
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecovery and separation of phosphorus as dicalcium phosphate dihydrate for fertilizer and livestock feed additive production from a low-grade phosphate ore†
John Anawati a and
Gisele Azimi*ab
a and
Gisele Azimi*ab
aLaboratory for Strategic Materials, Department of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College Street, Toronto, Ontario M5S 3E5, Canada. E-mail: g.azimi@utoronto.ca
bDepartment of Materials Science and Engineering, University of Toronto, 184 College Street, Toronto, Ontario M5S 3E4, Canada
First published on 21st October 2020
Abstract
With the rapid increase in the world population, the global demand for food production has been increasing steeply. This increase has resulted in an increased demand for phosphorus crop fertilizers and livestock feed additives. Considering recent predictions that the global reserves of high-grade phosphorus resources would deplete within 15 years, new initiatives have begun to utilize low-grade resources to ensure sustainable supply of this essential nutrient. The main challenge with the use of low-grade resources is the difficulty with the efficient and economical separation of phosphorus from the other constituent elements, such as iron, aluminum, and magnesium. Most previous studies on the adoption of low-grade phosphate ores have focussed on ore beneficiation processes which are expensive, complex, and in some cases inefficient. In this study, we develop an integrated process for the direct recovery and separation of dicalcium phosphate dihydrate for fertilizer and livestock feed additive production from a low-grade (2.0 wt% P) iron-rich (19.7 wt% Fe) phosphate ore. The process combines leaching using dilute sulfuric acid (0.29 M) and selective precipitation using calcium oxide. During selective precipitation, ethylenediaminetetraacetic acid (EDTA) is used as a stabilizing agent to prevent iron and phosphorus co-precipitation. This process can be operated as a closed loop, allowing the recovery and recycling of both water and EDTA, while eliminating the production of liquid waste. The developed process achieves around 70% phosphorus recovery as an industrial-grade (19 wt% P) dicalcium phosphate dihydrate product with minimal iron, magnesium, and aluminum contamination, while also producing value-added calcium sulfate dihydrate (gypsum) and iron/magnesium byproducts. This process enables economical and sustainable recovery of phosphorus from low-grade ores, which can address the rising global demand for food production.
Introduction
Phosphorus is a key nutrient for living organisms due to its role in several key bioprocesses. In agriculture, phosphorus-containing fertilizers are employed to achieve sufficient crop growth and phosphorus-rich feed additives are used to enhance livestock nutrition. Phosphorus supplements are usually produced from high-grade phosphate rock concentrates (28–40 wt% P2O5).1 The sources for this phosphate rock are either sedimentary phosphorites (29–35 wt% P2O5) or igneous apatites (∼27 wt% P2O5), with the phosphorus occurring as apatite minerals (Ca5(PO4)3(F, OH, Cl)).2 The phosphate rock is converted to phosphoric acid (H3PO4) which is used to produce phosphate fertilizers. In the wet phosphoric acid process, the phosphate rock is reacted with sulfuric acid (H2SO4), producing gypsum (CaSO4·2H2O) and aqueous phosphoric acid (reaction (1)).3| Ca5(PO4)3[F, OH, Cl](s) + 5H2SO4(aq) + 2H2O(l) → 3H3PO4(aq) + 5CaSO4·2H2O(s) + [HF, H2O, HCl](aq) | (1) |
The main role of phosphate supplements is to deliver soluble and bioavailable phosphorus to plants and livestock as crop fertilizers or animal feed additives.1 Crop fertilizers come in different forms such as monoammonium phosphate (NH4H2PO4), diammonium phosphate ((NH4)2HPO4), single superphosphate (Ca(H2PO4)2 + CaSO4·2H2O, 15–21 wt% P2O5) and triple superphosphate (Ca(H2PO4)2 > 40% P2O5), while livestock feed additives are usually in the form of dicalcium phosphate (CaHPO4), monocalcium phosphate (Ca(H2PO4)2), or defluorinated phosphate (Ca3(PO4)2).1,4–6 The P2O5 grade, mineral form, and calcium content of these products are critical for their effective performance in their target applications and their market value. Given their bulk use, their efficient and economical production is essential.5,7,8 In particular, these products should be produced with minimal reagent and energy consumption and ideally produced near their point of use to minimize transportation costs.
The global demand for phosphorus is continuously increasing. Between 2016 and 2020, the demand has increased by 7% and it is predicted that by 2022, the demand will exceed the supply in several regions of the world, such as Latin America, South Asia, Europe, and Oceania.9 Considering that phosphate rock is a non-renewable resource, high-grade phosphorus deposits which are primarily located in Morocco, China, USA, Jordan, and Tunisia are depleting and the depletion is predicted to become critical around 2033.10,11
Considering the increasing depletion of high P2O5 grade resources, industrialists are highly interested on alternative phosphorus resources including low phosphorus grade ores,12 industrial waste products,13 and municipal waste.14 The low P2O5 grade and high impurity concentration in these resources can cause operational challenges in the wet phosphoric acid process as follows:12,15
(a) feeds with less than 30 wt% P2O5 and high concentrations of calcium (CaO to P2O5 ratio > 1.6) increase transportation costs and lead to excessive sulfuric acid consumption,
(b) excessive magnesium in the ore (MgO > 1 wt%) can lead to filter blockage and problematic viscosities within the process,
(c) aluminum and iron (Al2O3 + Fe2O3 > 3 wt%) can affect plant capacity, P2O5 recovery, filtration rates, and introduce post-precipitation issues,
(d) carbonates and organics (>5–6 wt%) can affect ore flotation, filtration rates, product color, and create foaming issues,
(e) other minor impurities such as chlorine, fluorine, strontium, and heavy metals can cause various problems including corrosion, product safety, and crystallization issues.
In Mato Grosso, one of the major agricultural regions of Brazil, a low-grade phosphate ore deposit containing 4.7 wt% P2O5 has been discovered which offers the potential for crop fertilizer and livestock feed additive production in close proximity to phosphate consuming farms. In this ore, phosphorus occurs as apatite (hydroxyapatite and chlorapatite) which is contained as a minor phase within the rock primarily composed of FeO(OH) and SiO2 with small amounts of other minerals including MgCO3, MgSiO3, H2Si6O13, and Al2H4Si2O9. Detailed characterization of the ore is provided in the Results and discussion section. The major challenge with this ore is the low phosphorus grade and high concentration of impurities such as iron, aluminum, and magnesium, which must be separated from the phosphorus to achieve acceptable P2O5 grades in the product. Developing a P2O5-containing product from this ore can help eliminate the need for transporting other ores to this agricultural region of the world, and the associated environmental and economic costs.16
There are several beneficiation techniques to increase the P2O5 content of ore concentrate to make a suitable feed for the wet phosphoric acid process. These include froth flotation of silicates and carbonates, attrition scrubbing and desliming of clays, electrostatic separation of silica, magnetic separation of ferriferous minerals, gravity separation of coarse phosphate minerals, calcination of calcareous minerals, and dilute organic acid leaching of carbonate minerals.12 These processes, although promising, are complex and expensive, and they are far from being adequate in achieving high-grade P2O5 products. Moreover, some of these processes are only applicable to phosphate ores with specific characteristics, e.g., presence of ferriferous or carbonate minerals.
The separation of P2O5 from the other ore materials can also be achieved with solvent extraction after acid leaching; however, this process is expensive and reagent intensive because of the large number of mixer-settler stages required, and produces large volumes of organic waste.17,18 An alternative approach is selective precipitation after leaching. A previous study utilized this approach for incinerated wastewater treatment sludge, which is similar to a low-grade phosphate ore; however, their process relied on the high aluminum content of the ash (18 wt% Al2O3) which resulted in phosphorus recovery as aluminum phosphate (AlPO4), which is not a suitable for crop fertilizer or livestock feed additive production.19
In this study, a simple closed loop, low cost, and sustainable process was developed to recover the phosphorus content of the Brazilian low-grade ore and separate the impurities without using complex and costly beneficiation or costly solvent extraction circuits. In this process, the crushed ore was leached with sulfuric acid, and phosphorus along with impurities were extracted into the leachate. To separate phosphorus from impurities, a selective precipitation step with lime (CaO) was utilized. In the first trial, this process led to co-precipitation of iron with phosphorus. To avoid this problem, chelation stabilization with ethylenediaminetetraacetic acid (EDTA, H4C10H12N2O8/[C10H12N2O8]4−) was utilized after leaching, before selective precipitation.
The complexation formation constant of several chelating agents such as DTPA (C14H23N3O10), EDDHA (C18H20N2O6), and DOTA (C16H28N4O8) with Fe3+ is given in ESI Table S1.†20–22 The EDTA was selected for this study because it has the best compromise between high complexation formation constant and low cost.
Since EDTA is an expensive reagent, compared to sulfuric acid and lime,23 its recovery and recycling is essential to make the process economical. Furthermore, in typical hydrometallurgical processes, the generation of hazardous waste, particularly liquid waste, can cause negative environmental impacts,24 which can be compounded by consumption and depletion of water resources. The developed process in this study involves water and EDTA recycling steps to ensure that the production of waste products is minimized. The process produces three solid products: (1) high-grade dicalcium phosphate dihydrate (main product), (2) gypsum, which can be used for manufacturing of wallboards, cement, and plaster of paris, or for soil conditioning (byproduct), and (3) iron hydroxide (Fe(OH)3)-rich product which can be used for pigment production (byproduct). The solid residue after the leaching step can be washed to remove excess acid and returned to the mine site as backfill. As noted above, EDTA was recovered and recycled to reduce reagent costs. Dissolved EDTA is known to precipitate from solution as its fully protonated acid form (H4EDTA) upon acidification below pH 2.0.25,26 In this process, the recovery of EDTA by acidification with H2SO4 offers the dual benefits of reducing the EDTA consumption and enabling water recycling because the remaining liquor can be further acidified to be used in the ore leaching step.
The operating principle of the proposed process is similar to the approach used in previous studies on the regeneration of EDTA for the removal of heavy metals, such as cadmium, copper, nickel, lead, and zinc from contaminated soils.27–29 In these processes, solutions containing EDTA chelated with divalent cations (Me2+ = Cd2+, Cu2+, Ni2+, Pb2+, Zn2+) were mixed with soluble iron(III) salts (such as FeCl3 or Fe(NO3)3) to substitute the chelated divalent cations with trivalent Fe3+, which is bound more strongly. The Fe–EDTA + Me2+ solutions were then mixed with disodium phosphate (Na2HPO4) to precipitate Me2+ ions as phosphates. The iron was then removed by hydrolysis using either NaOH or calcium hydroxide (Ca(OH)2) to raise the pH such that Fe(OH)3 precipitates, allowing the EDTA solution to be re-used for soil flushing after pH adjustment.
This study was focused on the development and validation of the closed loop phosphorous recovery process consisting of leaching with H2SO4 followed by stabilization with EDTA, and selective precipitation with CaO. By employing thorough chemical and physical characterization techniques, thermodynamic simulations, and experimental testing, a sustainable and economical road map is proposed. We expect our findings will help move towards addressing the increasing global demand for crop fertilizers and livestock feed additives without causing significant negative environmental impacts.
Experimental
Materials
Phosphate ore from the Jauru municipality of Mato Grosso, Brazil was provided by BEMISA Group. Sulfuric acid (H2SO4, 95.0–98.0 wt%, VWR), nitric acid (HNO3, 68.0–70.0 wt%, VWR), hydrochloric acid (HCl, 36.5–38.0 vol%, VWR), calcium oxide (CaO, 99.5 wt%, Materion), disodium ethylenediaminetetraacetic acid dihydrate (Na2H2C10H12N2O8·2H2O, 99.5 wt%, EM Science), and deionized water (0.055 μS, Millipore) were used to perform the experiments. Certified multi-element standard stock solutions (Inorganic Ventures) were used for calibration of analytical instruments. Please note that for readability, and to differentiate the different forms of the polyprotic acid, ethylenediaminetetraacetic acid in general (including the various ionized, bound, and/or protonated forms) was abbreviated as EDTA, H4C10H12N2O8 as H4EDTA, the [C10H12N2O8]4− ion as [EDTA]4−, and Na2H2C10H12N2O8·2H2O as Na2EDTA·2H2O.The EDTA is an organic polyprotic acid complexing agent (reaction (2)) that binds to dissolved metal ions (Mx+), forming a soluble metallo–organic complex (reaction (3), Fig. 1). The [EDTA]4− ligands around the metal ions prevent anions from interacting with the metal ion; hence, the formation of undesired precipitating species (e.g., FePO4·2H2O) is avoided. As a result, the phosphate product could be selectively precipitated without iron contamination.
| H4C10H12N2O8(s) + 4H2O(l) ⇌ [H3C10H12N2O8](aq)− + H3O(aq)+ + 3H2O(l) ⇌ [H2C10H12N2O8](aq)2 + 2H3O(aq)+ + 2H2O(l) ⇌ [HC10H12N2O8](aq)3− + 3H3O(aq)+ + H2O(l) ⇌ [C10H12N2O8](aq)4− + 4H3O(aq)+ | (2) |
| M(aq)x+ + [C10H12N2O8](aq)4− ⇌ [MC10H12N2O8](aq)(x−4) | (3) |
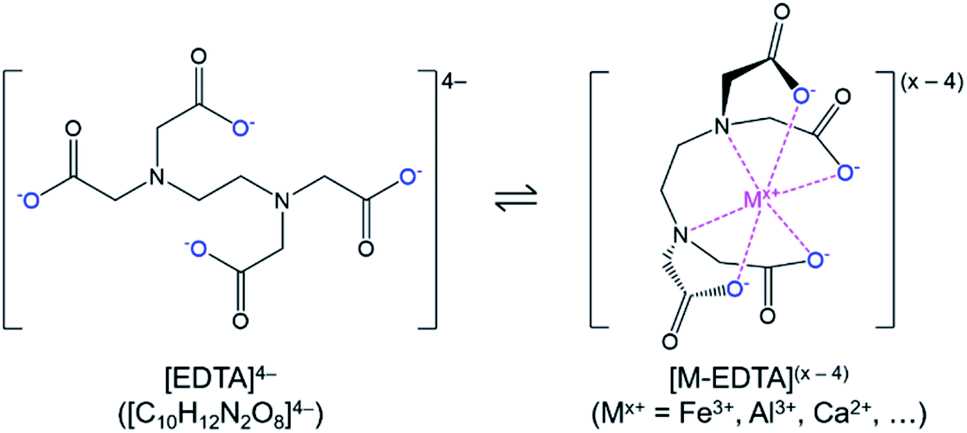 | ||
| Fig. 1 Chemical structures of [EDTA]4− and [M–EDTA](x−4) ions. The protonatable carboxylate groups are highlighted in blue and the bound metal ion (such as Fe3+, Al3+, Ca2+) is highlighted in magenta. | ||
The EDTA is a common and readily available chelating agent which can be used for stabilizing and solubilizing various metal ions. It has been used for the selective extraction of dysprosium during the solvent extraction of rare earth magnet leachates,30 for preventing calcium and magnesium phosphate precipitation in wastewater treatment,31 for preventing the precipitation of iron and arsenic during the transportation of drinking water samples,32 and the recovery of nickel from spent industrial catalysts,33 among other uses. The EDTA has also been utilized during separation of iron and titanium by selective precipitation; it was used to stabilize Fe3+ ions while TiO2+ ions were precipitated as titanium dioxide (TiO2) via hydrolysis using sodium hydroxide (NaOH).34
Compositional, mineralogical, and elemental mapping characterization
The elemental composition of the as-received Jauru Ore (JO) was characterized by microwave assisted aqua regia digestion (MW-AR) followed by inductively coupled plasma optical emission spectrometry (ICP-OES, PerkinElmer Optima 8000). In MW-AR, the solid ore sample was digested in 3HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1HNO3 (0.3–0.4 gsolids in 20 mL) at 200 °C (ramp up: 9 °C min−1, hold: 20 min, ramp down: 5 °C min−1 – MARS6 Xpress microwave digestion system). The digested samples were filtered with 0.45 μm nylon syringe filters and diluted with 5 wt% HNO3 prior to measurement to ensure the concentrations were within the calibrated range (0–10.0 mg L−1). The concentrations of the elements of interest (sodium, magnesium, aluminum, silicon, phosphorus, sulfur, calcium, titanium, manganese, and iron) were quantified by ICP-OES. To prevent the precipitation of supersaturated solution components after sampling, the samples were diluted immediately after being taken.
:1HNO3 (0.3–0.4 gsolids in 20 mL) at 200 °C (ramp up: 9 °C min−1, hold: 20 min, ramp down: 5 °C min−1 – MARS6 Xpress microwave digestion system). The digested samples were filtered with 0.45 μm nylon syringe filters and diluted with 5 wt% HNO3 prior to measurement to ensure the concentrations were within the calibrated range (0–10.0 mg L−1). The concentrations of the elements of interest (sodium, magnesium, aluminum, silicon, phosphorus, sulfur, calcium, titanium, manganese, and iron) were quantified by ICP-OES. To prevent the precipitation of supersaturated solution components after sampling, the samples were diluted immediately after being taken.
The crystal structure of the starting ore and solid products was characterized using X-ray diffraction (XRD – 2θ: 5–75°, scan rate: 1.25° min−1, Rigaku MiniFlex 600 Diffractometer). Elemental mapping of the ore was conducted by electron probe X-ray microanalysis (EPMA – JEOL JXA8230 5-WDS Electron Microprobe). Prior to EPMA analysis, the samples were fixed in epoxy resin (EpoKwick™ FC resin and hardener, Buehler) and cross-sectioned by polishing.
Leaching trials
Leaching experiments were performed to determine the leaching kinetics and leaching percentage and to make feeds for the precipitation step. Extraction efficiency was defined as the mass ratio in the liquor and the solid ore (eqn (4)).
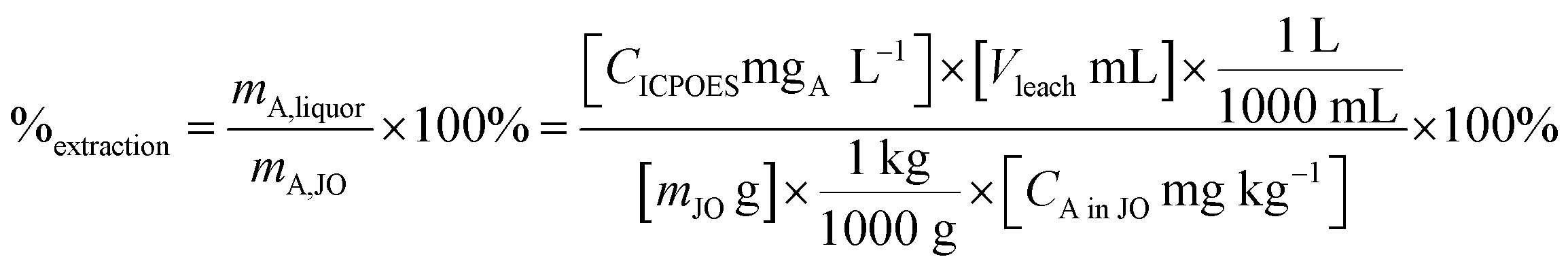 | (4) |
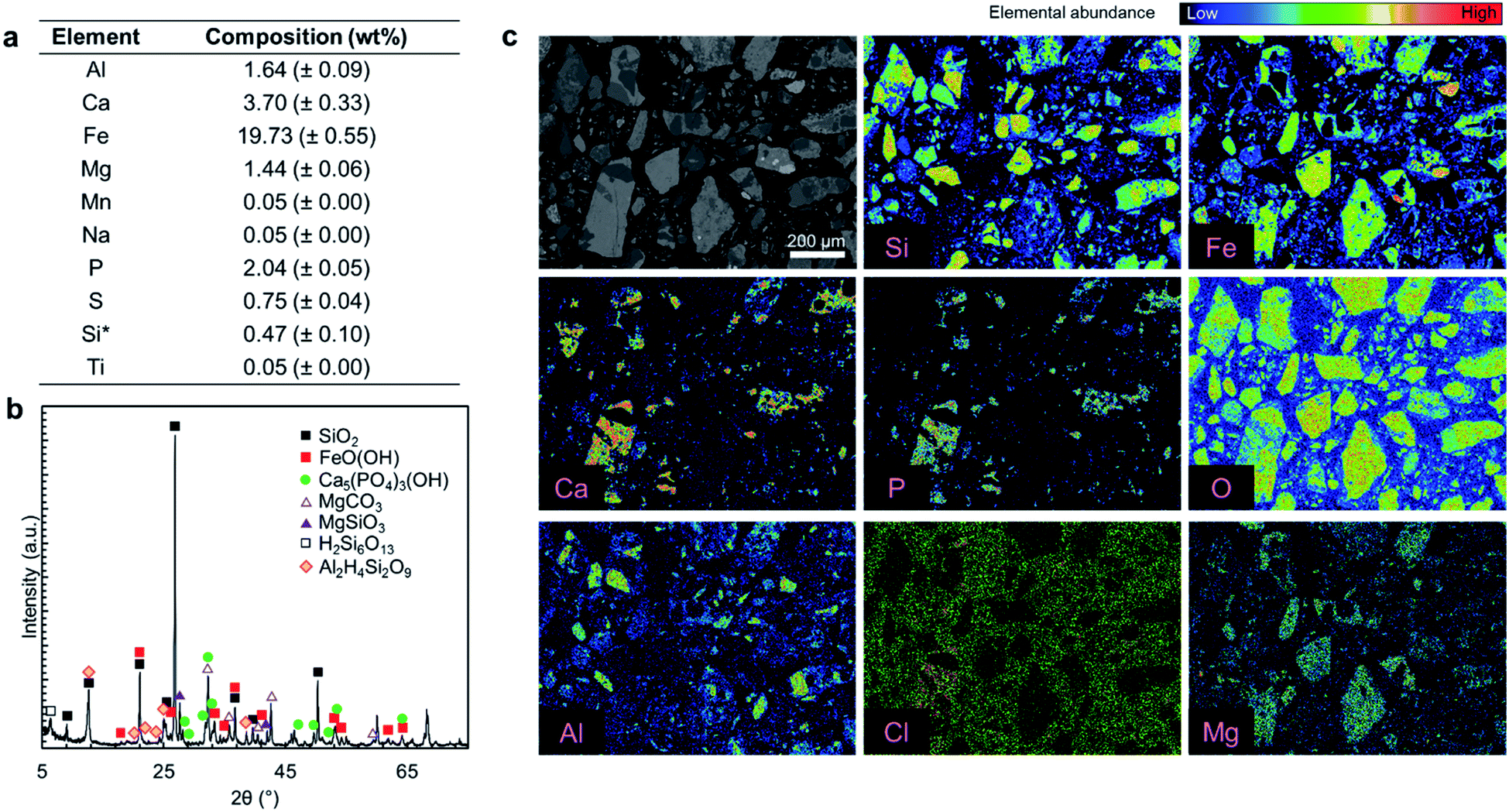 | ||
| Fig. 2 Ore characterization results. (a) Elemental composition obtained by aqua regia digestion followed by ICP-OES (average of four measurements (n = 4)). The Si concentration corresponds to the acid-soluble silicon content. (b) X-ray diffractogram. (c) EPMA elemental mapping; the color scale for the EPMA maps show the relative elemental abundance and the scale is unique for each element. The ore particles were fixed in epoxy which contained chlorine and oxygen, leading to a high background reading for Cl and O. | ||
Unless otherwise noted, the ore was mixed with deionized water (0.30 gJO mL−1) and concentrated H2SO4 solution (0.1 gH2SO4 gJO−1) in a glass beaker equipped with a magnetic stirrer. In the trials using regenerated acid solution, the regenerated acid was mixed with make-up water and H2SO4 prior to ore addition. In the kinetic leaching trials, the slurry was sampled at regular time intervals and the samples were immediately filtered with a 0.45 μm nylon syringe filter. In the non-kinetic trials, leaching was stopped after 5 min by vacuum filtering the slurry using a 6 μm paper filter.
Precipitation simulations
Thermodynamic calculations were performed with OLI Studio 9.6 and OLI Flowsheet 9.6 (OLI Systems) using the Mixed Solvent Electrolyte (MSE) model. The input concentrations for the calculations were set to approximate the experimentally measured liquor concentrations, using H2O, CaO, Fe2(SO4)3, Al2(SO4)3, CaSO4, Na2SO4, MgSO4, P2O5, H2SO4, H4C10H12N2O8, and Na2H2C10H12N2O8 as the input species.Precipitation trials
Two different types of precipitation trials were performed: single-step precipitation to determine the pH levels at which each element precipitates, and a multi-step precipitation, which aimed to replicate the industrial process by including each of the proposed precipitation and separation steps.In the single-step precipitation trials, the filtered liquor from the leaching step was placed in a beaker equipped with a pH probe and magnetic stirring. First, Na2EDTA·2H2O was added to achieve a concentration of 30 mM EDTA in the solution. Then, solid CaO was slowly added to the solution to raise the pH and induce precipitation. At approximately regular pH intervals up to pH = 11, the solution was sampled by a syringe and immediately filtered with a 0.45 μm nylon syringe filter. The sample was immediately diluted with 5 wt% HNO3 and analyzed with ICP-OES to determine the elemental composition. The CaO was added slowly such that pH remained approximately stable for 5 min prior to making another addition.
The multi-step precipitation trials were performed using the above approach; however, the process was carried out in a way that approximates the proposed industrial process, in which CaO is added to the solution to reach the target pH and a full solid–liquid separation is performed at each processing step. The target pH levels were selected on the basis of the single-step precipitation trials to maximize the separation of various solid products (P2O5 solids, impurities, regenerated reagents). The list of target pH levels and estimated required CaO or H2SO4 addition to reach the target pH are shown in Table 1. In the simplified process trial, steps 2 and 3 were combined to produce a single mixed P2O5 containing product, and steps 4 and 5 were combined to produce a single solid containing impurity elements.
| Separation step | Target pH | Estimated CaO/H2SO4 addition | Expected product |
|---|---|---|---|
| 1 | 3.0 | 6.12 gCaO L−1 | CaSO4·2H2O |
| 2 | 4.8 | 3.02 gCaO L−1 | CaHPO4 |
| 3 | 6.5 | 1.90 gCaO L−1 | Ca5(PO4)3OH |
| 4 | 9.5 | 2.24 gCaO L−1 | Fe(OH)3 |
| 5 | 11.0 | 0.55 gCaO L−1 | Mg(OH)2 |
| 6 | 2.0 | 4.09 gH2SO4 L−1 | CaSO4·2H2O + H4EDTA |
| 7 | 0.6 | 15.19 mLH2SO4 L−1 | Regenerated acid solution |
In each step, the estimated amount of CaO (by OLI simulation) was added to the solution and it was left to stir for >2 h, until the pH had stabilized. If the target pH was not reached, the simulation was used to recalculate a new amount of CaO to add, and this process was repeated until the target pH was reached. This approach of using long equilibration times and controlled CaO additions was used to ensure that CaO was not in excess and sufficient time was allowed for all species to precipitate. Separations were conducted by vacuum filtration with 6 μm filter paper and the collected solid products were dried at 40 °C for 24 h and weighed to allow calculation of the product composition.
The leachate solutions were brought to pH > 11 to precipitate the co-extracted impurities and the remaining P2O5 content. Sulfuric acid was then added to the calcium- and EDTA-containing solution to precipitate the EDTA as H4EDTA, which was collected. Then, H2SO4 was further added to produce an acidic solution analogous to the acid solution used in the initial leaching of the ore.
In the trials in which recovered H4EDTA from a previous trial was used as the EDTA source, the dried H4EDTA solids were added to the process and the difference required to achieve 30 mM EDTA was added as Na2EDTA·2H2O. The H4EDTA content of the solids was estimated from the molar composition by assuming that the solids were a combination of CaSO4·2H2O (from the molar concentration of sulfur), Ca(OH)2 (from the molar concentration of calcium minus the calcium in CaSO4·2H2O), and H4EDTA (from the remaining mass). The estimated recovery was also corrected to account for solution losses due to sampling and filter cake humidity. The XRD analysis of the EDTA product showed that it is primarily composed of CaSO4·2H2O and H4EDTA. The amount of regenerated EDTA to add was calculated from the estimated H4EDTA content of the recovered solids from the previous trial.
Results and discussion
Characterization results
Fig. 2a presents the elemental composition of the as-received ore, while Fig. 2b and c present the XRD diffractogram and elemental mapping obtained by EPMA characterization. As shown in Fig. 2a, iron (19.73 wt%), calcium (3.7 wt%), aluminum (1.64 wt%), and magnesium (1.44 wt%) are the major constituents of the ore and phosphorus content is 2.04 wt%. The major crystal phases in the ore are FeO(OH) and SiO2 while it has a P2O5 grade of 4.67 wt%, primarily in the form of hydroxyapatite (Ca5(PO4)3OH) with some chlorapatite (Ca5(PO4)3Cl). The elemental analysis in Fig. 2a does not account for 100% of the mass because the constituent metals are present in oxide form or carbonate form (magnesium). Also SiO2 that is one of the major ore components, is insoluble in aqua regia.Typically, fluorine from fluorapatite (Ca5(PO4)3F) is a problematic component of phosphate ores because its presence can lead to serious corrosion issues and cause serious health and safety risks.15 In this study, fluorine was not detected in the EPMA analysis (ESI Fig. S1†); thus, it was concluded that the fluorapatite content of this ore is negligible. As a result, fluorine extraction was not considered to be a major concern for this ore. If fluorine were present in this ore, the high content of SiO2 would likely result in the formation of silica gels which can cause operational challenges in the solid–liquid separation, rather than dangerous free hydrofluoric acid (HF);15 it is important to follow the fluorine fate if a fluorapatite-rich ore is used.
Overall, this ore is similar to typical phosphate ores that have apatite as the primary phosphorus bearing phase; however, the P2O5 grade of this ore is at the lower extreme of the reported phosphate ores, which contain 5–39 wt% P2O5.1,2
Since this ore is rich in iron, aluminum, and magnesium contaminants (collectively RxOy) that are known to interfere with the quality of the precipitated fertilizer products,12,36,37 the primary goal of the proposed multi-step precipitation process is to extract the P2O5 content of the ore and separate it from the impurities, in order to produce crop fertilizer or livestock feed additive products.
Acid leaching results
Leaching experiments were conducted at three different H2SO4 concentrations, i.e., 0.0 M, 0.29 M, 1.45 M, corresponding to 0.0, 0.1, and 0.5 gH2SO4 gJO−1. In all cases, a solid to liquids (S/L) ratio (S/L) of 0.3 g mL−1 was used and temperature was set at 25 °C. Fig. 3a–c present the extraction efficiency of phosphorus and impurity elements at three acid levels as a function of time. The no-acid baseline resulted in negligible phosphorus, iron, aluminum, magnesium, and calcium and some sodium (10%) extraction. Increasing acid content resulted in increased extraction. Phosphorus extraction was almost the same for 0.29 and 1.45 M acid concentration, achieving around 70% after 25 min with the majority of extraction occurring within the first 5 min. On the contrary, increasing acid concentration resulted in increased impurity co-extraction. The different leaching behavior of phosphorus compared with other elements is due to the fact that phosphorus occurs as a distinct Ca5(PO4)3OH phase which reacts independently from the impurity-bearing phases. This means phosphorus can be selectively extracted by using low acid concentrations. Calcium extraction was approximately stable in both cases, with the solution first achieving slight supersaturation, then slowly decreasing in concentration as calcium precipitated, primarily as CaSO4·2H2O, confirmed by the co-localization of calcium and sulfur in the leached residue (ESI Fig. S2†).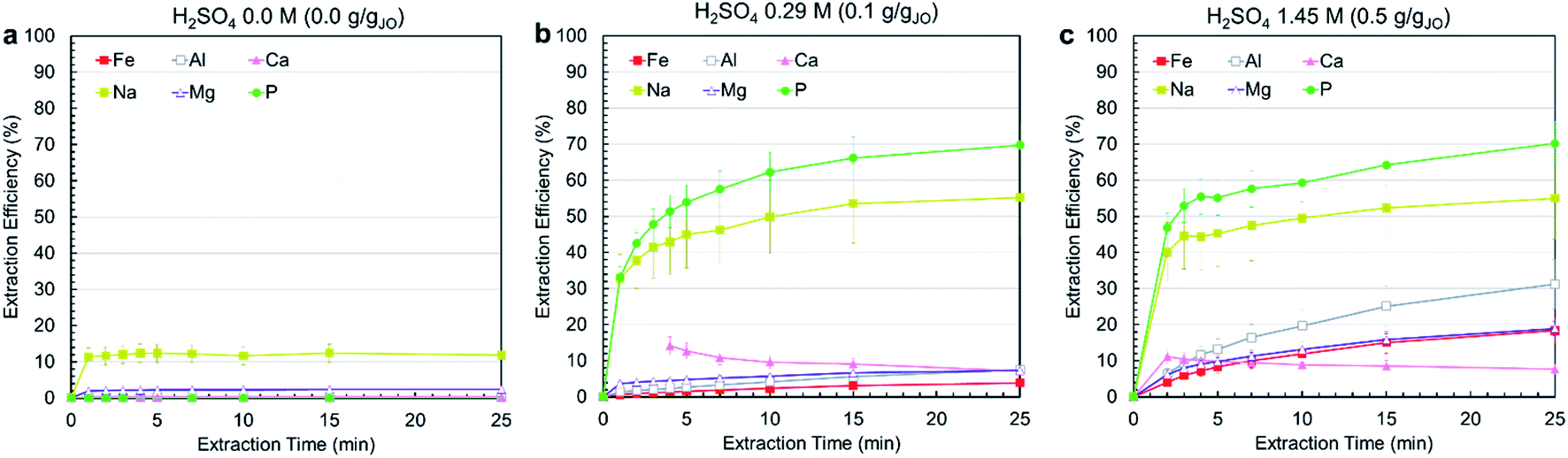 | ||
| Fig. 3 Ore leaching kinetics. (a) 0.0 M, (b) 0.29 M, and (c) 1.45 M H2SO4. All trials were conducted at 25 °C, with a solid/liquids ratio of 0.3 gJO mL−1. Please note that calcium measurements for 1, 2, and 3 min for the 0.29 M trial were not quantified as the concentration was outside the calibration range of the instrument. | ||
Considering the effect of acid concentration on extraction, to minimize impurity co-extraction, 0.29 M H2SO4 concentration was selected as the best condition and was used in the process. To further minimize impurity co-extraction and the residence time for the leaching step, a leaching duration of 5 min was selected for the subsequent trials. It is important to note that the subsequent precipitation steps were performed immediately after filtering the leachate solution because a time-delayed formation of precipitate solids was observed in filtered leachate solution that was left undisturbed for several hours (ESI Fig. S3†); this solid was confirmed to be AlP, AlPO4 and Al(H2P3O10)(H2O)2 by XRD.
Although the process reached only 50–70% phosphorus extraction, the combination of short residence time (5–25 min), low acid concentration (0.29 M), high S/L of 0.3 g mL−1 and low temperature (25 °C) makes the process economical and practical. Comparing this process with other studies on the leaching of phosphorus from phosphate rocks show that higher phosphorus extractions can only be achieved at very high acid concentrations (25–35 wt% H3PO4), low S/L ratios (0.05–0.1 g mL−1) and long leaching durations (3–4 h)38,39 which justifies the advantages of the developed process in this study, since conditions were selected such that they are applicable at the larger scale.
Each precipitation trial in the next step of the process used leachate from its own individual leaching trial; hence, the leachate composition was slightly different. Table 2 presents the average concentration of elements of interest for two different scenarios: (1) the case in which water and H2SO4 were used as the leaching agent, (2) the case in which regenerated leachate with make-up water and H2SO4 were used as the leaching agent. The primary difference between these leachate solutions is the sodium content, which was considerably higher in the second case due to added sodium upon addition of Na2EDTA·2H2O.
| Element | Fe | Al | Ti | Ca | Na | Mg | Mn | P | S | pH |
|---|---|---|---|---|---|---|---|---|---|---|
| Water + H2SO4 (n = 4) | ||||||||||
| Concentration (mg L−1) | 1358 | 196 | 3 | 1117 | 46 | 245 | 8 | 3270 | 4200 | 1.20 |
| Standard deviation | ±60 | ±15 | ±3 | ±46 | ±32 | ±12 | ±2 | ±302 | ±564 | ±0.12 |
|
||||||||||
| Regenerated leachate + make-up water & H2SO4 (n = 2) | ||||||||||
| Concentration (mg L−1) | 1080 | 154 | 4 | 1299 | 848 | 240 | 8 | 3655 | 4303 | 1.26 |
| Standard deviation | ±123 | ±10 | ±4 | ±133 | ±13 | ±4 | ±5 | ±160 | ±580 | ±0.04 |
Precipitation results
A pH-mediated selective precipitation step was developed to separate P2O5-containing products from the co-extracted impurities, in particular iron. The CaO was selected as the precipitating agent because of its abundance, low cost, and sufficient alkalinity. In addition to controlling pH, CaO provides a calcium source for the direct precipitation of calcium phosphate-based products, such as high-value livestock feed grade dicalcium phosphate (CaHPO4). Another benefit of CaO is the low solubility of gypsum, i.e., the neutralization product, which eliminates introducing large amounts of contaminating soluble ions during precipitation as NaOH or Na2CO3 would.As shown in Fig. 4a, using CaO alone did not result in efficient separation of phosphorus from iron because these elements precipitate in the same pH range. In a follow-up test using NaOH, the overlap in precipitation ranges was also observed; however, the phosphates did not fully precipitate due to the lack of an appropriate counterion (ESI Fig. S4†).
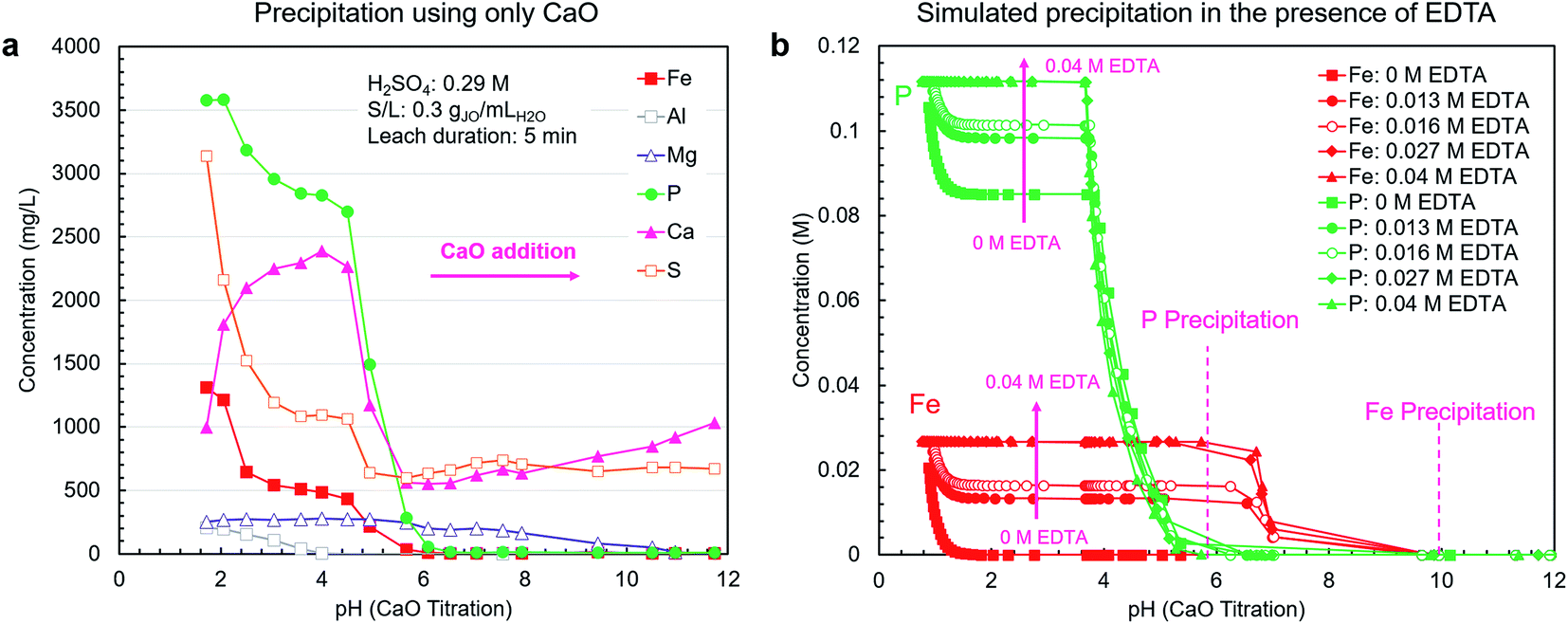 | ||
| Fig. 4 Precipitation of extracted elements from the leachate with CaO. (a) The leachate solution was neutralized by adding solid CaO and sampled at various pH levels for analysis. (b) Thermodynamic simulation results (OLI Studio, v. 9.6) for the effect of EDTA addition on the precipitation of phosphorus and iron. A representative solution containing water, iron, aluminum, calcium, sodium, magnesium, phosphorus, and H2SO4 was used as the modeling basis and H4EDTA was added in the range of 0–0.04 M. | ||
As mentioned previously, to prevent the co-precipitation of phosphorus and iron, likely as iron phosphate dihydrate (FePO4·2H2O) (reaction (5)), EDTA was utilized to stabilize the Fe3+ ions as Fe[EDTA]− according to reaction (6). By forming a stable soluble complex with EDTA (Kf = 1025.1),40 Fe3+ ions do not form a precipitate with PO43− ions. To estimate the stabilization capability of EDTA, thermodynamic simulations were performed using OLI Studio (v. 9.6). As shown in Fig. 4b, the simulation results showed that increasing EDTA concentration results in reduced iron–phosphorus co-precipitation up to a threshold level of EDTA after which the stabilization was maximized. Through EDTA stabilization, phosphorus and iron precipitation was decoupled, with the phosphorus precipitation completing at approximately pH = 6, primarily as either CaHPO4 (reaction (7)) at pH 3.7–4.9 or hydroxyapatite (Ca5(PO4)3OH) (reaction (8)) at pH 4.9–5.7. Iron started to precipitate as Fe(OH)3 (reaction (9)) after all the phosphorus had precipitated, at pH around 7. This precipitation would begin to occur at elevated pH values because at low H3O+ concentration levels, the hydrolysis reaction (reaction (9)) is favored over the chelation reaction (reaction (6)).
| Fe(aq)3+ + PO4(aq)3− + 2H2O(l) ⇌ FePO4·2H2O(s) | (5) |
| Fe(aq)3+ + [C10H12N2O8](aq)4− ⇌ [FeC10H12N2O8](aq)− | (6) |
| Ca2+ + PO43− + H3O+ ⇌ CaHPO4(s) + H2O | (7) |
| 5Ca2+ + 3PO43− + 2H2O ⇌ Ca5(PO4)3OH(s) + H3O+ | (8) |
| Fe3+ + 6H2O ⇌ Fe(OH)3(s) + 3H3O+ | (9) |
The simulation results presented in Fig. 4b only consider the thermodynamics of the system and do not account for the kinetics. Hence, to gain a clear picture of the system's behavior, the EDTA-mediated precipitation trends were measured experimentally. The EDTA concentration of 30 mM was selected on the basis of the simulation and preliminary testing results.
The experimental results (Fig. 5a) were in agreement with simulation results, but the measured pH thresholds were about 1 pH higher than their estimated counterparts. In the pH range of 1.2–3.1, the concentration of sulfur dropped sharply due to the precipitation of CaSO4·2H2O (reaction (10)). In the pH range of 3.7–4.9, small phosphorus precipitation was observed, but the major phosphorus precipitation was observed at pH range of 4.9–7.3. In this range, no iron or magnesium precipitation was observed with minimal aluminum precipitation. Iron was primarily precipitated between pH 8.9 and 9.9, coinciding with the precipitation of magnesium as magnesium hydroxide (Mg(OH)2) (reaction (11)). The precipitation of aluminum as aluminum hydroxide (Al(OH)3) started earlier at pH 8.0. Over the entire pH range, sodium concentration (introduced by the addition of Na2EDTA·2H2O) was constant because sodium salts are highly soluble.
| Ca2+ + SO42− + 2H2O ⇌ CaSO4·2H2O(s) | (10) |
| Mg2+ + 4H2O ⇌ Mg(OH)2(s) + 2H3O+ | (11) |
| Al3+ + 6H2O ⇌ Al(OH)3(s) + 3H3O+ | (12) |
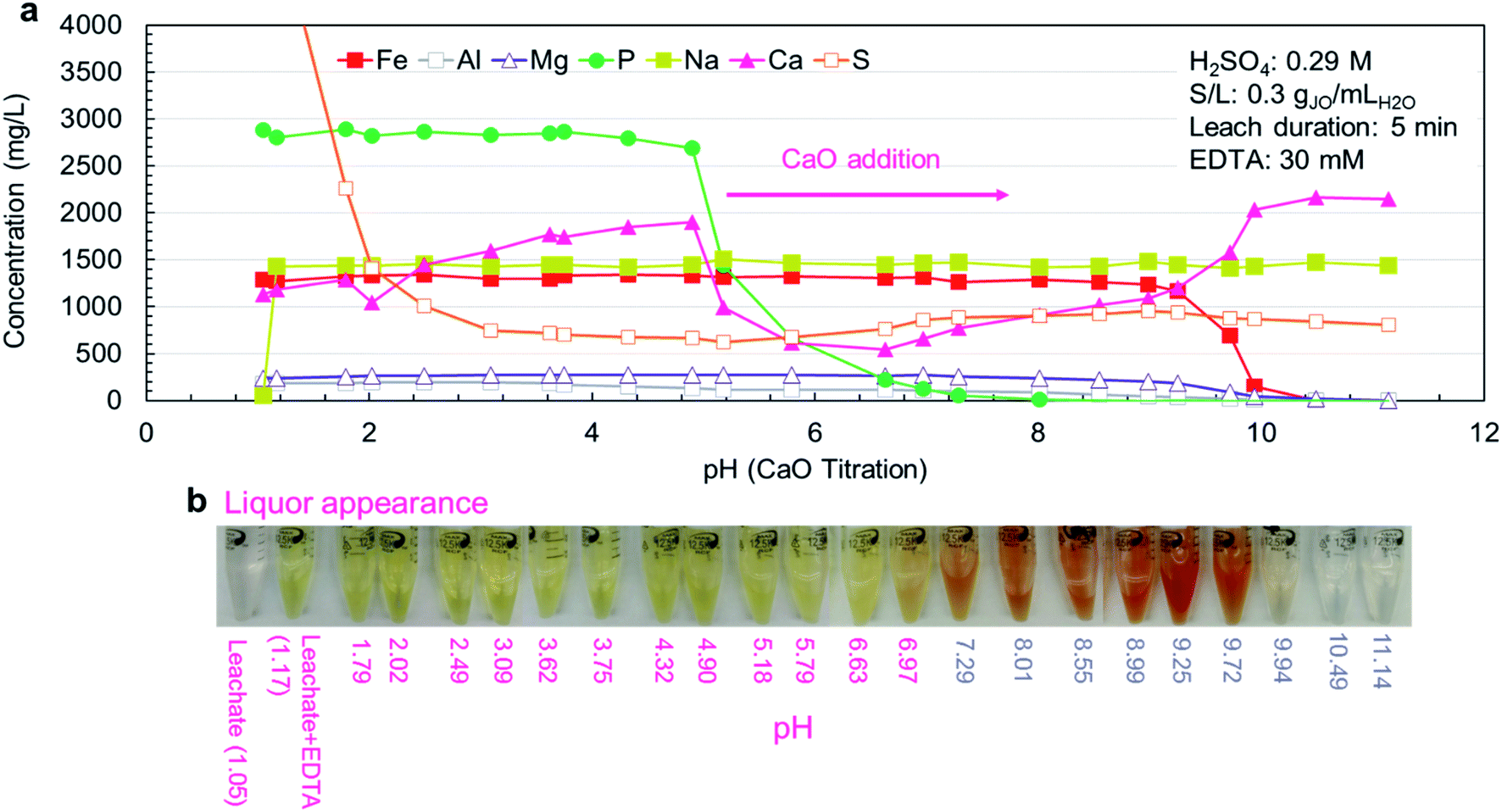 | ||
| Fig. 5 EDTA-stabilized precipitation of extracted elements from the leachate with CaO. Jauru ore direct leaching solution (DLJO) was stabilized with 30 mM Na2EDTA·2H2O, and pH was increased by adding CaO. (a) Solution concentration at various pH levels. (b) Photographs of liquor appearance at each pH level. | ||
The physical appearance of the leachate solutions at various pH points is presented in Fig. 5b. Upon addition of Na2EDTA·2H2O, the clear leachate solution immediately turned yellow due to the formation of [Fe–EDTA]−, which is known to have a yellow-brown color.41 As the phosphorus precipitated out of the solution, the yellow color shifted to red and as the iron precipitated, the red color was lost, leaving behind a clear solution. Since the observed color changes coincide with the various precipitation events, colorimetry can potentially be utilized for real time process monitoring at the industrial scale.
The primary separation optimization objective for this process was the production of high-grade P2O5 solids with minimal iron, aluminum, and magnesium contamination. On the basis of the above precipitation investigation, further simulation and additional preliminary testing, the following precipitation steps were selected, constituting the full selective precipitation train:
(1) leachate → pH 3.0: precipitation of CaSO4·2H2O. This byproduct must be removed because it reduces the P2O5 grade in the precipitate. It can be sold for manufacturing of wallboards, cement, and plaster of paris, or for soil conditioning.
(2) pH 3.0 → pH 4.8: precipitation of CaHPO4. This product represents the phosphate fertilizer product with higher P2O5 content and can potentially be used as a livestock feed additive.
(3) pH 4.8 → pH 6.5: precipitation of Ca5(PO4)3OH. This product represents the phosphate fertilizer product with lower P2O5 content.
(4) pH 6.5 → pH 9.5: precipitation of Fe(OH)3-rich solids. This product, which may contain undissolved CaO as Ca(OH)2 (CaO readily hydrates to Ca(OH)2 upon contact with water), represents the primary impurities that were co-extracted from the ore. These iron-rich solids could potentially be sold as an iron ore or be used as a feedstock for the production of specialty Fe-containing products, such as iron oxide pigments.
(5) pH 9.5 → pH > 11: this impurity product is expected to be relatively rich in Mg(OH)2 compared with the previous product and could potentially be sold as a precursor to fused magnesia (MgO) for construction materials.
The solution remaining after this precipitation train principally contains dissolved Ca2+, Na+, and SO42−, alongside soluble EDTA, present primarily as Ca[EDTA]2− complexes. Since EDTA is an expensive reagent, the economical operation of this process requires the recovery and recycling of EDTA. Furthermore, minimizing water consumption and wastewater production is crucial for the environmental sustainability of the process. Considering that neutral H4EDTA salt can be precipitated from aqueous solution by acidification with H2SO4,25,26 and that the liquor remaining after filtration is relatively free of contaminating ions (see details below), the acidification of the filtered liquor can serve the dual purposes of recovering EDTA and regenerating the wastewater into an acidic leaching agent for the initial leaching step. The target acid regeneration pH of 0.6 corresponds to the pH of 0.29 M H2SO4 solution. As such, the following acid addition steps were added to the process:
(6) pH > 11 → pH 2.0: precipitation of H4EDTA. This EDTA can be used to replace the Na2EDTA·2H2O used in the initial cycle (ESI Fig. S5†). This H4EDTA product contained CaSO4·2H2O solids; however, this contaminant did not affect the efficacy of the recycled EDTA and was captured during precipitation step 1 (at pH = 3).
(7) pH 2.0 → pH 0.6: regeneration of the acidic leaching solution. Upon acidification, the liquor can be recycled for the initial ore leaching step.
Complete process trials (leaching, stabilization, precipitation)
The complete precipitation train outlined above was repeated, and at each precipitation step and complete solid–liquid separations were performed. Two complete process trials were performed:(A) an initial start-up cycle in which deionized water and H2SO4 were used as the leaching agent and EDTA was added as Na2EDTA·2H2O.
(B) A continuous cycle, in which regenerated leachate and H4EDTA were used. Make-up water and H2SO4 solution were added to maintain a consistent S/L ratio. Make-up Na2EDTA·2H2O was added to maintain an EDTA concentration of 30 mM.
As shown in concentration-pH curves in Fig. 6a and b, the phosphorus and iron precipitations are well separated, consistent with the precipitation results shown in Fig. 5a. In trial A, a small iron-phosphorus co-precipitation was observed, but this did not happen in trial B. The decrease in iron co-precipitation in trial B (recycle trial) could be related to the addition of EDTA as H4EDTA rather than Na2EDTA·2H2O, which suggests that operating the process as a closed loop could reduce iron impurity in the product. Additionally, the bulk of aluminum precipitation occurred in the phosphate precipitation steps, on the contrary to the observed precipitation curve, but consistent with the simulation results. The discrepancy between the two trials could be due to the slow precipitation kinetics of Al(OH)3, which has previously been observed to precipitate at pH > 4.5 in a two-step reaction (a rapid step and a slow step).42 The extended equilibration times used in the complete process trials were sufficient for the Al(OH)3 to fully precipitate, whereas, the sampling in the precipitation curve trials (Fig. 5a) was too rapid to observe aluminum precipitation at lower pH values. In the complete process trials, the process equilibration time was slow because the calcium phosphate precipitation was limited by the release of Ca2+ ions from CaO/Ca(OH)2 dissolution. Because of the low solubility of Ca(OH)2, long equilibration times were needed to prevent the contamination of the solid products with excess Ca(OH)2 which would have reduced the P2O5 grade. The importance of CaO/Ca(OH)2 dissolution on the precipitation process also justifies the room temperature (25 °C) operation of the process, because Ca(OH)2 exhibits retrograde solubility (a decrease in solubility with increasing temperature); thus, operating the process at low temperature is more beneficial to obtain a higher grade product.
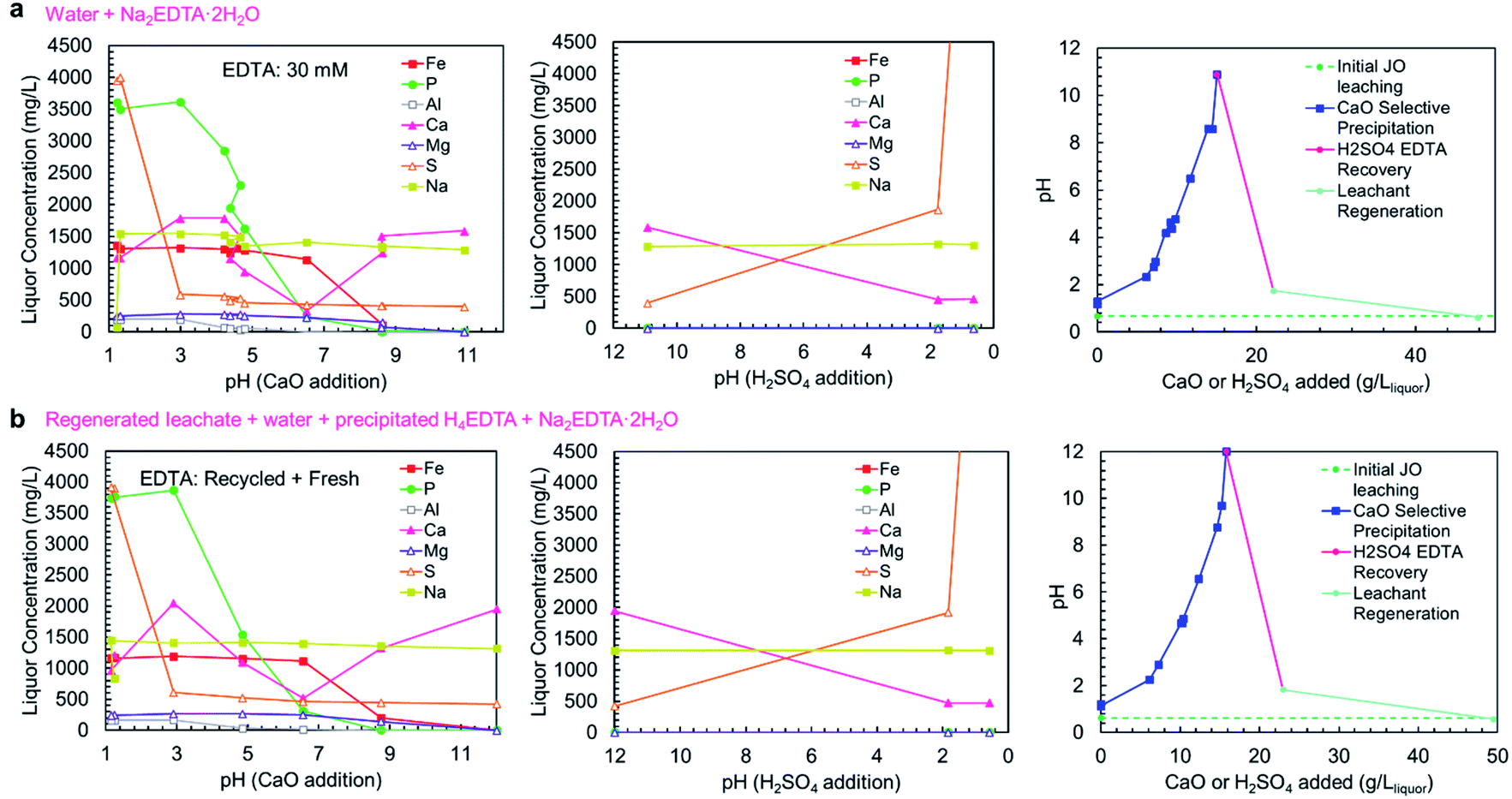 | ||
| Fig. 6 Concentration-pH curve for the complete selective precipitation process trials. The separations outlined in Table 1 were performed using (a) newly mixed leaching solution and Na2EDTA·2H2O, and (b) regenerated leachate solution and precipitated H4EDTA alongside make-up water and EDTA. The left panels show the liquor concentrations during the CaO addition steps. The middle panels show the liquor concentrations during the H2SO4 addition steps. The right panels show the amounts of CaO (solid) and H2SO4 (95–98 wt%) added to achieve the reported pH changes. | ||
In both trials A and B, the EDTA recovery and acid regeneration steps were consistent with calcium precipitating in the pH range of >11 → 2 as CaSO4·2H2O and no observable precipitation in the pH range of 2 → 0.6. In the first precipitation step, the expected precipitation of H4EDTA could not be verified by ICP-OES; however, XRD characterization of the precipitate confirmed H4EDTA presence. In both trials, the CaO and H2SO4 consumptions were nearly identical, indicating that the reagent consumptions are not negatively impacted by recycling of EDTA and water.
The compositions of the solid products (both elemental and oxide-equivalent mass compositions) produced in these trials and the molar elemental recoveries for each step (accounting for the relative amounts of solid products produced) are presented in Fig. 7a and b. As expected, the pH 3 product was primarily composed of calcium and sulfur in the form of CaSO4·2H2O, with negligible co-precipitation of phosphorus, iron, or aluminum. Together, the phosphate products (pH 4.8 and 6.5) represent 94–95% of the total extracted phosphate in the leachate. These products had nearly equimolar calcium and phosphorus, indicating that a high proportion of the phosphate precipitated as the higher-value CaHPO4 (1P:1Ca molar ratio), rather than Ca5(PO4)3OH (3P:5Ca molar ratio); however, the P2O5 grades of the products (36.9–41.9 wt%P2O5) were lower than the expected value for CaHPO4 (theoretical P2O5 = 52.2 wt%). During the neutralization of phosphoric acid with calcium hydroxide at temperatures below 60 °C, CaHPO4 is known to precipitate as its dihydrate (CaHPO4·2H2O).43 The measured P2O5 grades for the pH 4.8 and 6.5 products were consistent with the theoretical P2O5 grade of CaHPO4·2H2O (41.2 wt%). The formation of CaHPO4·2H2O was confirmed by XRD (Fig. 8g). The observed molar excess of calcium relative to phosphorus can be attributed to unreacted Ca(OH)2.
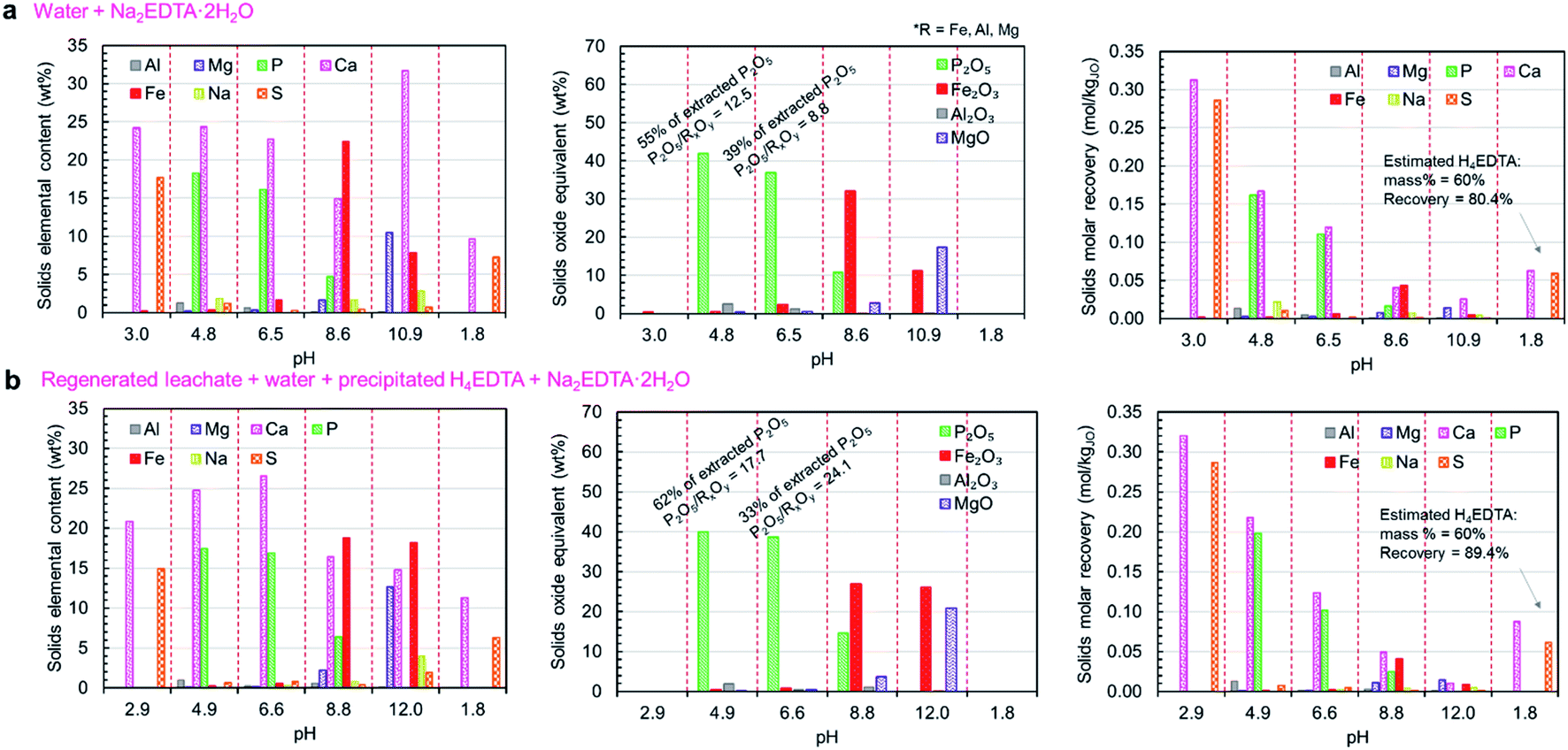 | ||
| Fig. 7 Solid product characteristics for the complete precipitation process trials. The separations outlined in Table 1 were performed using (a) fresh H2O + H2SO4 leaching agent and Na2EDTA·2H2O, and (b) regenerated leachate solution and precipitated H4EDTA alongside make-up water and EDTA. The left panels show the elemental composition of each solid, alongside the recovered dry mass of each product. The middle panels show the oxide-equivalent mass of P2O5, Fe2O3, Al2O3, and MgO in the solids. The right panels show the molar recovery of each precipitation step. | ||
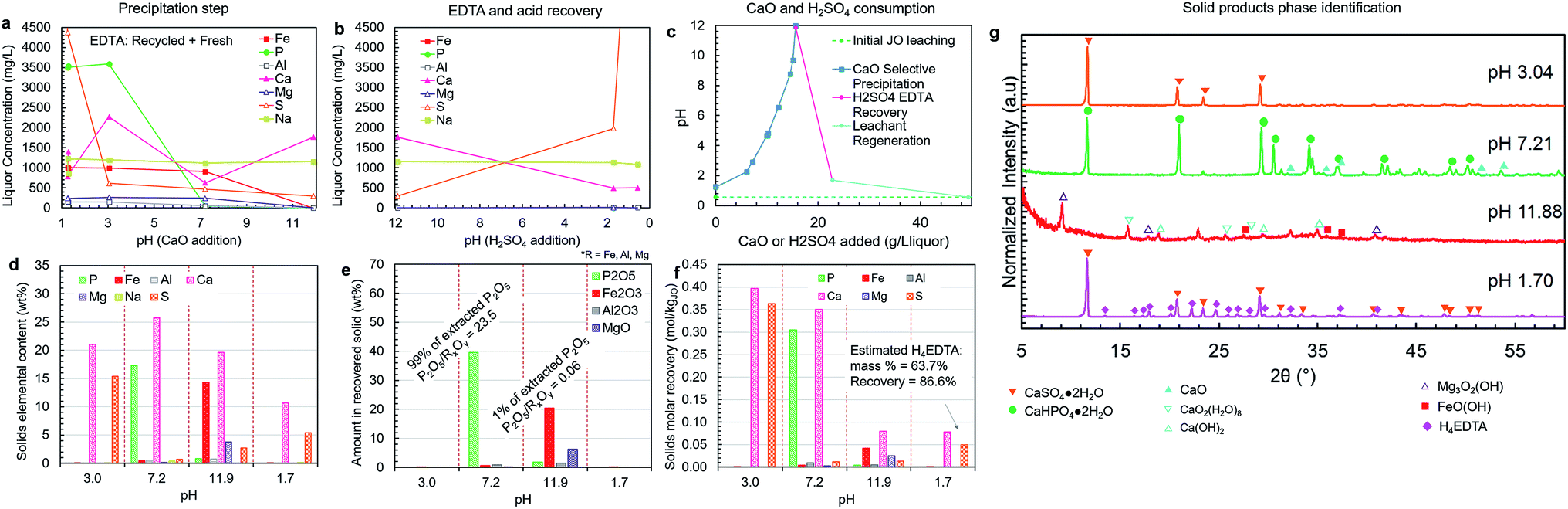 | ||
| Fig. 8 Liquor concentration and solid product composition for the simplified selective precipitation trials. The separations were carried out as outlined in Table 1; however, steps 2–3 and 4–5 were combined into single steps. The trial was performed using regenerated leachate solution and precipitated H4EDTA alongside make-up water and EDTA. (a) Liquor concentrations during the CaO addition steps. (b) Liquor concentrations during the H2SO4 addition steps. (c) The amounts of CaO (solid) and H2SO4 (95–98 wt%) added to achieve the reported pH changes. (d) Elemental composition of each solid. (e) The oxide-equivalent mass of P2O5, Fe2O3, Al2O3, and MgO in the solids. (f) Molar recovery of each precipitation step. (g) Solid product XRD diffractograms. The peak intensities were normalized to aid visualization. | ||
The observed predominance of CaHPO4·2H2O precipitation over Ca5(PO4)3OH was contrary to the results predicted by thermodynamic simulation; however, the simulation did not account for the kinetics of precipitation. Previous work has shown that calcium phosphate precipitation kinetics and the resulting mineral phases are highly pH dependent, i.e., the nucleation of crystalline CaHPO4·2H2O is favored over octacalcium phosphate (Ca8(HPO4)2(PO4)4·5H2O) and calcium deficient apatite at low pH, which was aided by the natural increase in pH driven by the H3O+-consuming precipitation reactions (reaction (7)).44 Because of the slow dissolution of CaO upon addition to the leachate solution, the majority of precipitation was able to occur at lower pH levels, resulting in higher-than-predicted formation of CaHPO4·2H2O.
In the impurity precipitation steps for both trials, the pH stabilized in the range of 8.5–8.7 after adding the prescribed amount of CaO and the impurity split was made at this pH rather than further adjusting the pH. The majority of the impurity solids were precipitated at pH < 8.5–8.7, comprising iron, as Fe(OH)3, some magnesium as Mg(OH)2, any unreacted Ca(OH)2 (as predicted by the simulations and observed by XRD – Fig. 8g), and the remaining 5–6% of unprecipitated P2O5. The pH 11 impurity solids were enriched in magnesium, while having a high content of iron and calcium. Since the processes are open to the atmosphere, it is possible that dissolution of atmospheric CO2 could cause some of the calcium to precipitate as CaCO3; however, this effect was likely small because CO2 was not actively injected into the solution, leading to a low gas–liquid interfacial area, and thus a slow rate of CO2 uptake.45
The pH 4.8 and pH 7.0 products were recovered separately because they were expected to have considerably different P2O5 grades and calcium to phosphorus (Ca/P) ratios; however, the results of these trials indicated that the properties of these two products are relatively similar; thus, it does not justify the use of two separation steps. Similarly, the two impurity products were collected separately to achieve iron and magnesium separation; however, both impurity solids contained both species; thus, process complexity could be avoided by collecting these two solids as a single product, as described below in the simplified process trials section. Compared with the primary phosphate product, these impurity products are produced in considerably smaller amounts and contain a large amount of unreacted Ca(OH)2; however, these products may still be used productively. One possible use is as a low grade iron ore, i.e., while the iron content (7.8–22.4 wt% iron) is considerably lower than that of typical iron ores, the primary impurity components are magnesium and calcium, which are typically mixed with iron ore as fluxing agents in blast furnace operations. The obtained precipitate could potentially be sold as an ore-flux mixture. Alternatively, this product could be sold as feed material for the production of iron oxide pigments because it is enriched in Fe(OH)3.46
The H4EDTA product yield was similar in both trials, with solids having an estimated 60 wt% H4EDTA content with 80–89% EDTA recovery (corrected to account for losses to sampling and filter cake moisture). This recovered EDTA can be used as a substitute for fresh Na2EDTA·2H2O, which was verified by conducting a precipitation experiment (similar to the experiment shown in Fig. 5a) using only recovered H4EDTA as the EDTA source, the results of which are presented in ESI Fig. S5.†
Simplified process trial (leaching, stabilization, precipitation)
A simplified version of the process was tested in which precipitation steps 2 and 3 were combined into a single step at approximately pH 7 (pH 7.2), and the impurity precipitation steps 4 and 5 were combined into a single step at pH > 11 (pH 11.9) (Fig. 8a–g). This simplified process employed recycled H4EDTA and leaching solution. In this process configuration, similar to previous trials, clear separation of phosphorus and iron was achieved, and the reagent consumption was nearly identical. The pH 7.2 product contained 99% of the extracted phosphorus, achieving a P2O5 grade of 39.6 wt% and a P2O5/RxOy ratio of 23.5 (8% of the extracted iron, 66% of the extracted aluminium, and 9% of the extracted magnesium were co-precipitated). Thus, by reducing process complexity, the product recovery and quality were both improved, potentially due to the presence of crystalline CaHPO4·2H2O precipitate in the slurry that was formed in the lower pH range at the start of the precipitation step, which act as a seed for CaHPO4·2H2O crystallization near the end of the step at higher pH. Only 1% of the extracted phosphorus was lost to the impurity products, which were primarily composed of calcium and iron with some magnesium and sulfur. This impurity product removed nearly all the remaining extracted iron (91%), aluminium (32%), and magnesium (91%) in the liquor.As mentioned above, the phosphate product occurred primarily as hydrated CaHPO4·2H2O, which has a lower P2O5 grade than the anhydrous form. Anhydrous CaHPO4 is known to precipitate directly at temperatures above 70 °C;47 however, as mentioned above, the retrograde solubility of Ca(OH)2 would further reduce the rate of phosphate precipitation; thus, requiring longer residence times.
The CaHPO4·2H2O is known to thermally dehydrate at temperatures above 135 °C;48 therefore, the product can be heat treated to increase the its P2O5 grade. By drying the product at 150 °C, the P2O5 grade could be further increased to 43.4 wt%, consistent with a partial dehydration of the dihydrate product to anhydrous dicalcium phosphate. No major crystal phase change was detected by XRD in the 150 °C sample; however, the dihydrate to anhydrous phase transition has previously been reported to not produce strong X-ray diffraction pattern changes when the heating is conducted in dry air.48 Further optimization of the product drying to achieve complete dehydration was deemed outside the scope of this study, but can be addressed in future work. Overall, the dicalcium phosphate dihydrate (DCP) product composition is comparable to commercially available DCP products.49
Upon re-acidification at pH 1.70, the EDTA was precipitated as a mixed H4EDTA + CaSO4·2H2O solid (Fig. 8g). The estimated EDTA recovery in this process was 87%. This indicates that the presence of sulfur does not negatively affect the binding of EDTA–Fe. The EDTA loss is due to the liquor held by the wet filter cakes during the solid–liquid separation steps. In practice at the industrial scale, this lost EDTA can be recovered by washing the filter cake with water, which can also supply the make-up water that must be added to compensate for the humidity losses in the product drying steps. Furthermore, the EDTA loss could partially be due to entrainment of EDTA during various precipitation steps. Given the critical nature of EDTA consumption to the overall operating costs, further optimization at the pilot scale should be undertaken to quantify and reduce EDTA losses at each step of the process.
The simplified version of the process was effective at efficiently producing the target products with fewer processing steps and complexity. As such, the simplified process, incorporating separations at pH 3, 7.2, 11.9, and 1.7 was selected as the best process configuration and its block flow diagram and mass balance is presented in the next section.
Block flow diagram and mass balance of the simplified process
The cyclical leaching and selective precipitation trials demonstrated the potential of operating the developed process continuously, in a closed loop, as illustrated in Fig. 9. In this process, fresh ore is first selectively leached in relatively dilute (0.29 M) sulfuric acid, producing a phosphate- and iron-rich leachate. The solid residues of the leaching step can potentially be washed with water to remove any residual acidity, allowing the ore to be landfilled or backfilled (the wash water could then be used as make-up water in the process). The iron content of the leachate is then stabilized using EDTA as a chelating agent, allowing selective precipitation of phosphorus using CaO.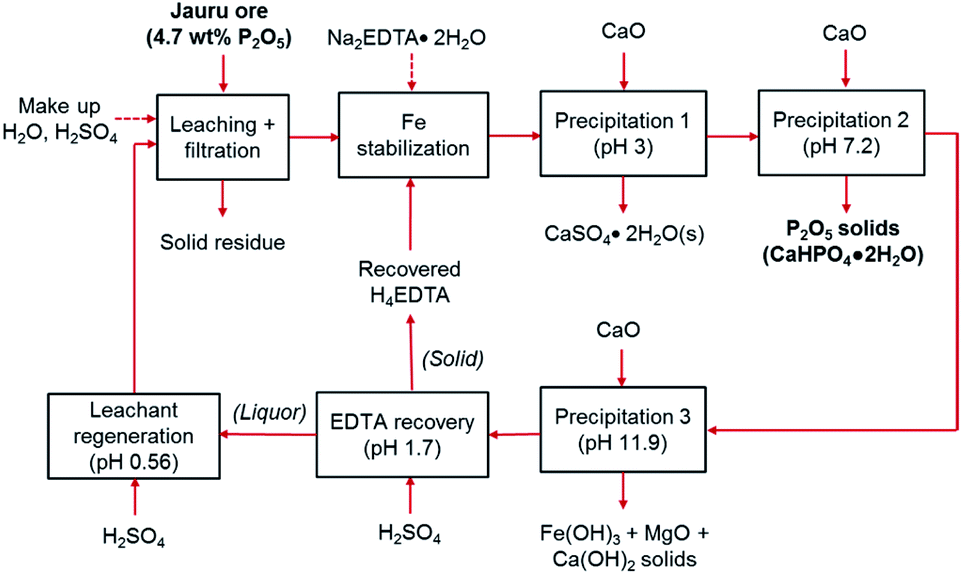 | ||
| Fig. 9 Block flow diagram of the overall process for P2O5 recovery from low-grade phosphate ore. | ||
The selective precipitation sequentially produces three value-added solid products. (1) At pH 3, CaSO4·2H2O is produced, which can be used for manufacturing of wallboards, cement, and plaster of paris, or for soil conditioning. Similar to phosphogypsum, the solid CaSO4·2H2O byproduct of the wet phosphoric acid process, can be used directly for various applications, although it may require some purification if the final product proves to have unacceptable levels of fluoride, organic matter, or radionuclides, such as Ra226, Pb210, Po210, U238 or U234.50 (2) At pH 7, the primary product, dicalcium phosphate dihydrate is precipitated. As mentioned above, the phosphorus and impurities content of this product is consistent with commercially employed DCP products.49 (3) At pH > 11, the co-extracted iron and magnesium are precipitated, providing a potential low-grade iron ore product or a feedstock for pigment production. Since these various products can be employed in several different applications, the properties and specifications should be verified according to the specific end-use in future pilot-scale testing, and appropriate pretreatment or purification steps should be specified and employed if necessary. This process can operate as a closed loop because the alkaline EDTA-bearing solution remaining after selective precipitation can be re-acidified with H2SO4, precipitating recyclable H4EDTA and CaSO4·2H2O solids at pH 1.7. After solid–liquid separation, the liquor can be employed as regenerated leachant for the initial leaching step at pH ∼ 0.6. The physical appearance of the slurries, filtered solutions, and wet filter cakes are shown in ESI Fig. S6.†
Because of the cyclical nature of the process, majority of the water and EDTA are recycled; hence, it only requires small make-up additions to account for lost water to humidity in the solid filter cakes and lost EDTA as an impurity in the solid products. Hence, the only major reagents required for the process are H2SO4 and CaO. The mass balance for the system at an industrial scale (1000 kg ore) was estimated on the basis of the laboratory scale results for the simplified process trial and the results are presented in Table 3. These estimations were made based on the following assumptions:
| Process step | pH | Reagent consumption | Solid products recovery |
|---|---|---|---|
| Ore leaching and filtration | 1.3 | Ore: 1000 kg, regenerated leachant: 3.3 m3 (from previous cycle) | Solid residue: 1020 kg |
| Precipitation 1 (gypsum) | 3.0 | Regenerated H4EDTA: 44 kg (from previous cycle), Na2EDTA·2H2O: 4 kg, CaO: 30 kg | CaSO4·2H2O product: 89 kg, 0 wt% P2O5 |
| Precipitation 2 (dicalcium phosphate dihydrate) | 7.2 | CaO: 17 kg | DCP product: 62 kg (dried at 150 °C), 43 wt% P2O5, 23.5 P2O5/RxOy |
| Precipitation 3 (impurities) | 11.9 | CaO: 11 kg | Impurity product: 21 kg, 2 wt% P2O5, 0.06 P2O5/RxOy |
| EDTA recovery | 1.7 | H2SO4: 24 kg (95–98 wt%) | Regenerated H4EDTA: 40 kg (for next cycle), 64 wt% H4EDTA |
| Leachant regeneration | 0.6 | H2SO4: 89 kg, make-up water: 725 kg | Regenerated leachant: 3.3 m3 (for next cycle) |
(1) the laboratory results can be scaled to the pilot/industrial scale.
(2) The solid–liquid separations are 100% efficient at separating the liquor and the precipitated solids. This could potentially be achieved by using the make-up water for the process to wash each solid product.
(3) The effect of water-containing solids such as CaSO4·2H2O on the water content of the system is negligible.
(4) The total liquid volume of the system is approximately constant at 3.3 m3. The addition of CaO, H2SO4 and EDTA does not significantly affect the liquid volume in the system.
The large-scale production rates and reagent consumptions are only estimates based on the laboratory scale results and require validation with pilot-scale testing, in particular for further understanding the liquor losses in solid–liquid separation, the requirements for water and EDTA make-up, and potential scale-related effects.
The EDTA consumption is a key process performance indicator because it is considerably more expensive than the other reagents.
The economic feasibility of this process was preliminarily assessed by estimating the reagents costs and revenue from the products (Table S2†). The net revenue for the as-is process was estimated to be $3.50 (USD) per tonne of processed ore.
One of the major contributors to the cost was the 4 kg per tonne make-up Na2EDTA; however, since the process could be optimized such that 100% of EDTA is recovered, a revenue of up to $8.81 (USD) per tonne of ore could be achieved. Hence, there is a potential for this process to be operated profitably. Since ore transportation costs are a major cost contributor for phosphate fertilizer production in Brazil,16 this process is an economically attractive option of mining and processing of low-grade phosphate ore directly in the agricultural region of Mato Grosso, where it will ultimately be used.
Conclusions
In this work, a dicalcium phosphate dihydrate (CaHPO4·2H2O) product was produced from a low-grade phosphate ore rich in FeO(OH) and SiO2. The process consisted of leaching with H2SO4 followed by stabilization with EDTA, and selective precipitation with CaO. This process can be operated in a closed loop to recycle water and EDTA, allowing for efficient separation of P2O5 from co-extracted iron without a solvent extraction step. The EDTA forms a complex with Fe3+ ions, stabilizing them in the solution, which prevents their co-precipitation with phosphorus. This process produces three primary products, i.e., CaSO4·2H2O, CaHPO4·2H2O, and an iron and magnesium rich precipitate, without producing any major liquid waste. The solid residue after leaching step can potentially be washed and backfilled to reduce the environmental impacts. The CaHPO4·2H2O product (43 wt% P2O5) contains minimal impurities (P2O5/RxOy = 23.5) and can be used directly for fertilizer or livestock feed additive production.The developed process enables production of value-added products from a low-grade ore with minimal reagent consumption and minimal wastewater generation. The processing steps proposed in this work, i.e., leaching, ion stabilization, selective precipitation, and reagent recovery can potentially be adapted for other low-grade phosphorus resources or even secondary resources such as sewage or municipal waste.14,51 As an example, a phosphorus enrichment process for municipal solid waste incineration fly ash has been reported which produces a solid product relatively similar to the low-grade phosphate ore used in this process.52 The results of this work can help address global nutritional demand without imposing negative impacts on the environment.
Author contributions
John Anawati: designing and performing experiments, data analysis, writing – original draft, writing – review & editing. Gisele Azimi: conceiving the research, supervision, project administration, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms Jihye Kim for help with EPMA and Dr Raiden Acosta for help with XRD.References
- R. W. Scholz, A. H. Roy, D. T. Hellums, A. E. Ulrich and F. S. Brand, Sustainable phosphorus management: a global transdisciplinary roadmap, 2014 Search PubMed.
- M. B. Hocking, Handbook of Chemical Technology and Pollution Control, 3rd edn, 2005 Search PubMed.
- J. G. Speight, Industrial Inorganic Chemistry, 2017 Search PubMed.
- N. P. Cheremisinoff and P. E. Rosenfeld, in Handbook of Pollution Prevention and Cleaner Production: Best Practices in the Agrochemical Industry, Elsevier, 2011, pp. 1–24 Search PubMed.
- G. De Groote and G. Huyghebaert, Anim. Feed Sci. Technol., 1997, 69, 329–340 CrossRef CAS.
- A. E. Lamp, A. Mereu, I. Ruiz-Ascacibar and J. S. Moritz, J. Appl. Poult. Res., 2020, 29, 559–572 CrossRef.
- United States Department of Agriculture, Fertilizer Use and Price, https://www.ers.usda.gov/data-products/fertilizer-use-and-price/ Search PubMed.
- P. W. Waldroup, Poult. Sci., 1999, 78, 683–691 CrossRef CAS.
- Food and Agriculture Organization of the United Nations (FAO), World fertilizer trends and outlook to 2022, 2019 Search PubMed.
- D. Cordell, J. O. Drangert and S. White, Glob. Environ. Change, 2009, 19, 292–305 CrossRef.
- J. J. Elser, Curr. Opin. Biotechnol., 2012, 23, 833–838 CrossRef CAS.
- Y. Ruan, D. He and R. Chi, Minerals, 2019, 9, 253 CrossRef CAS.
- S. Kataki, H. West, M. Clarke and D. C. Baruah, Waste Manag., 2016, 49, 437–454 CrossRef CAS.
- S. Donatello and C. R. Cheeseman, Waste Manag., 2013, 33, 2328–2340 CrossRef CAS.
- T. F. Al-Fariss, H. O. Ozbelge, F. A. A. Aleem and S. M. Abdulrazik, J. King Saud Univ., Eng. Sci., 1992, 4, 33–44 CAS.
- G. A. Da Silva and L. A. Kulay, J. Cleaner Prod., 2005, 13, 1321–1325 Search PubMed.
- J. A. Hinkebein and E. L. Koerner, US Pat., US3361527, United States Patent and Trademark Office, 1964.
- J. Q. Jiang and O. Mwabonje, Sep. Sci. Technol., 2009, 44, 3258–3266 CrossRef CAS.
- M. Takahashi, S. Kato, H. Shima, E. Sarai, T. Ichioka, S. Hatyakawa and H. Miyajiri, Chemosphere, 2001, 44, 23–29 CrossRef CAS.
- A. E. Martell, R. J. Motekaitis, D. Chen, R. D. Hancock and D. McManus, Can. J. Chem., 1996, 74, 1872–1879 CrossRef CAS.
- G. Anderegg, F. Arnaud-Neu, R. Delgado, J. Felcman and K. Popov, Pure Appl. Chem., 2005, 77, 1445–1495 CAS.
- M. A. Sierra, M. Gómez-Gallego, R. Alcázar, J. J. Lucena, F. Yunta and S. García-Marco, Dalton Trans., 2004, 3741–3747 RSC.
- Independent Commodity Intelligence Services, Indicative Chemical Prices A-Z, http://www.icis.com/chemicals/channel-info-chemicals-a-z/ Search PubMed.
- I. Park, C. B. Tabelin, S. Jeon, X. Li, K. Seno, M. Ito and N. Hiroyoshi, Chemosphere, 2019, 219, 588–606 CrossRef CAS.
- Z. L. Kardos, US Pat., US5225087, 1993.
- D. Voglar and D. Lestan, Chemosphere, 2013, 91, 76–82 CrossRef CAS.
- L. Di Palma, P. Ferrantelli, C. Merli and F. Biancifiori, J. Hazard. Mater., 2003, 103, 153–168 CrossRef CAS.
- I. M. Lo and W. Zhang, J. Environ. Eng., 2005, 131, 1507–1513 CrossRef CAS.
- T. T. Lim, P. C. Chui and K. H. Goh, Chemosphere, 2005, 58, 1031–1040 CrossRef CAS.
- S. Riaño and K. Binnemans, Green Chem., 2015, 17, 2931–2942 RSC.
- H. Sabbag, A. Brenner, A. Nikolski and E. J. C. Borojovich, Desalin. Water Treat., 2015, 55, 61–69 CrossRef CAS.
- P. A. Gallagher, C. A. Schwegel, X. Wei and J. T. Creed, J. Environ. Monit., 2001, 3, 371–376 RSC.
- K. R. Vuyyuru, K. K. Pant, V. V. Krishnan and K. D. P. Nigam, Ind. Eng. Chem. Res., 2010, 49, 2014–2024 CrossRef CAS.
- M. Wang, K. Do Woo, I. Y. Kim, Woong-Ki and Z. Sui, Hydrometallurgy, 2007, 89, 319–322 CrossRef CAS.
- OLI Studio 9.6.2, OLI Systems Inc., 2019 Search PubMed.
- C. W. Weston, J. W. Wen and F. S. Mandel, US Pat., US4485078, United States Patent and Trademark Office, 1983.
- R. Hill, US Pat., US4039624, United States Patent and Trademark Office, 1975.
- F. Soltani, M. Abdollahy, J. Petersen, R. Ram, S. M. Javad Koleini and D. Moradkhani, Hydrometallurgy, 2019, 184, 29–38 CrossRef CAS.
- S. Wu, L. Zhao, L. Wang, X. Huang, Y. Zhang, Z. Feng and D. Cui, J. Rare Earths, 2019, 37, 652–658 CrossRef CAS.
- A. E. Martell, R. M. Smith and R. J. Motekaitis, in NIST Standard Reference Database 46, NIST, 2001 Search PubMed.
- W. Jiang, X. Wang, Q. Xu, J. Xiao and X. Wei, RSC Adv., 2019, 9, 132–138 RSC.
- E. Lydersen, B. Salbu, A. B. S. Polèo and I. P. Muniz, Water Resour. Res., 1991, 27, 351–357 CrossRef CAS.
- C. Oliveira, A. Ferreira and F. Rocha, Chem. Eng. Res. Des., 2007, 85, 1655–1661 CAS.
- O. Mekmene, S. Quillard, T. Rouillon, J. M. Bouler, M. Piot and F. Gaucheron, Dairy Sci. Technol., 2009, 89, 301–316 CrossRef CAS.
- M. Al-Hindi and F. Azizi, Can. J. Chem. Eng., 2018, 96, 274–284 CrossRef CAS.
- G. Buxbaum, H. Printzen, M. Mansmann, D. Räde, G. Trenczek, V. Wilhelm, S. Schwarz, H. Wienand, J. Adel, G. Adrian, K. Brandt, W. B. Cork, H. Winkeler, W. Mayer and K. Schneider, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009, vol. 61, pp. 59–64 Search PubMed.
- M. Frèche and J. C. Heughebaert, J. Cryst. Growth, 1989, 94, 947–954 CrossRef.
- J. G. Rabatin, R. H. Gale and A. E. Newkirk, J. Phys. Chem., 1960, 64, 491–493 CrossRef CAS.
- L. W. de O. Souza, A. de S. Moretti, F. M. Tucci, N. H. de Souza, P. A. M. Leal and N. H. Anzai, Rev. Bras. Zootec., 2009, 38, 90–98 CrossRef.
- A. M. Rashad, J. Cleaner Prod., 2017, 166, 732–743 CrossRef CAS.
- J. J. Weeks and G. M. Hettiarachchi, J. Environ. Qual., 2019, 48, 1300–1313 CAS.
- Y. Kalmykova and K. Karlfeldt Fedje, Waste Manag., 2013, 33, 1403–1410 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental Fig. S1–S6, Tables S1 and S2. See DOI: 10.1039/d0ra07210a |
| This journal is © The Royal Society of Chemistry 2020 |