
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of chiral 1,4-oxazepane-5-carboxylic acids from polymer-supported homoserine†
Petra Králováa,
Barbora Lemrováb,
Michal Maloň c and
Miroslav Soural*ac
c and
Miroslav Soural*ac
aDepartment of Organic Chemistry, Faculty of Science, Palacký University, 771 46 Olomouc, Czech Republic. E-mail: miroslav.soural@upol.cz
bJEOL (U.K.) Ltd., JEOL House, Silver Court, Watchmead, Welwyn Garden City, Hertfordshire AL7 1LT, UK
cInstitute of Molecular and Translational Medicine, Faculty of Medicine and Dentistry, Palacký University, Hněvotínská 5, 779 00 Olomouc, Czech Republic
First published on 30th September 2020
Abstract
The preparation of novel 1,4-oxazepane-5-carboxylic acids bearing two stereocenters is reported in this article. Fmoc-HSe(TBDMS)-OH immobilized on Wang resin was reacted with different nitrobenzenesulfonyl chlorides and alkylated with 2-bromoacetophenones to yield N-phenacyl nitrobenzenesulfonamides. Their cleavage from the polymer support using trifluoroacetic acid (TFA) led to the removal of the silyl protective group followed by spontaneous lactonization. In contrast, TFA/triethylsilane (Et3SiH)-mediated cleavage yielded 1,4-oxazepane derivatives as a mixture of inseparable diastereomers. The regioselectivity/stereoselectivity depended on the substitution of the starting 2-bromoacetophenones and was studied in detail. Catalytic hydrogenation of the nitro group improved the separability of the resulting diastereomeric anilines, which allowed us to isolate and fully characterize the major isomers.
Introduction
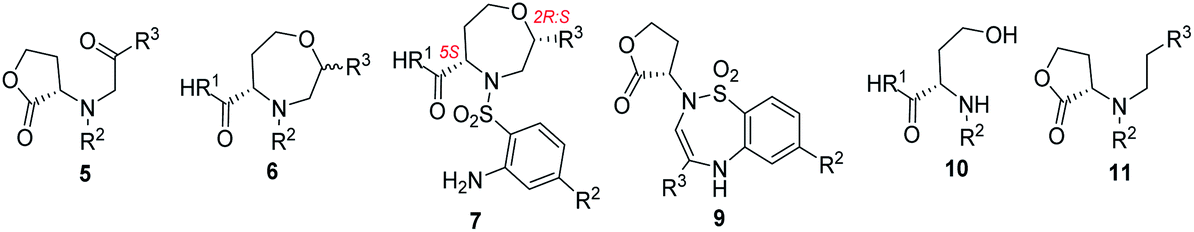
Chiral seven-membered heterocycles bearing one or more heteroatoms in their skeleton are an interesting group of compounds with unique physico-chemical and biological properties. The prominent heterocyclic scaffold within this group is represented by 1,4-oxazepanes, which occur in both synthetic compounds and natural products (Fig. 1).1–6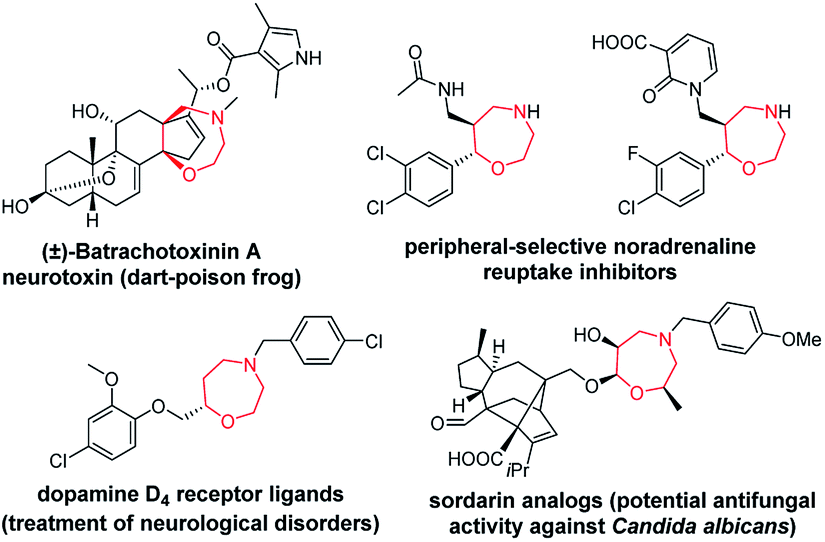 | ||
| Fig. 1 Selected pharmacologically relevant compounds bearing a 1,4-oxazepane scaffold.1–6 | ||
Compounds bearing 1,4-oxazepane scaffolds have been reported as potent anticonvulsants7 and antifungal agents5,8 or agents to treat inflammatory bowel disease,9 lupus nephritis10 and respiratory diseases, including asthma and bronchiectasis.11,12 Over the past decade, synthetic chemists have struggled to develop different strategies to access 1,4-oxazepanes from various starting materials.
The most robust synthetic approaches reported to date are based on intramolecular cyclization of alkenols,13,14 alkynols15,16 or hydroxyketones,16 typically using Brönsted or Lewis acids; however, some alternative methods of limited applicability have also been described recently.17–25
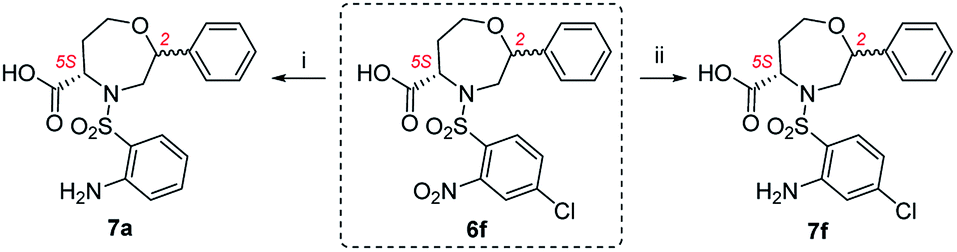
Although the synthetic availability of chiral 1,4-oxazepanes has already been determined, the decoration of the scaffold with reactive functional groups amenable to further diversification remains a challenging task due to the limited applicability of previously developed procedures for functionalizing starting materials. In our previous contribution, we reported the simple synthesis of chiral morpholines starting from resin-bound serine.26 Using either TFA- or TFA/Et3SiH-induced cleavage of the corresponding polymer-supported intermediates (Fig. 2), we synthesized either dihydrooxazine-3-carboxylic acids or morpholine-3-carboxylic acids with full control of the newly formed stereocenter. Later, we extended this method to the simple synthesis of fused [6 + 7]27,28 or [6 + 6]29,30 morpholines. To eventually synthesize the corresponding homological compounds, we decided to use polymer-supported homoserine to access the 1,4-oxazepane-5-carboxylic acids suitable for further modification. In this article, we report on the applicability, regioselectivity and stereoselectivity of the proposed method.
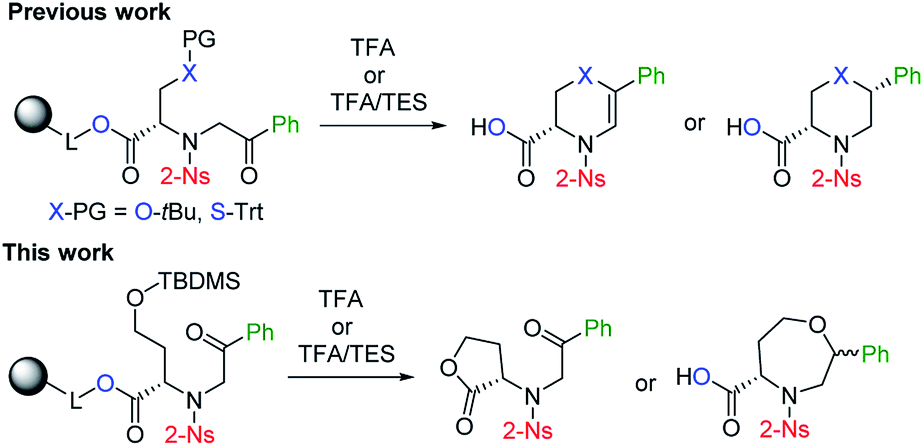 | ||
| Fig. 2 Previously reported stereoselective synthesis of chiral (thio)morpholines and the application of the method to homological starting material. | ||
Results and discussion
Fmoc-HSe(TBDMS)-OH was prepared from homoserine in two steps31,32 and immobilized on Wang resin using the 1-hydroxybenzotriazole (HOBt)/diisopropylcarbodiimide (DIC) technique to suppress racemization. The key intermediate 3a was synthesized according to our previous protocols26 consisting of Fmoc-protective group cleavage, reaction with 2-nitrobenzenesulfonyl chloride (2-Ns-Cl) and alkylation using 2-bromoacetophenone (Scheme 1). Inspired by the smooth cyclization of serine-based analogs to 3,4-dihydro-1,4-oxazine-3-carboxylic acids,26 we hypothesized that exposure of resin 3a to TFA could yield the corresponding homologous product, i.e., 4,5,6,7-tetrahydro-1,4-oxazepine-5-carboxylic acid 4a. The reaction yielded a single product with 87% crude purity (calculated from HPLC-UV traces at 205–400 nm) and 74% overall yield (calculated from the 1H NMR spectrum of the purified product). Although HRMS analysis corresponded to the molecular mass of suggested product 4a, NMR analysis (see ESI for details†) revealed preferential lactonization, which yielded compound 5a. Interestingly, when Et3SiH was added to the cleavage cocktail, a different course of reaction was observed. We received two chromatographically inseparable compounds with identical molecular masses (as indicated by HPLC-UV-MS analysis, the combined crude purity was 85%). To eventually improve the separability, we performed catalytic hydrogenation using palladium on carbon (Pd/C) in 2-isopropanol (IPA),28 which again afforded a mixture of two isomers (the combined crude purity was 91%, and the ratio was 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44, as calculated from HPLC-UV traces at 205–400 nm); however, in this case, reverse-phase chromatography (RP-HPLC) indicated possible separation. Consequently, the major isomer was successfully isolated using semipreparative RP-HPLC at 34% overall yield (calculated from the 1H NMR spectrum of the purified product) and subjected to detailed NMR investigation.
:44, as calculated from HPLC-UV traces at 205–400 nm); however, in this case, reverse-phase chromatography (RP-HPLC) indicated possible separation. Consequently, the major isomer was successfully isolated using semipreparative RP-HPLC at 34% overall yield (calculated from the 1H NMR spectrum of the purified product) and subjected to detailed NMR investigation.
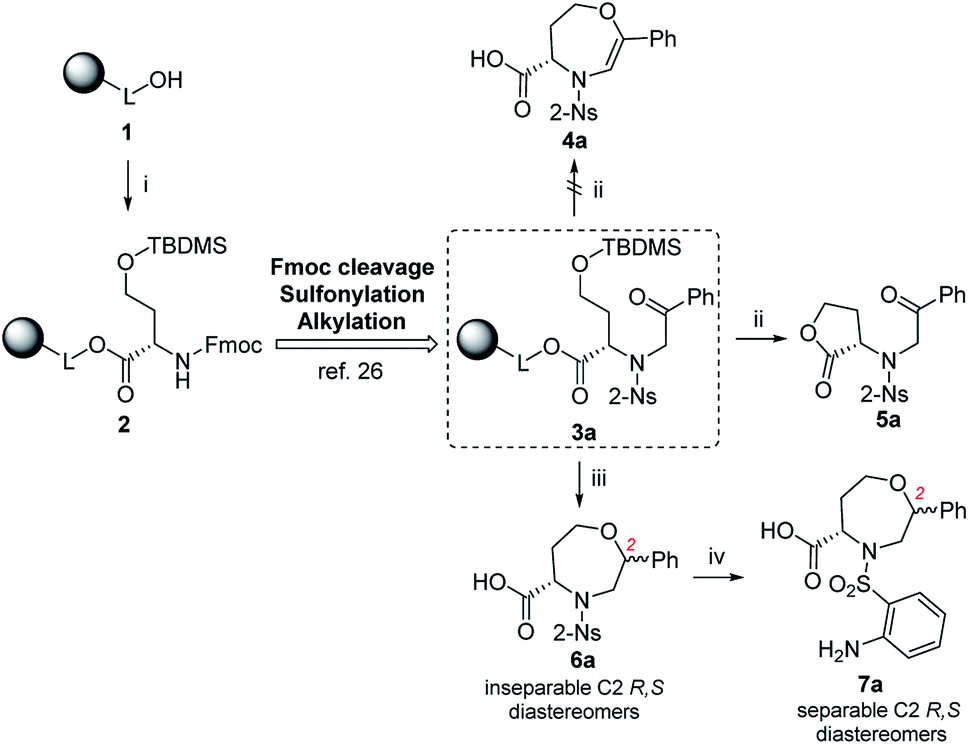 | ||
| Scheme 1 The reactivity of resin-bound intermediate 3a under different reaction conditions. Reagents and conditions: (i) 1-hydroxybenzotriazole (HOBt), 4-(dimethylamino)pyridine (DMAP), diisopropylcarbodiimide (DIC), N,N-dimethylformamide (DMF), CH2Cl2, 24 h, rt; (ii) 50% trifluoroacetic acid (TFA)/CH2Cl2, 1 h, rt; (iii) TFA/triethylsilane (Et3SiH)/CH2Cl2 (10:1:9), 30 min, rt; (iv) H2, 10% palladium on carbon (Pd/C) or platinum(IV) oxide (PtO2), 2-isopropanol (IPA), 24 h, rt. | ||
We recorded and analyzed 1H, 13C{1H}, APT, 1H–1H COSY, 1H–1H NOESY, 1H–13C HMQC, 1H–13C HMBC and 1H–15N HMBC NMR data to determine the constitution. Complete assignment of the 1H, 13C and 15N signals was possible and is shown in the ESI (Fig. S17–S19 and Table S2†). In brief, by means of the homonuclear and heteronuclear correlation data, we identified the 1,4-oxazepane-5-carboxylic acid, phenyl and 2-aminobenzenesulfonyl moieties indicating the structure of 7a. The connectivity between the oxazepane and phenyl rings was confirmed by three long-range 1H–13C correlations (see ESI, Fig. S18†). Finally, the planar structure was established by the 1H–1H NOESY spectrum, which gave the key correlations between oxazepane protons and aminobenzenesulfonyl proton H19 and correlations between oxazepane protons and phenyl protons H9,13 (Fig. 3 and S19†). All the 1D and 2D NMR spectra can be found in the ESI (Fig. S20–S27†).
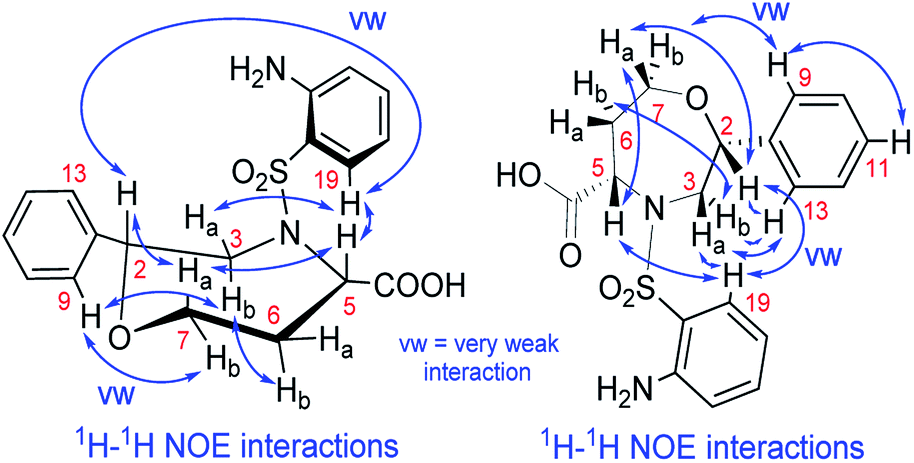 | ||
| Fig. 3 The NOE correlations prove the constitution and configuration of oxazepane derivative 7a. | ||
To determine the conformation and relative configuration of 7a, we analyzed the 1H–1H coupling constants on the oxazepane ring and the NOE correlations in detail. Despite the spatial flexibility of seven membered rings leading to multiple possible conformational states, the analysis of vicinal 1H–1H couplings indicated that the scaffold existed in the most energetically favourable chair conformation (Fig. 3). Since the configuration of the C5 stereocenter was defined by the configuration of the starting material (S), the configuration of the newly formed C2 stereocenter was assigned as R.
Although we did not isolate and analyze the minor isomer of 7a, in the case of 7b, RP-HPLC purification enabled the separation and isolation of both isomers. Thorough NMR structural analysis indicated the formation of C2 R,S diastereomers 7b2R and 7b2S (Table 1). The 1H NMR spectrum of 7b2R was fairly similar to the spectrum of 7a; hence, we concluded that this compound had the same configuration as 7a. Compound 7b2S was analyzed by 1D and 2D NMR spectroscopy (see ESI, Fig. S34–S41 and Table S3†) to verify the planar structure, assign all the 1H and 13C signals and determine the relative configuration. Finally, we confirmed the configuration of 7b2S by analyzing the vicinal 1H–1H couplings on the oxazepane ring and NOE correlations (Fig. 4).
|
||||||
|---|---|---|---|---|---|---|
| Position | C2 R isomer | C2 S isomer | ||||
| 1H NMR δH [ppm] | Splitting pattern | J [Hz] | 1H NMR δH [ppm] | Splitting pattern | J [Hz] | |
| H2 | 4.28 | dd | 9.3, 1.2 | 4.64 | dd | 8.7, 1.6 |
| Ha3 | 3.82 | ddd | 16.2, 1.2, 1.2 | 3.63 | dd | 14.2, 1.6 |
| Hb3 | 3.53 | dd | 16.2, 9.6 | 3.43 | dd | 14.2, 8.7 |
| H5 | 4.49 | ddd | 10.8, 7.1, 1.0 | 4.59 | dd | 4.5, 4.5 |
| Ha6 | 2.46 | dddd | 15.7, 7.1, 6.3, 1.0 | 2.30 | dddd | 15.8, 4.5, 4.5, 1.6 |
| Hb6 | 2.21 | dddd | 15.7, 10.8, 9.3, 1.5 | 2.10 | dddd | 15.8, 11.0, 4.5, 3.0 |
| Ha7 | 3.67 | ddd | 12.9, 9.3, 1.0 | 3.78 | ddd | 12.8, 11.0, 1.6 |
| Hb7 | 4.05 | ddd | 12.9, 6.3, 1.5 | 4.02 | ddd | 12.8, 4.5, 3.0 |
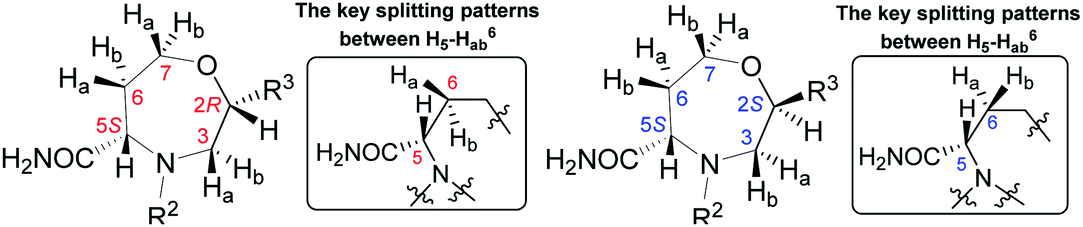
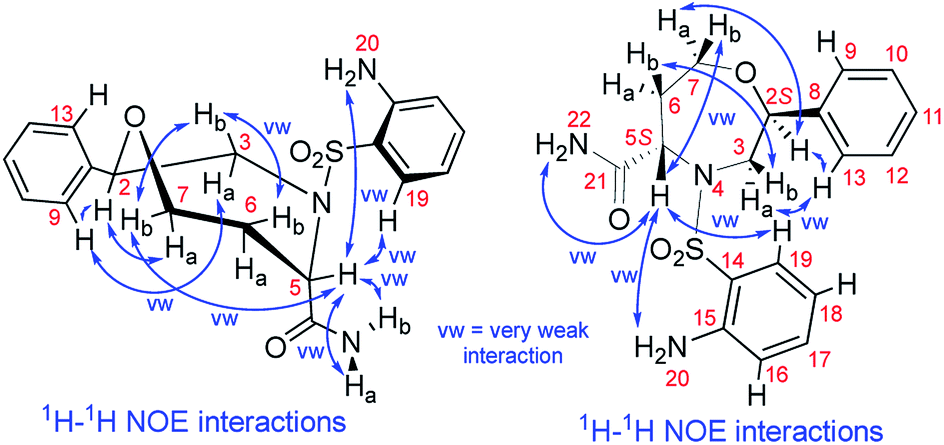 | ||
| Fig. 4 The NOE correlations proving the configuration of oxazepane derivative 7b2S. | ||
Table 1 summarizes the 1H chemical shifts, splitting patterns, and 2J and 3J homonuclear couplings observed in 7b2R and 7b2S. While the chemical shifts differ rather insignificantly between the two diastereomers, the splitting pattern of signal H5 and 3J(H5–Ha6) and 3J(H5–Hb6) can be used to differentiate between 7b2R and 7b2S. In the C2 R isomer 7b2R, two relatively large couplings and one very small long-range coupling leading to a doublet of doublet of doublets are observed. However, in the case of the C2 S isomer 7b2S, the vicinal coupling constants are smaller and equal or nearly equal, and hence, the signal takes the shape of a pseudotriplet (dd). The same trend was observed in all derivatives 6–7.
After proving the structure and formation of the two diastereomers, we tried to improve the stereoselectivity by using a lower reaction temperature (0 °C or −20 °C); however, a nearly equal ratio of isomers was obtained. Furthermore, we tested the use of TMSOTf/Et3SiH as reported earlier for an analogical starting material,17 but a complex mixture of unknown compounds was obtained.
To explain the different reaction outcomes depending on the composition of the cleavage cocktail, we suggested a reaction mechanism (Scheme 2). The protonation of intermediate 3a cleaved from the resin can be followed by intramolecular attack of ketone or carboxylic acid with the hydroxy group as the internal nucleophile. With respect to the higher electrophilicity of the ketone, we presume that the formation of intermediate B over E is preferred. Intermediate B can be further stabilized by the formation of intermediates C and D; however, due to their limited stability, all the reactions in pathway A–D are reversible, which can lead to regeneration of starting material A. The same is true for the formation of E from A. In contrast, the conversion of E to 5a is irreversible, as lactone does not undergo hydrolysis under the conditions used. In the presence of Et3SiH, the preferential formation of B is considered again; however, the subsequently formed intermediates C and D are attacked by triethylsilane as the external nucleophile, which leads to the formation of stable compound 6a. Consequently, the formation of lactone 5a was not observed in this case.
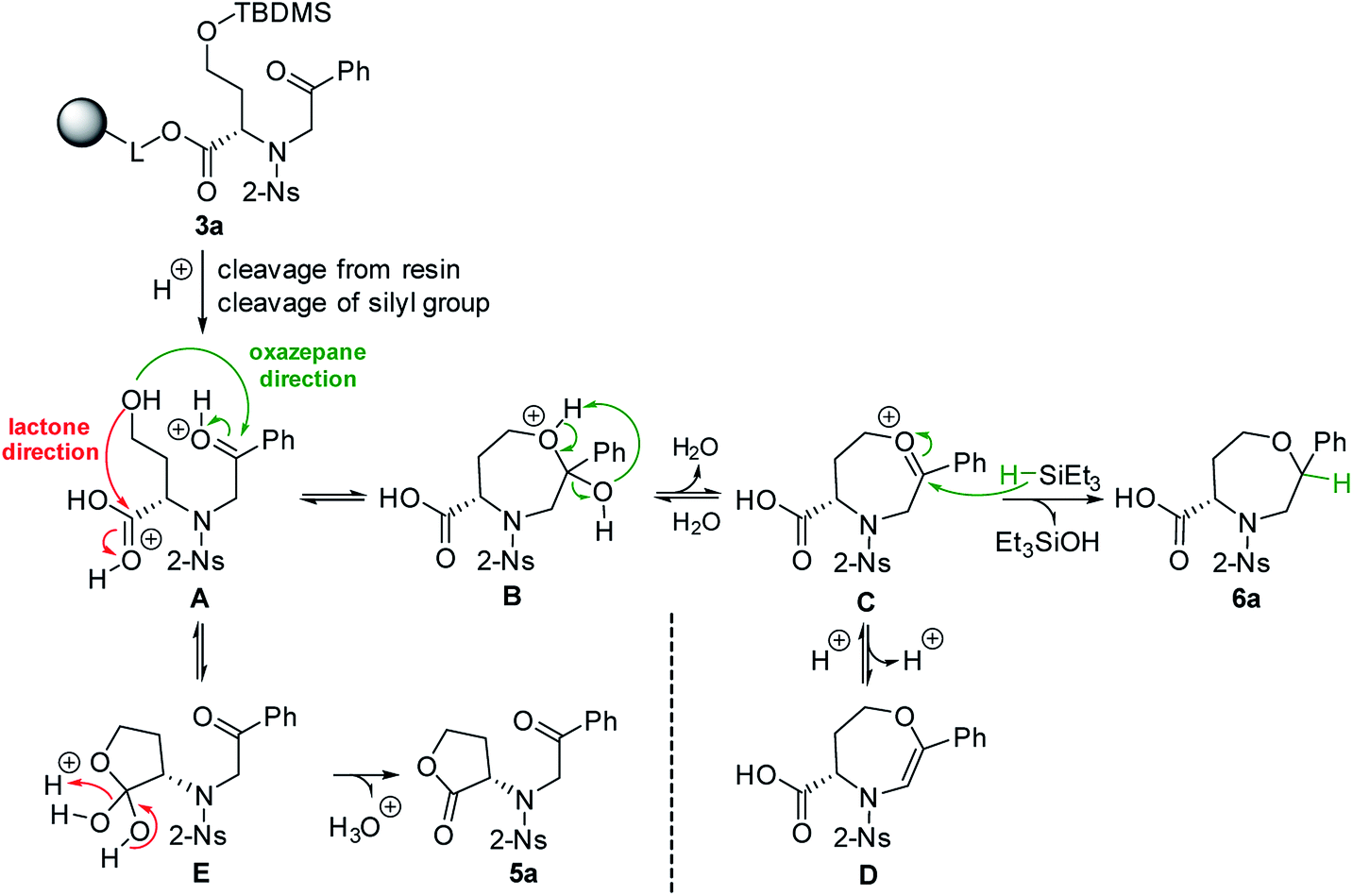 | ||
| Scheme 2 The hypothetical mechanism explaining the different reactivity of intermediate 3a. | ||
After the determination of the reaction outcome, we used different starting materials to evaluate the limitations and scope of the methodology and to reveal the structure–regioselectivity and structure–stereoselectivity relationships. For this purpose, Wang resin was replaced with Rink amide resin, Wang-piperazine resin and BAL resin with immobilized propylamine26 to alter the carboxylic group to carboxamides. Furthermore, variously substituted sulfonyl chlorides and fourteen 2-bromoacetophenones bearing electron-donating or electron-withdrawing groups were selected, including one heterocyclic derivative (thienyl) and another aliphatic derivative (Me; Fig. 5).
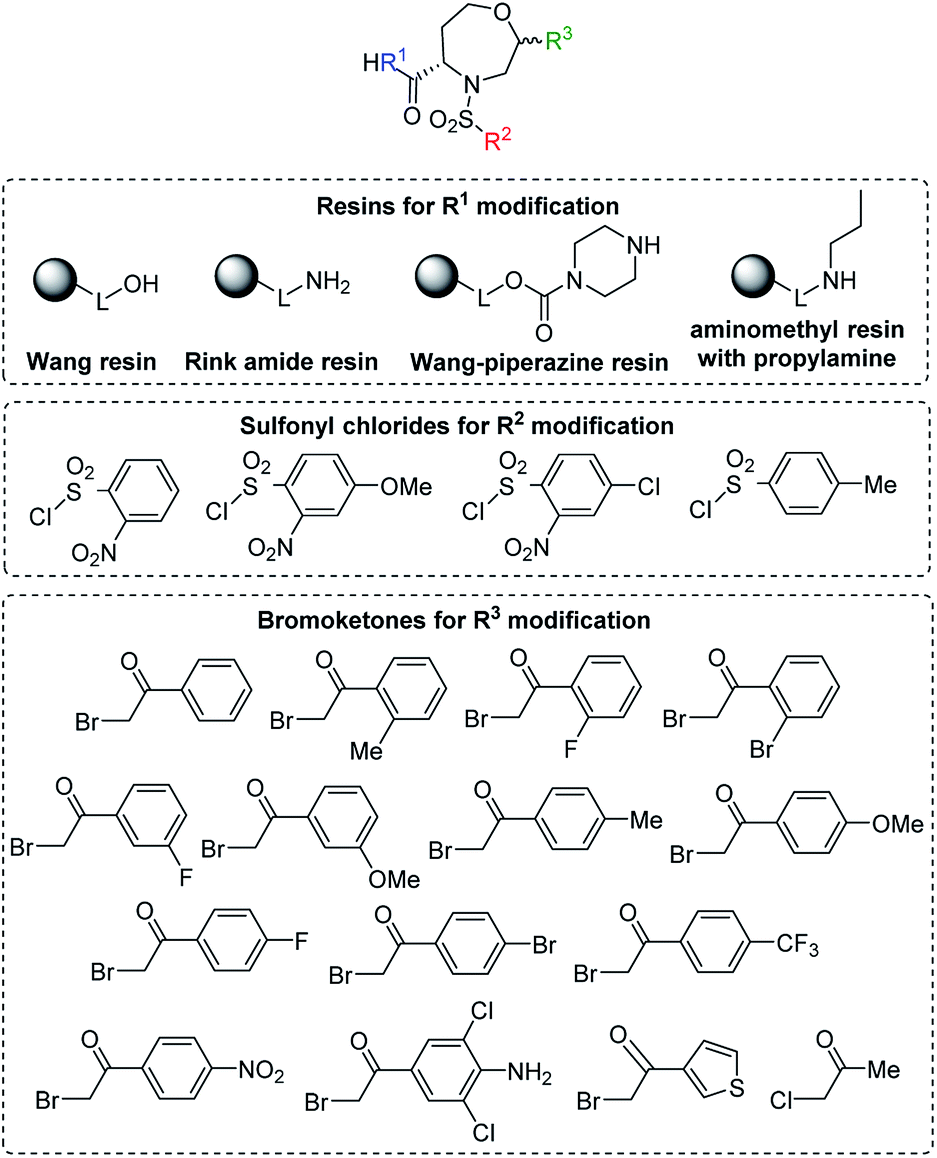 | ||
| Fig. 5 General structure of target compounds and the list of the tested starting synthons (see Table 2 for the list of target compounds). | ||
First, we tested the combination of 2-Ns-Cl and 2-bromoacetophenone with different resins. The use of Rink amide resin (R1 = NH2) required repetition of the alkylation step and yielded the final aniline 7b in 89% crude purity as a mixture of C2 R,S diastereomers in a ratio of 45:55. Its RP-HPLC enabled the separation of both isomers in 9% (C2 R isomer) and 4% overall yields (C2 S isomer, Table 2). In the case of the Wang-piperazine resin (R1 = piperazin-1-yl), the TFA/Et3SiH-mediated cleavage of the corresponding sulfonamide from the resin unexpectedly caused the cleavage of the piperazine moiety, and oxazepane-5-carboxylic acid derivative 7a was obtained as a mixture of C2 R,S isomers in a ratio of 72:28. The major diastereomer was isolated in 10% overall yield. The use of BAL resin-immobilized propylamine failed in the alkylation step stage, which is in accordance with our previous results.26
|
|||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | Crude diastereomeric ratio of C2 R:S stereoisomersa [%] |
Crude combined purityb [%] | Final purity of major purified isomerc,d [%] | Overall yield of major isomere [%] |
| a Ratio of diastereomers prior to RP HPLC purification calculated from HPLC-UV traces at 205–400 nm.b Combined crude purity of diastereomers after the entire reaction sequence calculated from HPLC-UV traces at 205–400 nm.c Calculated from HPLC-UV traces at 205–400 nm after RP-HPLC purification.d Ratio of C2 R,S diastereomers calculated from 1H NMR of the purified product.e Calculated from the 1H NMR spectrum of the purified product.f Product prepared from Wang-piperazine resin. | |||||||
| 5a | — | 2-Ns | Ph | — | 87 | 99c | 74 |
| 6g | O | 4-Me-Ph | Ph | 62:38 |
73 | 97d | 18 |
| 7a | O | H | Ph | 56:44 |
91 | 98d | 34 |
| 7af | O | H | Ph | 72:28 |
76 | 98d | 10 |
| 7b2R | NH | H | Ph | 45:55 |
89 | 100d | 9 |
| 7b2S | 100d | 4 | |||||
| 7e | O | MeO | Ph | 69:31 |
91 | 100d | 21 |
| 7f2RS | O | Cl | Ph | 70:30 |
86 | 78d | 20 |
| 7i2R | O | H | 2-F-Ph | 29:71 |
69 | 100d | 4 |
| 7i2S | 93d | 10 | |||||
| 7k | O | H | 3-F-Ph | 64:36 |
70 | 100d | 10 |
| 7m | O | H | 4-Me-Ph | 77:23 |
90 | 100d | 19 |
| 7o | O | H | 4-F-Ph | 67:33 |
77 | 94d | 10 |
| 7q | O | H | 4-CF3-Ph | 61:39 |
33 | 96d | 8 |
| 7t | O | H | 3-Thienyl | 93:7 |
89 | 98d | 22 |
| 9h | — | H | 2-Me-Ph | — | 50 | 99c | 19 |
| 9j | — | H | 2-Br-Ph | — | 49 | 99c | 13 |
| 10r | O | 2-Ns | — | — | 82 | 99c | 50 |
| 11n | — | 2-Ns | 4-MeO-Ph | — | 77 | 99c | 33 |
After that, various sulfonylating agents (Fig. 5) were tested, starting from intermediate 2 and using 2-bromoacetophenone as the alkylating agent. In the case of sulfonamides 3e and 3g bearing 4-methoxy-2-nitrobenzenesulfonyl group or tosyl group as R2, the alkylation required a longer reaction time (2 and 6 days, respectively) to completion. The TFA/Et3SiH cleavage of tosyl intermediate 3g yielded the desired oxazepane in 73% crude purity as a partially separable mixture of C2 R,S isomers in a ratio of 62:38. Its RP-HPLC purification yielded the major isomer C2 R in 18% overall yield. In the case of 3e and 3f bearing 4-methoxy and 4-chloro-2-nitrobenzene-sulfonyl as R2 substituents, the nitro-oxazepane derivatives 6e and 6f were obtained as inseparable mixtures of C2 R,S diastereoisomers in 81–90% crude purities. Similar to previously reported results,28 the hydrogenation of 6f led to undesired hydrogenolysis of the C–Cl bond (Scheme 3). For this reason, Pd/C was replaced with PtO2, which yielded aniline 7f in 86% crude purity as a mixture of C2 R,S diastereomers in a ratio of 70:30. In this case, the isolation of the major isomer from the diastereomeric mixture was problematic and furnished the product C2 R in only 78% diastereomeric purity. In the case of 6e bearing a 4-methoxy group, hydrogenation using PtO2 suppressed the previously reported demethylation,28 and the final compound 7e was obtained in 91% crude purity as a mixture of C2 R,S diastereomers in a ratio of 69:31. RP-HPLC purification enabled the separation of the major C2 R isomer in 21% overall yield.
 | ||
| Scheme 3 Dehalogenation of oxazepane derivative 6f during hydrogenation. Reagents and conditions: (i) H2, Pd/C, IPA, 24 h, rt; (ii) H2, PtO2, IPA, 24 h, rt. | ||
Finally, we tested various 2-haloketones bearing electron-donating and electron-withdrawing groups in the o-, m- and p-positions, and one heterocyclic and aliphatic derivative was included. All these building blocks were tested in combination with intermediate 2 and 2-Ns-Cl. In the case of 7j and 7p bearing 2-Br-Ph or 4-Br-Ph as R3, PtO2 had to be used to avoid undesired debromination. The preferential formation of oxazepane was observed in each case. In the case of 7p, inseparable C2 R,S diastereomers were isolated in 14% overall yield as diastereomeric anilines in a ratio of 42:58. In the case of 3i bearing 2-F-Ph as R3, the regioselectivity was compromised, and the TFA/Et3SiH reaction yielded 1,4-oxazepane 6i accompanied by lactone 5i (26% according to HPLC). The following hydrogenation of the reaction mixture yielded the corresponding diastereomeric aniline 7i (69% combined crude purity) and amino-lactone 8i, which spontaneously cyclized to benzothiadiazepine 1,1-dioxide 9i (26% according to HPLC-UV-MS analysis) (Scheme 4).33 In this case, RP-HPLC purification enabled the separation of both diastereomers. Intermediates 3h (R3 = 2-Me-Ph) and 3j (R3 = 2-Br-Ph) yielded lactones 5h and 5j as the major products (72% and 64% crude purities). Their hydrogenation yielded the corresponding anilines 8h and 8j, which were cyclized to benzothiadiazepine 1,1-dioxides 9h and 9j.33 Derivatives 9h and 9j were isolated using semipreparative RP-HPLC at 19% overall yield and fully characterized. Interestingly, in contrast to previously reported results,33 compounds 9 were fully stable and did not undergo ring contraction to benzothiadiazine 1,1-dioxides.
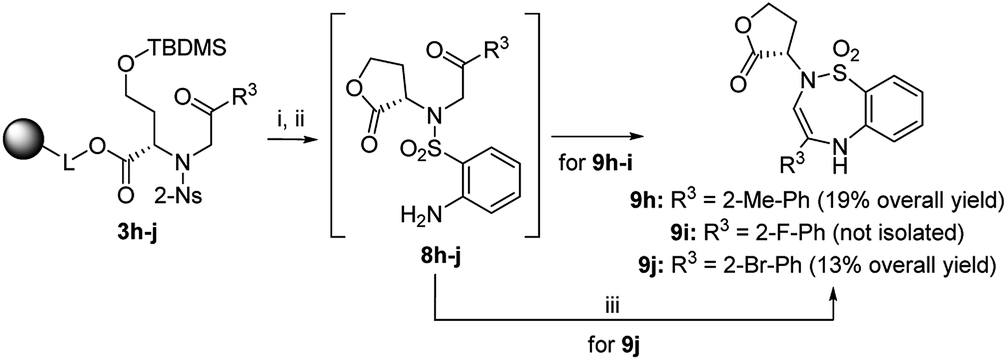 | ||
| Scheme 4 Preferential lactonization and formation of benzothiadiazepine 1,1-dioxide 9. Reagents and conditions: (i) TFA/Et3SiH/CH2Cl2 (10:1:9), 30 min, rt; (ii) H2, 10% Pd/C (for derivatives 9h–i) or PtO2 (for derivative 9j), IPA, 24 h, rt; (iii) 5% TFA/CH2Cl2, 24 h, rt. | ||
In the case of 3k (3-F-Ph as R3), the TFA/Et3SiH reaction and the subsequent hydrogenation yielded diastereomeric aniline 7k in a ratio of 64:36 and 70% combined crude purity, and the major isomer was isolated and fully characterized. On the other hand, intermediate 3l (3-MeO-Ph as R3) yielded a mixture of oxazepane derivative 6l and lactone 11l in a ratio of approximately 1:1, and for this reason, the products were not isolated (Scheme 6). Intermediates 3m, 3o–q, t were synthesized from p-substituted 2-bromoacetophenones (4-Me-Ph, 4-F-Ph, 4-Br-Ph and 4-CF3-Ph as R3) and 3-thienyl derivative yielded oxazepanes 7m, 7o–q, t as separable C2 R,S diastereomers in variable ratios (Table 2) with combined crude purities in a range of 33–91%. In the case of oxazepane derivative 6r (4-NO2-Ph as R3), hydrogenation using either Pd/C or PtO2 led to cleavage of the heterocyclic scaffold, and compound 10r was isolated (Scheme 5) in 82% crude purity and 50% overall yield (Table 2).
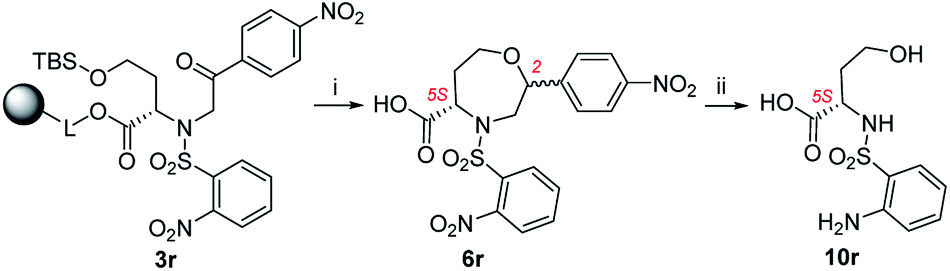 | ||
| Scheme 5 Cleavage of oxazepane derivative 6r during hydrogenation. Reagents and conditions: (i) TFA/Et3SiH/CH2Cl2 (10:1:9), 30 min, rt; (ii) H2, Pd/C, IPA, 24 h, rt. | ||
In the cases of 3n (4-MeO-Ph as R3) and 3s (4-NH2-3,5-diCl-Ph as R3), we observed reversed regioselectivity, and after TFA/Et3SiH cleavage, lactone derivatives 11n and 11s were formed exclusively; however, 11s was accompanied with oxazepane 6s as the minor product (30% crude purity). Due to the excess triethylsilane in the reaction mixture, the abovementioned derivatives were obtained as phenylethyl derivatives 11 (Scheme 6). The exposure to TFA/Et3SiH had to be prolonged to make the reduction quantitative. In combination with the outcome obtained from intermediate 3l (3-MeO-Ph as R3), we can state that electron-donating R3 groups in the m- or p-positions contribute to the formation of lactones, as they probably diminish the reactivity of adjacent ketones toward nucleophilic addition, which suppresses the formation of intermediate C (Scheme 2).
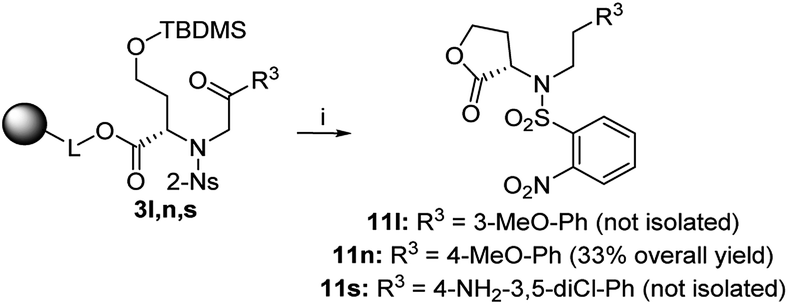 | ||
| Scheme 6 Preferential lactonization followed by ketone reduction to products 11. Reagents and conditions: (i) TFA/Et3SiH/CH2Cl2 (10:1:9), 24 h, rt. | ||
Similar to previously reported results,34 the alkylation step with 2-chloroacetone provided the corresponding oxazepane 6u in only 50% conversion. For this reason, the product was not isolated.
Conclusions
To conclude, we developed a simple methodology to prepare 2-phenyl-substituted-1,4-oxazepane-5-carboxylic acid derivatives. Although the formation of the oxazepane scaffold was nonstereoselective, as in the case of serine-based analogs leading to chiral morpholines,26 the separation and full characterization of major diastereomers was feasible. The developed strategy provided rather minor limitations, e.g., competitive lactonization in the cases of m- and p-electron-donating groups as R3 substituents. Importantly, the developed protocols and corresponding intermediates can be applied for the synthesis of differently fused 1,4-oxazepanes based on previously reported approaches targeted to fused morpholines.27–30Experimental section
General information
Solvents and chemicals were purchased from Sigma-Aldrich (Milwaukee, WI, http://www.sigmaaldrich.com), Acros Organic (Geel, Belgium, http://www.acros.com) and Fluorochem (Hadfield, United Kingdom, http://www.fluorochem.co.uk). Wang resin (100–200 mesh, 1% DVB, 1.4 mmol g−1), Rink resin (100–200 mesh, 1% DVB, 0.4 mmol g−1) and aminomethyl resin (100–200 mesh, 1% DVB, 0.98 mmol g−1) were obtained from AAPPTec (Louisville, KY, http://www.aapptec.com). Solid-phase synthesis was carried out in plastic reaction vessels (syringes, each equipped with a porous disk) using a manually operated synthesizer (Torviq, Niles, MI, http://www.torviq.com). All reactions were carried out at ambient temperature (23 °C) unless stated otherwise. The synthesis of Fmoc-HSe(TBDMS)-OH,31,32 immobilization of Fmoc-HSe(TBDMS)-OH on the resin26 and N-phenacyl sulfonamides 3a–u26 was performed according to these reported protocols. The LC-MS analyses were carried out on UHPLC-MS system consisting of UHPLC chromatograph Acquity with photodiode array detector and single quadrupole mass spectrometer (Waters), using X-Select C18 column with the mobile phase consisting of 10 mM ammonium acetate (AmAc) in H2O and MeCN. The ESI source operated at discharge current of 5 μA, vaporizer temperature of 350 °C and capillary temperature of 200 °C. For the LC/MS analysis, a sample of resin (∼5 mg) was treated with TFA in CH2Cl2, the cleavage cocktail was evaporated under a stream of nitrogen, and cleaved compounds extracted into MeCN/H2O (20% or 50%; 1 mL). Purification was carried out on C18 semipreparative RP-HPLC with the gradient of 10 mM aqueous AmAc and MeCN, flow rate 15 mL min−1 or by normal phase by silica gel chromatography. Residual solvents (H2O and AmAc buffer) were lyophilized by the ScanVac Coolsafe 110-4 working at −110 °C. All 1D and 2D NMR experiments were performed with using ECX500 spectrometer (JEOL RESONANCE, Tokyo, Japan) at magnetic field strength of 11.75 T corresponding to 1H and 13C resonance frequencies of 500.16 MHz and 125.77 MHz at 27 °C. Chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are reported in Hertz (Hz). The signals of MeCN-d3 were set at 1.94 ppm in 1H NMR spectra and at 118.26 ppm in 13C NMR spectra. 15N chemical shifts were referenced to external 90% formamide in DMSO-d6 at 112.00 ppm.35 The assignment of 1H, 13C{1H} and 15N signals was done by APT, 1H–1H COSY, 1H–1H NOESY, 1H–13C HMQC, 1H–13C HMBC and 1H–15N HMBC. Abbreviations in NMR spectra: br. s – broad singlet, br. d – broad doublet, s – singlet, d – doublet, dd – doublet of doublets, ddd – doublet of doublets of doublets, dddd – doublet of doublets of doublets of doublets, m – multiplet. Acetate salt (residual agent from the semipreparative HPLC purification) exhibited a singlet at 2.09–2.53 ppm in 1H NMR spectra and two resonances at 41.00 ppm and 171.00 ppm in 13C NMR spectra. HRMS analysis was performed using LC-MS (Dionex Ultimate 3000, Thermo Fischer Scientific, MA, USA) with Exactive Plus Orbitrap high-resolution mass spectrometer (Thermo Exactive Plus, Thermo Fischer Scientific, MA, USA) operating at positive or negative full scan mode (120000 FWMH) in the range of 100–1000 m/z with electrospray ionization working at 150 °C and the source voltage of 3.6 kV. Chromatographic separation was performed on column Phenomenex Gemini (C18, 50 × 2 mm, 3 μm particle) with isocratic elution and mobile phase (MP) containing MeCN/10 mM AmAc (80:20; v/v). The samples were dissolved in the initial MP. The acquired data were internally calibrated with phthalate as a contaminant in MeOH (m/z 297.15909). IR spectra were measured by DRIFT (Diffuse Reflectance Infrared Fourier Transform) on a Thermo Nicolet AVATAR 370 FTIR spectrometer. Absorbance peaks (wavenumbers) are reported in reciprocal centimeters (cm−1) and transmittances (T) are reported in percentages (%). Specific optical rotations were measured on Automatic Compact Polarimeter POL-1/2 (ATAGO, Japan) with LED Light Source and 589 nm interference filter at 24 °C. The length of cuvette was 2 cm and specific optical rotations are reported as follows: [α]TD, concentration (g mL−1) and solvent.
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3101, 3953, 2930, 2918, 1780, 1595, 1544, 1378, 1340, 1301, 1206, 1157, 1080, 1063, 760, 742, 700 cm−1. [α]25D = −21.0° (c = 0.00067 g mL−1 in MeCN).:1:9) for 30 min (except for 3n and 3s) or 24 h (for derivatives 3n and 3s) at room temperature. Then the resin was washed three times with fresh cleavage cocktail (5 mL) and the combined fractions were evaporated using a stream of nitrogen and lyophilized overnight. The crude products were purified using RP-HPLC.:S diastereomers in a ratio of 62:38, the isolation of major C2 R epimer was performed. White amorphous solid (16.1 mg, 0.043 mmol, 18%). HPLC purity 97%. NMR: mixture with 3% of C2 S isomer. 1H NMR (500 MHz, MeCN-d3): δ = 7.75 (d, J = 8.3 Hz, 2H), 7.23–7.37 (m, 7H), 4.81 (br. s, 5H, residual water), 4.55 (dd, J = 10.6, 7.1 Hz, 1H), 4.32 (d, J = 9.1 Hz, 1H), 4.02 (ddd, J = 12.7, 6.3, 1.5 Hz, 1H), 3.78 (ddd, J = 15.9, 1.0, 1.0 Hz, 1H), 3.47–3.55 (m, 1H), 3.49 (ddd, J = 12.7, 9.5, 1.2 Hz, 1H), 2.47–2.54 (m, 1H), 2.39 (s, 3H), 2.13 (dddd, J = 15.9, 10.6, 9.1, 1.6 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 175.7, 144.8, 141.0, 138.9, 130.7, 129.3, 128.7, 128.2, 127.0, 83.2, 67.7, 59.5, 54.4, 35.7, 21.5. HRMS (ESI, pos.): m/z calcd for C19H25N2O5S [M + H]+ 393.1479 found 393.1482. IR (DRIFT): = 3062, 3029, 2950, 2862, 11917, 1723, 1596, 1450, 1334, 1306, 1171, 1152, 761, 746, 699 cm−1. [α]24D = −36.2° (c = 0.00062 g mL−1, MeCN). = 2932, 2836, 1783, 1542, 1512, 1367, 1342, 1250, 1241, 1159, 785, 752, 708 cm−1. [α]24D = −76.9° (c = 0.00029 g mL−1, MeCN).
= 3101, 3953, 2930, 2918, 1780, 1595, 1544, 1378, 1340, 1301, 1206, 1157, 1080, 1063, 760, 742, 700 cm−1. [α]25D = −21.0° (c = 0.00067 g mL−1 in MeCN).:1:9) for 30 min (except for 3n and 3s) or 24 h (for derivatives 3n and 3s) at room temperature. Then the resin was washed three times with fresh cleavage cocktail (5 mL) and the combined fractions were evaporated using a stream of nitrogen and lyophilized overnight. The crude products were purified using RP-HPLC.:S diastereomers in a ratio of 62:38, the isolation of major C2 R epimer was performed. White amorphous solid (16.1 mg, 0.043 mmol, 18%). HPLC purity 97%. NMR: mixture with 3% of C2 S isomer. 1H NMR (500 MHz, MeCN-d3): δ = 7.75 (d, J = 8.3 Hz, 2H), 7.23–7.37 (m, 7H), 4.81 (br. s, 5H, residual water), 4.55 (dd, J = 10.6, 7.1 Hz, 1H), 4.32 (d, J = 9.1 Hz, 1H), 4.02 (ddd, J = 12.7, 6.3, 1.5 Hz, 1H), 3.78 (ddd, J = 15.9, 1.0, 1.0 Hz, 1H), 3.47–3.55 (m, 1H), 3.49 (ddd, J = 12.7, 9.5, 1.2 Hz, 1H), 2.47–2.54 (m, 1H), 2.39 (s, 3H), 2.13 (dddd, J = 15.9, 10.6, 9.1, 1.6 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 175.7, 144.8, 141.0, 138.9, 130.7, 129.3, 128.7, 128.2, 127.0, 83.2, 67.7, 59.5, 54.4, 35.7, 21.5. HRMS (ESI, pos.): m/z calcd for C19H25N2O5S [M + H]+ 393.1479 found 393.1482. IR (DRIFT): = 3062, 3029, 2950, 2862, 11917, 1723, 1596, 1450, 1334, 1306, 1171, 1152, 761, 746, 699 cm−1. [α]24D = −36.2° (c = 0.00062 g mL−1, MeCN). = 2932, 2836, 1783, 1542, 1512, 1367, 1342, 1250, 1241, 1159, 785, 752, 708 cm−1. [α]24D = −76.9° (c = 0.00029 g mL−1, MeCN).Prepared from Wang resin. Cleaved from 785 mg of resin 3a (0.140 mmol g−1, 0.110 mmol of substrate). The separable mixture of C2 R
:S diastereomers in a ratio of 56:44, the isolation of the major C2 R epimer was performed. White amorphous solid (14.2 mg, 0.038 mmol, 34%). HPLC purity 97%. NMR: mixture with 2% of C2 S isomer. 1H NMR (500 MHz, MeCN-d3): δ = 7.66 (dd, J = 8.1, 1.5 Hz, 1H, HC19), 7.34 (ddd, J = 8.3, 7.2, 1.5 Hz, 1H, HC17), 7.23–7.30 (m, 3H, HC10–12), 7.15 (m, 2H, HC9,13), 6.84 (dd, J = 8.3, 1.0 Hz, 1H, HC16), 6.75 (ddd, J = 8.1, 7.2, 1.0 Hz, 1H, HC18), 4.66 (dd, J = 10.4, 7.1 Hz, 1H, HC5), 4.16 (d, J = 9.5 Hz, 1H, HC2), 4.02 (ddd, J = 12.7, 6.3, 1.1 Hz, 1H, HbC7), 3.70 (d, J = 16.1 Hz, 1H, HaC3), 3.65 (dd, J = 12.7, 8.9 Hz, 1H, HaC7), 3.50 (dd, J = 16.1, 9.5 Hz, 1H, HbC3), 2.54 (ddd, J = 15.4, 7.1, 6.3 Hz, 1H, HaC6), 2.16 (dddd, J = 15.4, 10.4, 8.9, 1.1 Hz, 1H, HbC6). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 174.8 (C21), 147.5 (C15), 141.0 (C8), 135.4 (C17), 131.0 (C19), 129.3 (C10,12), 128.7 (C11), 127.1 (C9,13), 121.9 (C14), 118.6 (C16), 117.7 (C18), 82.7 (C2), 67.5 (C7), 59.1 (C5), 54.1 (C3), 35.6 (C6). 15N NMR (51 MHz, MeCN-d3): δ = 99.9 (N4), 63.0 (N20). HRMS (ESI, pos.): m/z calcd for C18H21N2O5S [M + H]+ 377.1166 found 377.1167. IR (DRIFT): = 2950, 2864, 1719, 1617, 1483, 1452, 1317, 1169, 1140, 751, 698 cm−1. [α]24D = −64.1° (c = 0.00020 g mL−1, MeCN).
Prepared from Wang-piperazine resin. Cleaved from 669 mg of resin 3a (0.601 mmol g−1, 0.402 mmol of substrate). The separable mixture of C2 R
:S diastereomers in a ratio of 72:28, the isolation only of the major C2 R epimer was performed. White amorphous solid (14.5 mg, 0.039 mmol, 10%). HPLC purity 99%. NMR: mixture with 2% of C2 S isomer. The analytical data (1H and 13C NMR, HRMS, IR (DRIFT) and [α]24D) corresponded with 7a prepared from Wang resin.
:S diastereomers in a ratio of 45:55, the isolation of C2 R epimer was performed. White amorphous solid (7.2 mg, 0.019 mmol, 9%). HPLC purity 99%. 1H NMR (500 MHz, MeCN-d3): δ = 7.70 (dd, J = 8.2, 1.5 Hz, 1H), 7.39 (ddd, J = 8.4, 7.1, 1.5 Hz, 1H), 7.26–7.33 (m, 3H), 7.20–7.21 (m, 2H), 6.89 (dd, J = 8.4, 1.1 Hz, 1H), 6.80 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 6.34 (br. s, 1H), 5.68 (br. s, 1H), 5.38 (br. s, 2H), 4.49 (ddd, J = 10.8, 7.1, 1.0 Hz, 1H), 4.28 (dd, J = 9.3, 1.2 Hz, 1H), 4.05 (ddd, J = 12.9, 6.3, 1.5 Hz, 1H), 3.82 (ddd, J = 16.2, 1.2 Hz, 1.2, 1H), 3.67 (ddd, J = 12.9, 9.3, 1.0 Hz, 1H), 3.53 (dd, J = 16.2, 9.6 Hz, 1H), 2.46 (dddd, J = 15.7, 7.1, 6.3, 1.0 Hz, 1H), 2.21 (dddd, J = 15.7, 10.8, 9.3, 1.5 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 174.2, 147.4, 140.9, 135.7, 131.1, 129.3, 128.7, 127.1, 121.7, 118.8, 118.1, 82.7, 67.4, 59.3, 54.7, 35.6. HRMS (ESI, pos.): m/z calcd for C18H22N3O4S [M + H]+ 376.1326 found 376.1326. IR (DRIFT): = 3447, 3352, 3205, 3062, 3030, 2952, 2864, 1675, 1616, 1483, 1452, 1318, 1301, 1140, 748, 698 cm−1. [α]24D = −62.5° (c = 0.00024 g mL−1, MeCN).:S diastereomers in a ratio of 45:55, the isolation of C2 S epimer was performed. White amorphous solid (3.2 mg, 0.009 mmol, 4%). HPLC purity 99%. 1H NMR (500 MHz, MeCN-d3): δ = 7.58 (dd, J = 8.1, 1.6 Hz, 1H, HC19), 7.35 (ddd, J = 8.5, 7.2, 1.6 Hz, 1H, HC17), 7.28–7.32 (m, 2H, HC10,12), 7.24–7.28 (m, 1H, HC11), 7.17–7.20 (m, 2H, HC9,13), 6.85 (dd, J = 8.5, 1.1 Hz, 1H, HC16), 6.75 (br. s, 1H, HbN22), 6.70 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H, HC18), 5.99 (br. s, 1H, HaN22), 5.62 (br. s, 2H, HN20), 4.64 (dd, J = 8.7, 1.6 Hz, 1H, HC2), 4.59 (dd, J = 4.5, 4.5 Hz, 1H, HC5), 3.94 (ddd, J = 12.8, 4.5, 3.0 Hz, 1H, HbC7), 3.78 (ddd, J = 12.8, 11.0, 1.6 Hz, 1H, HaC7), 3.63 (dd, J = 14.2, 1.6 Hz, 1H, HaC3), 3.43 (dd, J = 14.2, 8.7 Hz, 1H, HbC3), 2.30 (dddd, J = 15.8, 4.5, 4.5, 1.6 Hz, 1H, HaC6), 2.10 (dddd, J = 15.8, 11.0, 4.5, 3.0 Hz, 1H, overlap with water, HbC6). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 174.4 (C21), 148.0 (C15), 141.9 (C8), 135.7 (C17), 131.3 (C19), 129.3 (C10,12), 128.6 (C11), 126.6 (C9,13), 119.1 (C14), 118.7 (C16), 117.5 (C18), 81.4 (C2), 67.0 (C7), 59.4 (C5), 56.8 (C3), 33.0 (C6). HRMS (ESI, pos.): m/z calcd for C18H22N3O4S [M + H]+ 376.1326 found 376.1325. IR (DRIFT): = 3448, 3350, 3216, 3061, 3029, 2969, 2954, 2920, 1676, 1616, 1483, 1452, 1318, 1303, 1142, 752, 699 cm−1. [α]24D = +271.4° (c = 0.00007 g mL−1, MeCN).:S diastereomers in a ratio of 69:31, the isolation of major C2 R epimer was performed. White amorphous solid (20.1 mg, 0.050 mmol, 21%). HPLC purity 99%. 1H NMR (500 MHz, MeCN-d3): δ = 7.58 (d, J = 8.9 Hz, 1H), 7.22–7.30 (m, 3H), 7.15–7.16 (m, 2H), 6.31–6.35 (m, 2H), 5.02 (br. s, 2H), 4.61 (dd, J = 10.9, 7.1 Hz, 1H), 4.20 (d, J = 9.5 Hz, 1H), 4.02 (ddd, J = 12.7, 6.4, 1.2 Hz, 1H), 3.76 (s, 3H), 3.58–3.39 (m, 2H), 3.50 (dd, J = 16.1, 9.5 Hz, 1H), 2.46–2.54 (m, 1H), 2.16 (dddd, J = 15.4, 10.9, 9.2, 1.2 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 175.8, 165.5, 149.5, 141.1, 133.1, 129.3, 128.6, 127.0, 114.3, 105.4, 101.5, 82.7, 67.6, 59.5, 56.1, 54.1, 35.7. HRMS (ESI, pos.): m/z calcd for C19H23N2O6S [M + H]+ 407.1271 found 407.1269. IR (DRIFT): = 3469, 3370, 3228, 3029, 2939, 2864, 1736, 1725, 1605, 1450, 1303, 1133, 752, 699, 689 cm−1. [α]24D = −16.8° (c = 0.00030 g mL−1, MeCN).:S diastereomers in a ratio of 70:30, the interpretation of major C2 R epimer was performed. White amorphous solid (21.0 mg, 0.051 mmol, 20%). HPLC purity 78%. NMR: mixture with 22% of C2 S isomer. 1H NMR (500 MHz, MeCN-d3): δ = 7.71 (d, J = 8.5 Hz, 1H), 7.39 (s, 1H), 7.29–7.30 (m, 2H), 7.20–7.26 (m, 3H), 6.90 (d, J = 8.3 Hz, 1H), 6.01 (br. s, 2H), 4.66 (dd, J = 10.3, 7.2 Hz, 1H), 4.41 (d, J = 9.6 Hz, 1H), 4.06 (ddd, J = 12.6, 6.3, 1.5 Hz, 1H), 3.71 (ddd, J = 12.6, 8.9, 1.0 Hz, 1H), 3.65 (d, J = 16.0 Hz, 1H), 3.46 (dd, J = 16.0, 9.6 Hz, 1H), 2.51–2.60 (m, 1H), 2.19 (dddd, J = 15.6, 10.5, 8.9, 1.5 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 175.7, 151.0, 148.6, 141.0, 132.6, 129.3, 128.7, 127.0, 121.4, 119.8, 114.7, 82.9, 67.6, 59.4, 54.5, 35.7. HRMS (ESI, pos.): m/z calcd for C18H20ClN2O5S [M + H]+ 411.0776 found 411.0777. IR (DRIFT): = 3303, 3064, 3029, 2970, 2950, 2868, 1736, 1727, 1587, 1450, 1317, 1292, 1163, 758, 699 cm−1. [α]24D = −17.3° (c = 0.00098 g mL−1, MeCN).:S diastereomers in a ratio of 29:71, the isolation of major C2 R epimer was performed. White amorphous solid (1.6 mg, 0.004 mmol, 4%). HPLC purity 94%. 1H NMR (500 MHz, MeCN-d3): δ = 7.64 (dd, J = 8.2, 1.5 Hz, 1H), 7.28–7.37 (m, 3H), 7.14 (ddd, J = 8.6, 7.5, 1.1 Hz, 1H), 7.06 (ddd, J = 10.6, 8.3, 1.1 Hz, 1H), 6.82 (dd, J = 8.2, 1.1 Hz, 1H), 6.72 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 4.69 (dd, J = 10.1, 7.2 Hz, 1H), 4.64 (dd, J = 9.7, 0.9 Hz, 1H), 4.03 (ddd, J = 12.7, 6.4, 1.9 Hz, 1H), 3.85 (ddd, J = 15.8, 0.9, 0.9 Hz, 1H), 3.68 (ddd, J = 12.7, 8.9, 1.4 Hz, 1H), 3.55 (ddd, J = 15.8, 9.7 Hz, 1H), 2.54 (dddd, J = 15.6, 7.2, 6.4, 1.4 Hz, 1H, overlap with water), 2.19 (dddd, J = 15.6, 10.1, 8.9, 1.9 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 175.5, 160.4 (d, J = 243.2 Hz), 147.4, 135.3, 130.9, 130.6 (d, J = 9.1 Hz), 129.2, 129.2, 127.7 (d, J = 13.1 Hz), 125.3 (d, J = 3.0 Hz), 121.8, 117.7, 116.8 (d, J = 26.2 Hz), 76.9, 67.4, 58.4, 52.6, 35.4. HRMS (ESI, pos.): m/z calcd for C18H20FN2O5S [M + H]+ 395.1071 found 395.1072. IR (DRIFT): = 3462, 3372, 3214, 2926, 2867, 1724, 1617, 1567, 1483, 1453, 1317, 1142, 753 cm−1. [α]24D = −68.2° (c = 0.00011 g mL−1, MeCN).:S diastereomers in a ratio of 29:71, the isolation of C2 R epimer was performed. White amorphous solid (4.1 mg, 0.010 mmol, 10%). HPLC purity 93%. NMR: mixture with 7% of C2 R isomer. 1H NMR (500 MHz, MeCN-d3): δ = 7.57 (dd, J = 8.0, 1.5 Hz, 1H), 7.40 (ddd, J = 7.5, 7.5, 1.7 Hz, 1H), 7.26–7.31 (m, 2H), 7.14 (ddd, J = 8.6, 7.5, 1.1 Hz, 1H), 7.02 (ddd, J = 10.6, 8.2, 1.1 Hz, 1H), 6.79 (dd, J = 8.2, 1.1 Hz, 1H), 6.65 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 4.93 (dd, J = 8.9, 2.0 Hz, 1H), 4.78 (dd, J = 4.8, 4.8 Hz, 1H), 4.02 (ddd, J = 13.0, 3.8, 3.8 Hz, 1H), 3.78 (ddd, J = 13.0, 11.2, 2.0 Hz, 1H), 3.61 (dd, J = 13.8, 2.0 Hz, 1H), 3.43 (dd, J = 13.8, 8.9 Hz, 1H), 2.37 (dddd, J = 15.6, 4.8, 3.8, 2.0 Hz, 1H), 2.24 (dddd, J = 15.6, 11.2, 4.8, 3.8 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 174.3, 159.9 (d, J = 244.2 Hz), 148.0, 135.3, 131.1, 130.5 (d, J = 8.1 Hz), 128.8, 128.7, 128.6 (d, J = 4.0 Hz), 125.4 (d, J = 3.0 Hz), 119.7, 117.1, 116.0 (d, J = 21.2 Hz), 76.0, 68.0, 59.9, 54.7, 33.8. HRMS (ESI, pos.): m/z calcd for C18H20FN2O5S [M + H]+ 395.1071 found 395.1072. IR (DRIFT): = 3463, 3370, 2920, 2863, 1719, 1618, 1566, 1484, 1453, 1318, 1141, 753 cm−1. [α]24D = −93.0° (c = 0.00050 g mL−1, MeCN).:S diastereomers in a ratio of 64:36, the isolation of major C2 R epimer was performed. White amorphous solid (8.5 mg, 0.022 mmol, 10%). HPLC purity 99%. 1H NMR (500 MHz, MeCN-d3): δ = 7.68 (dd, J = 8.1, 1.5 Hz, 1H), 7.34 (ddd, J = 8.4, 7.1, 1.5 Hz, 1H), 7.29 (ddd, J = 7.9, 7.9, 6.0 Hz, 1H), 6.87–6.99 (m, 3H), 6.85 (dd, J = 8.3, 1.0 Hz, 1H), 6.75 (ddd, J = 8.1, 7.1, 1.0 Hz, 1H), 4.62 (dd, J = 10.8, 7.2 Hz, 1H), 4.19 (d, J = 9.2 Hz, 1H), 4.02 (ddd, J = 12.6, 6.3, 1.1 Hz, 1H), 3.70 (ddd, J = 15.8, 1.1, 1.1 Hz, 1H), 3.65 (ddd, J = 12.6, 9.2, 0.7 Hz, 1H), 3.50 (dd, J = 16.2, 9.6 Hz, 1H), 2.45–2.56 (m, 1H), 2.15 (dddd, J = 15.8, 10.8, 9.2, 1.2 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 175.4, 163.6 (d, J = 243.2 Hz), 147.5, 143.7 (d, J = 7.1 Hz), 135.4, 131.1 (d, J = 9.1 Hz), 131.0, 123.0 (d, J = 2.0 Hz), 121.8, 118.6, 117.6, 115.3 (d, J = 21.2 Hz), 113.8 (d, J = 23.2 Hz), 82.0, 67.6, 59.4, 54.0, 36.0. HRMS (ESI, pos.): m/z calcd for C18H20FN2O5S [M + H]+ 395.1071 found 395.1073. IR (DRIFT): = 3469, 3371, 2932, 2867, 1719, 1616, 1566, 1482, 1450, 1318, 1137, 89, 785, 748, 688 cm−1. [α]24D = −25.7° (c = 0.00070 g mL−1, MeCN).:S diastereomers in a ratio of 77:23, the isolation of major C2 R epimer was performed. White amorphous solid (8.0 mg, 0.021 mmol, 19%). HPLC purity 99%. 1H NMR (500 MHz, MeCN-d3): δ = 7.66 (dd, J = 8.1, 1.5 Hz, 1H), 7.34 (ddd, J = 8.4, 7.1, 1.5 Hz, 1H), 7.11 (d, J = 7.9 Hz, 2H), 7.04 (d, J = 7.9 Hz, 2H), 6.85 (dd, J = 8.2, 1.0 Hz, 1H), 6.75 (ddd, J = 8.1, 7.1, 1.0 Hz, 1H), 4.67 (dd, J = 10.8, 7.1 Hz, 1H), 4.14 (d, J = 9.0 Hz, 1H), 4.01 (ddd, J = 12.7, 6.3, 1.5 Hz, 1H), 3.69 (ddd, J = 16.2, 1.0, 1.0 Hz, 1H), 3.64 (ddd, J = 12.7, 9.0, 1.0 Hz, 1H), 3.49 (dd, J = 16.2, 9.6 Hz, 1H), 2.46–2.56 (m, 1H), 2.29 (s, 3H), 2.16 (dddd, J = 15.6, 10.8, 9.0, 1.5 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 174.6, 147.5, 138.4, 138.1, 135.4, 131.0, 129.9, 127.0, 122.0, 118.6, 117.6, 82.6, 67.4, 59.1, 54.1, 35.6, 21.1. HRMS (ESI, pos.): m/z calcd for C19H23N2O5S [M + H]+ 391.1322 found 391.1324. IR (DRIFT): = 3460, 3368, 3211, 2950, 2865, 1719, 1617, 1599, 1483, 1452, 1317, 1169, 814, 748, 690 cm−1. [α]24D = +34.8° (c = 0.00023 g mL−1, MeCN).:S diastereomers in a ratio of 67:33, the isolation of major C2 R epimer was performed. Pale yellow amorphous solid (6.3 mg, 0.016 mmol, 10%). HPLC purity 99%. NMR: mixture with 6% of C2 S isomer 1H NMR (500 MHz, MeCN-d3): δ = 7.66 (dd, J = 8.1, 1.6 Hz, 1H), 7.33 (ddd, J = 8.4, 7.2, 1.6 Hz, 1H), 7.14 (br. d, J = 8.6 Hz, 2H), 6.99 (br. d, J = 8.9 Hz, 2H), 6.84 (dd, J = 8.2, 1.0 Hz, 1H), 6.74 (ddd, J = 8.1, 7.1, 1.0 Hz, 1H), 4.58 (dd, J = 10.8, 7.0 Hz, 1H), 4.15 (d, J = 9.3 Hz, 1H), 3.99 (ddd, J = 12.6, 6.3, 1.3 Hz, 1H), 3.60–3.67 (m, 2H), 3.50 (dd, J = 16.2, 9.6 Hz, 1H), 2.44–2.55 (m, 1H), 2.14 (dddd, J = 15.6, 10.8, 9.2, 1.3 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 176.2, 163.0 (d, J = 244.2 Hz), 147.5, 137.2 (d, J = 3.0 Hz), 135.4, 131.0, 129.0 (d, J = 9.1 Hz), 121.9, 118.6, 117.5, 115.9 (d, J = 21.2 Hz), 81.9, 67.7, 59.9, 54.0, 35.7. HRMS (ESI, pos.): m/z calcd for C18H20FN2O5S [M + H]+ 395.1071 found 395.1073. IR (DRIFT): = 3460, 3368, 3210, 2929, 2863, 1717, 1601, 1566, 1509, 1483, 1453, 1317, 1300, 1140, 834, 802, 748, 689 cm−1. [α]24D = +35.3° (c = 0.00017 g mL−1, MeCN).:S diastereomers in a ratio of 61:39, the isolation of major C2 R epimer was performed. Yellow amorphous solid (8.3 mg, 0.017 mmol, 8%). HPLC purity 97%. NMR: mixture with 4% of C2 S isomer. 1H NMR (500 MHz, MeCN-d3): δ = 7.77 (dd, J = 7.9, 1.3 Hz, 1H), 7.63 (br. d, J = 8.2 Hz, 2H), 7.50–7.58 (m, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.39 (br. d, J = 8.2 Hz, 2H), 6.94–7.02 (m, 1H), 4.71 (dd, J = 9.1, 7.2 Hz, 1H), 4.41 (d, J = 9.4 Hz, 1H), 4.08 (ddd, J = 12.6, 6.1, 0.9 Hz, 1H), 3.76 (dd, J = 16.2 Hz, 1H), 3.74 (dd, J = 12.6, 9.1 Hz, 1H), 3.47 (dd, J = 16.0, 9.4 Hz, 1H), 2.51–2.63 (m, 1H), 2.13–2.25 (m, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 174.3, 149.9, 145.2, 135.6, 130.7, 130.1 (q, J = 32.1 Hz), 127.7, 126.2 (q, J = 3.9 Hz), 125.4 (q, J = 272.0 Hz), 123.2, 120.4, 115.3, 82.3, 67.5, 58.9, 54.1, 41.2, 35.5. HRMS (ESI, pos.): m/z calcd for C19H20F3N2O5S [M + H]+ 445.1040 found 445.1042. IR (DRIFT): = 3320, 2937, 2871, 1731, 1597, 1459, 1323, 1164, 1149, 835, 758 cm−1. [α]24D = −73.1° (c = 0.00039 g mL−1, MeCN).:S diastereomers in a ratio of 93:7, the isolation of major C2 R epimer was performed. White amorphous solid (10.8 mg, 0.028 mmol, 22%). HPLC purity 98%. NMR: mixture with 2% of C2 S isomer. 1H NMR (500 MHz, MeCN-d3): δ = 7.76 (dd, J = 7.8, 0.9 Hz, 1H), 7.52 (t, J = 7.8 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.32 (dd, J = 5.0, 3.0 Hz, 1H), 7.15–7.18 (m, 1H), 6.94 (dd, J = 5.0, 1.3 Hz, 2H), 4.64 (dd, J = 10.0, 7.3 Hz, 1H), 4.46 (d, J = 9.5 Hz, 1H), 4.02 (ddd, J = 12.7, 6.4, 1.5 Hz, 1H), 3.74 (d, J = 16.0 Hz, 1H), 3.69 (ddd, J = 12.7, 9.0, 1.0 Hz, 1H), 3.49 (dd, J = 16.0, 9.5 Hz, 1H), 2.47–2.56 (m, 1H), 2.15 (dddd, J = 15.6, 10.4, 9.0, 1.5 Hz, 1H). 13C{1H} NMR (126 MHz, MeCN-d3): δ = 175.2, 150.0, 142.1, 135.5, 130.7, 127.0, 126.9, 123.0, 122.3, 120.2, 115.2, 79.3, 67.5, 59.3, 53.5, 35.7. HRMS (ESI, neg.): m/z calcd for C16H17N2O5S2 [M − H]− 381.0573 found 381.0581. IR (DRIFT): = 3297, 3102, 2950, 2864, 1720, 1596, 1458, 1317, 1293, 1216, 1147, 1030, 955, 758 cm−1. [α]24D = −221.9° (c = 0.00029 g mL−1, MeCN). = 3345, 3066, 3016, 2920, 1775, 1663, 1474, 1335, 1159, 750 cm−1. [α]24D = −42.1° (c = 0.00063 g mL−1, MeCN). = 3476, 3375, 3261, 1770, 1619, 1482, 1453, 1318, 754 cm−1. [α]24D = +5.8° (c = 0.00119 g mL−1, MeCN). = 3349, 2921, 2851, 1776, 1662, 1594, 1475, 1338, 1162, 751 cm−1. [α]24D = −51.7° (c = 0.00029 g mL−1, MeCN).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an internal grant from Palacký University (IGA_PrF_2020_012).Notes and references
- Y. Du, D. P. Garden, L. Wang, B. S. Zhorov and K. Dong, J. Biol. Chem., 2011, 286, 13151–13160 CrossRef CAS.
- K. Ishimoto, K. Yamaguchi, A. Nishimoto, M. Murabayashi and T. Ikemoto, Org. Process Res. Dev., 2017, 21, 2001–2011 CrossRef CAS.
- T. Yukawa, Y. Nakada, N. Sakauchi, T. Kamei, M. Yamada, Y. Ohba, I. Fujimori, H. Ueno, M. Takiguchi, M. Kuno, I. Kamo, H. Nakagawa, Y. Fujioka, T. Igari, Y. Ishichi and T. Tsukamoto, Bioorg. Med. Chem., 2016, 24, 3716–3726 CrossRef CAS.
- K. Audouze, E. Ø. Nielsen and D. Peters, J. Med. Chem., 2004, 47, 3089–3104 CrossRef CAS.
- M. H. Serrano-Wu, D. R. S. Laurent, Y. Chen, S. Huang, K.-R. Lam, J. A. Matson, C. E. Mazzucco, T. M. Stickle, T. P. Tully, H. S. Wong, D. M. Vyas and B. N. Balasubramanian, Bioorg. Med. Chem. Lett., 2002, 12, 2757–2760 CrossRef CAS.
- S. Kaneko, M. Arai, T. Uchida, T. Harasaki, T. Fukuoka and T. Konosu, Bioorg. Med. Chem. Lett., 2002, 12, 1705–1708 CrossRef CAS.
- G. Sharma, J. Y. Park and M. S. Park, Arch. Pharmacal Res, 2008, 31, 838 CrossRef CAS.
- S. Kaneko, M. Arai, T. Uchida, T. Harasaki, T. Fukuoka and T. Konosu, Bioorg. Med. Chem. Lett., 2002, 12, 1705–1708 CrossRef CAS.
- Insmed Incorporated, USA, PCT Int. Appl., 2020, 82 Search PubMed.
- Insmed Incorporated, USA, PCT Int. Appl., 2020, 51 Search PubMed.
- AstraZeneca AB, Swed., PCT Int. Appl., 2019, 106 Search PubMed.
- Insmed Incorporated, USA and AstraZeneca AB, PCT Int. Appl., 2018, 61 Search PubMed.
- M. Bezanson, J. Pottel, R. Bilbeisi, S. Toumieux, M. Cueto and N. Moitessier, J. Org. Chem., 2013, 78, 872–885 CrossRef CAS.
- M. J. Deka, K. Indukuri, S. Sultana, M. Borah and A. K. Saikia, J. Org. Chem., 2015, 80, 4349–4359 CrossRef CAS.
- S. J. Gharpure, D. S. Vishwakarma and S. K. Nanda, Org. Lett., 2017, 19, 6534–6537 CrossRef CAS.
- J. K. Vandavasi, W.-P. Hu, H.-Y. Chen, G. C. Senadi, C.-Y. Chen and J.-J. Wang, Org. Lett., 2012, 14, 3134–3137 CrossRef CAS.
- S. J. Gharpure and J. V. K. Prasad, Eur. J. Org. Chem., 2013, 2013, 2076–2079 CrossRef CAS.
- J.-C. Castillo, J. Portilla, B. Insuasty, J. Quiroga and R. Abonia, Curr. Org. Chem., 2018, 15, 370–379 CAS.
- S. K. Das, A. K. Srivastava and G. Panda, Tetrahedron Lett., 2010, 51, 1483–1485 CrossRef CAS.
- P. Ghosh, M. J. Deka and A. K. Saikia, Tetrahedron, 2016, 72, 690–698 CrossRef CAS.
- J. Nonnenmacher, F. Grellepois and C. Portella, Eur. J. Org. Chem., 2009, 2009, 3726–3731 CrossRef.
- T. Bera, B. Singh, T. A. Hamlin, S. C. Sahoo and J. Saha, J. Org. Chem., 2019, 84, 15255–15266 CrossRef CAS.
- R. Zhou, J. Wang, C. Duan and Z. He, Org. Lett., 2012, 14, 6134–6137 CrossRef CAS.
- K. Samanta and G. Panda, Org. Biomol. Chem., 2011, 9, 7365–7371 RSC.
- M. K. Jackl, L. Legnani, B. Morandi and J. W. Bode, Org. Lett., 2017, 19, 4696–4699 CrossRef CAS.
- P. Králová, V. Fülöpová, M. Maloň, T. Volná, I. Popa and M. Soural, ACS Comb. Sci., 2017, 19, 173–180 CrossRef.
- P. Králová, M. Maloň, T. Volná, V. Ručilová and M. Soural, ACS Comb. Sci., 2017, 19, 670–674 CrossRef.
- P. Králová, M. Maloň and M. Soural, ACS Comb. Sci., 2017, 19, 770–774 CrossRef.
- V. Ručilová, P. Králová and M. Soural, Eur. J. Org. Chem., 2017, 2017, 7034–7039 CrossRef.
- M. Ručilová, V. Maloň and M. Soural, Eur. J. Org. Chem., 2018, 2018, 564–570 CrossRef.
- F. Filira, B. Biondi, L. Biondi, E. Giannini, M. Gobbo, L. Negri and R. Rocchi, Org. Biomol. Chem., 2003, 1, 3059–3063 RSC.
- C. R. Zwick and H. Renata, Tetrahedron, 2018, 74, 6469–6473 CrossRef CAS.
- V. Fülöpová, A. Krchňáková, E. Schütznerová, J. Zajíček and V. Krchňák, J. Org. Chem., 2015, 80, 1795–1801 CrossRef.
- V. Fülöpová, T. Gucký, M. Grepl and M. Soural, ACS Comb. Sci., 2012, 14, 651–656 CrossRef.
- G. E. Martin and C. E. Hadden, J. Nat. Prod., 2000, 63, 543–585 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07997a |
| This journal is © The Royal Society of Chemistry 2020 |