr Kim (Korea research Institute of Chemical Technology), and Professor JY Noh (Department of Internal Medicine, Korea University, Guro Hospital) for providing us with the plasmids containing the hemagglutinin and neuraminidase genes. We would also like to thank Eun Jin Park and Dr Sangmin Jeon of the Department of Chemical Engineering department, POSTECH for allowing us to use his lab facility to make the LFA strips.
References
- J. Kwon, C. Narayan, C. Kim, M. J. Han, M. Kim and S. K. Jang, J. Biomed. Nanotechnol., 2019, 15, 1609–1621 CrossRef PubMed
.
- C. Paules and K. Subbarao, Lancet, 2017, 390, 697–708 CrossRef
- S. Tong, X. Zhu, Y. Li, M. Shi, J. Zhang, M. Bourgeois, H. Yang, X. Chen, S. Recuenco, J. Gomez, L. M. Chen, A. Johnson, Y. Tao, C. Dreyfus, W. Yu, R. McBride, P. J. Carney, A. T. Gilbert, J. Chang, Z. Guo, C. T. Davis, J. C. Paulson, J. Stevens, C. E. Rupprecht, E. C. Holmes, I. A. Wilson and R. O. Donis, PLoS Pathog., 2013, 9, e1003657 CrossRef PubMed
- http://www.who.int/influenza/surveillance_monitoring/updates/latest_update_GIP_surveillance/en/ .
- S. Barik, BMC Med., 2012, 10, 104 CrossRef PubMed
- P. D. Reuman, D. I. Bernstein, M. C. Keefer, E. C. Young, J. R. Sherwood and G. M. Schiff, Antiviral Res., 1989, 11, 27–40 CrossRef CAS PubMed
- F. G. Hayden, J. J. Treanor, R. S. Fritz, M. Lobo, R. F. Betts, M. Miller, N. Kinnersley, R. G. Mills, P. Ward and S. E. Straus, J. Am. Med. Assoc., 1999, 282, 1240–1246 CrossRef CAS PubMed
- F. Shi, Y. Xie, L. Shi and W. Xu, Curr. Med. Chem., 2013, 20, 3923–3934 CrossRef CAS PubMed
- M. G. Ison, Expert Rev. Anti-Infect. Ther., 2015, 13, 417–425 CAS
- F. G. Hayden and A. J. Hay, Curr. Top. Microbiol. Immunol., 1992, 176, 119–130 CAS
- Centers for Disease Control and Prevention. influenza (flu), 2017. https://www.cdc.gov/flu/.
- World Health Organisation – http://www.who.int/mediacentre/factsheets/fs194/en/.
- S. Kashiwagi and N. Rinsho, Jpn J. Clin. Med., 2003, 61, 1963–1966 Search PubMed
- S. W. Younkin, R. F. Betts, F. K. Roth and R. G. Douglas Jr., Antimicrob. Agents Chemother., 1983, 23, 577–582 CrossRef CAS PubMed
- M. Rahman, M. F. Vandermause, B. A. Kieke and E. A. Belongia, Diagn. Microbiol. Infect. Dis., 2008, 62, 162–166 CrossRef CAS PubMed
- J. Barenfanger, C. Drake and G. Kacich, J. Clin. Microbiol., 1999, 37, 1415–1418 CAS
- P. C. Woo, S. S. Chiu, W. H. Seto and M. Peiris, J. Clin. Microbiol., 1997, 35, 1579–1581 CAS
- M. J. P. van Dongen, R. U. Kadam, J. Juraszek, E. Lawson, B. Brandenburg, F. Schmitz, W. B. G. Schepens, B. Stoops, H. A. van Diepen, M. Jongeneelen, C. Tang, J. Vermond, A. van Eijgen-Obregoso Real, S. Blokland, D. Garg, W. Yu, W. Goutier, E. Lanckacker, J. M. Klap, D. C. G. Peeters, J. Wu, C. Buyck, T. H. M. Jonckers, D. Roymans, P. Roevens, R. Vogels, W. Koudstaal, R. H. E. Friesen, P. Raboisson, D. Dhanak, J. Goudsmit and I. A. Wilson, Science, 2019, 363 DOI:10.1126/science.aar6221
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed
- H. Kaur, J. G. Bruno, A. Kumar and T. K. Sharma, Theranostics, 2018, 8, 4016–4032 CrossRef CAS PubMed
- M. Ye, J. Hu, M. Peng, J. Liu, J. Liu, H. Liu, X. Zhao and W. Tan, Int. J. Mol. Sci., 2012, 13, 3341–3353 CrossRef CAS PubMed
- C. J. Wei, L. Xu, W. P. Kong, W. Shi, K. Canis, J. Stevens, Z. Y. Yang, A. Dell, S. M. Haslam, I. A. Wilson and G. J. Nabel, J. Virol., 2008, 82, 6200–6208 CrossRef CAS PubMed
- J. Stevens, A. L. Corper, C. F. Basler, J. K. Taubenberger, P. Palese and I. A. Wilson, Science, 2004, 303, 1866–1870 CrossRef CAS PubMed
- H. Kaur, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 2323–2329 CrossRef CAS PubMed
- K. Sefah, D. Shangguan, X. Xiong, M. B. O'Donoghue and W. Tan, Nat. Protoc., 2010, 5, 1169–1185 CrossRef CAS PubMed
- H. Kaur, J. J. Li, B. H. Bay and L. Y. Yung, PLoS One, 2013, 8, e50964 CrossRef CAS PubMed
- P. Dua, S. Kim and D. K. Lee, Methods, 2011, 54, 215–225 CrossRef CAS PubMed
- M. Avci-Adali, M. Metzger, N. Perle, G. Ziemer and H. P. Wendel, Oligonucleotides, 2010, 20, 317–323 CrossRef CAS PubMed
- S. Ohuchi, BioRes. Open Access, 2012, 1, 265–272 CrossRef CAS PubMed
- M. Chen, Y. Yu, F. Jiang, J. Zhou, Y. Li, C. Liang, L. Dang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2016, 17 DOI:10.3390/ijms17122079
- Z. Tang, P. Parekh, P. Turner, R. W. Moyer and W. Tan, Clin. Chem., 2009, 55, 813–822 CrossRef CAS PubMed
- X. Y. Tao, J. Y. Choi, W. J. Kim, J. H. Lee, Q. Liu, S. E. Kim, S. B. An, S. H. Lee, S. D. Woo, B. R. Jin and Y. H. Je, J. Virol., 2013, 87, 8441–8450 CrossRef CAS PubMed
- T. A. Kost and J. P. Condreay, Trends Biotechnol., 2002, 20, 173–180 CrossRef CAS PubMed
- M. Cerutti and J. Golay, mAbs, 2012, 4, 294–309 CrossRef PubMed
- R. White, C. Rusconi, E. Scardino, A. Wolberg, J. Lawson, M. Hoffman and B. Sullenger, Mol. Ther., 2001, 4, 567–573 CrossRef CAS PubMed
- J. D. Vaught, C. Bock, J. Carter, T. Fitzwater, M. Otis, D. Schneider, J. Rolando, S. Waugh, S. K. Wilcox and B. E. Eaton, J. Am. Chem. Soc., 2010, 132, 4141–4151 CrossRef CAS PubMed
- L. Gold, D. Ayers, J. Bertino, C. Bock, A. Bock, E. N. Brody, J. Carter, A. B. Dalby, B. E. Eaton, T. Fitzwater, D. Flather, A. Forbes, T. Foreman, C. Fowler, B. Gawande, M. Goss, M. Gunn, S. Gupta, D. Halladay, J. Heil, J. Heilig, B. Hicke, G. Husar, N. Janjic, T. Jarvis, S. Jennings, E. Katilius, T. R. Keeney, N. Kim, T. H. Koch, S. Kraemer, L. Kroiss, N. Le, D. Levine, W. Lindsey, B. Lollo, W. Mayfield, M. Mehan, R. Mehler, S. K. Nelson, M. Nelson, D. Nieuwlandt, M. Nikrad, U. Ochsner, R. M. Ostroff, M. Otis, T. Parker, S. Pietrasiewicz, D. I. Resnicow, J. Rohloff, G. Sanders, S. Sattin, D. Schneider, B. Singer, M. Stanton, A. Sterkel, A. Stewart, S. Stratford, J. D. Vaught, M. Vrkljan, J. J. Walker, M. Watrobka, S. Waugh, A. Weiss, S. K. Wilcox, A. Wolfson, S. K. Wolk, C. Zhang and D. Zichi, PLoS One, 2010, 5, e15004 CrossRef CAS PubMed
- J. C. Rohloff, A. D. Gelinas, T. C. Jarvis, U. A. Ochsner, D. J. Schneider, L. Gold and N. Janjic, Mol. Ther. – Nucleic Acids, 2014, 3, e201 CrossRef CAS PubMed
- S. Gupta, M. Hirota, S. M. Waugh, I. Murakami, T. Suzuki, M. Muraguchi, M. Shibamori, Y. Ishikawa, T. C. Jarvis, J. D. Carter, C. Zhang, B. Gawande, M. Vrkljan, N. Janjic and D. J. Schneider, J. Biol. Chem., 2014, 289, 8706–8719 CrossRef CAS PubMed
- M. Kim, S. Y. Kim, H. W. Lee, J. S. Shin, P. Kim, Y. S. Jung, H. S. Jeong, J. K. Hyun and C. K. Lee, Antiviral Res., 2013, 100, 460–472 CrossRef CAS PubMed
- J. Bhardwaj, N. Chaudhary, H. Kim and J. Jang, Anal. Chim. Acta, 2019, 1064, 94–103 CrossRef CAS PubMed
- Y. Pang, Z. Rong, J. Wang, R. Xiao and S. Wang, Biosens. Bioelectron., 2015, 66, 527–532 CrossRef CAS PubMed
- H. Bai, R. Wang, B. Hargis, H. Lu and Y. Li, Sensors, 2012, 12, 12506–12518 CrossRef CAS PubMed
- J. Zhang, J. Tian, Y. He, S. Chen, Y. Jiang, Y. Zhao and S. Zhao, Analyst, 2013, 138, 4722–4727 RSC
- K. K. To, K. H. Chan, I. W. Li, T. Y. Tsang, H. Tse, J. F. Chan, I. F. Hung, S. T. Lai, C. W. Leung, Y. W. Kwan, Y. L. Lau, T. K. Ng, V. C. Cheng, J. S. Peiris and K. Y. Yuen, J. Med. Virol., 2010, 82, 1–7 CrossRef PubMed
- S. Meschi, M. Selleri, E. Lalle, L. Bordi, M. B. Valli, F. Ferraro, G. Ippolito, N. Petrosillo, F. N. Lauria and M. R. Capobianchi, BMC Infect. Dis., 2011, 11, 140 CrossRef PubMed
- L. Van Wesenbeeck, D. D'Haese, J. Tolboom, H. Meeuws, D. E. Dwyer, M. Holmes, M. G. Ison, K. Katz, A. McGeer, J. Sadoff, G. J. Weverling and L. Stuyver, Open Forum Infect. Dis., 2015, 2, ofv166 CrossRef PubMed
- D. A. Tyrrell and R. C. Valentine, J. Gen. Microbiol., 1957, 16, 668–675 CrossRef CAS PubMed
- A. Harris, G. Cardone, D. C. Winkler, J. B. Heymann, M. Brecher, J. M. White and A. C. Steven, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19123–19127 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an01943j |
| This journal is © The Royal Society of Chemistry 2020 |