
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnique protonation states of aspartate and topaquinone in the active site of copper amine oxidase†
Mitsuo Shoji *ab,
Takeshi Murakawac,
Mauro Boerod,
Yasuteru Shigetaa,
Hideyuki Hayashie and
Toshihide Okajimaef
*ab,
Takeshi Murakawac,
Mauro Boerod,
Yasuteru Shigetaa,
Hideyuki Hayashie and
Toshihide Okajimaef
aCenter for Computational Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan. E-mail: mshoji@ccs.tsukuba.ac.jp
bJST-PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cDepartment of Biochemistry, Osaka Medical College, 2-7 Daigakumachi, Takatsuki, Osaka 569-8686, Japan
dUniversity of Strasbourg, Institut de Physique et Chimie des Matériaux de Strasbourg, CNRS, UMR 7504, 23 rue du Loess, F-67034, France
eDepartment of Chemistry, Osaka Medical College, 2-7 Daigakumachi, Takatsuki, Osaka 569-8686, Japan
fInstitute of Scientific and Industrial Research, Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan
First published on 21st October 2020
Abstract
The oxidative deamination of biogenic amines, crucial in the metabolism of a wealth of living organisms, is catalyzed by copper amine oxidases (CAOs). In this work, on the ground of accurate molecular modeling, we provide a clear insight into the unique protonation states of the key catalytic aspartate residue Asp298 and the prosthetic group of topaquinone (TPQ) in the CAO of Arthrobacter globiformis (AGAO). This provides both extensions and complementary information to the crystal structure determined by our recent neutron diffraction (ND) experiment. The hybrid quantum mechanics/molecular mechanics (QM/MM) simulations suggest that the ND structure closely resembles a state in which Asp298 is protonated and the TPQ takes an enolate form. The TPQ keto form can coexist in the fully protonated state. The energetic and structural analyses indicate that the active site structure of the AGAO crystal is not a single state but rather a mixture of the different protonation and conformational states identified in this work.
Introduction
Copper amine oxidases (CAOs) are ubiquitous enzymes present in a wide variety of aerobic organisms from bacteria to yeast, plants, and mammals.1,2 The various functions of CAOs are exerted by the oxidative deamination of primary amines to their corresponding aldehydes followed by the reduction of molecular oxygen (O2) to generate hydrogen peroxide (H2O2). In bacteria, CAOs are required for assimilating amines being the only source of nitrogen and carbon in their metabolism. All CAOs so far characterized are homodimer, and each subunit contains 3–4 domains with a molecular mass of 70–95 kDa.3,4 The active site of CAO is located at the edge of the β-sandwich of the C-terminal domain. The CAO active site in each subunit contains one type II copper (Cu) ion and a quinone cofactor, 2,4,5-trihydroxyphenylalanine quinone (TPQ). The cofactor originates from a post-translational modification of a specific tyrosine residue through an autocatalytic oxidation process involving the Cu and dioxygen.5The overall catalytic reaction of CAO proceeds through a ping–pong Bi–Bi mechanism including the two half-reactions composed of the reductive and oxidative half-reactions sketched in Fig. 1.6 In the reductive half-reaction, an oxidative form of TPQ is nucleophilically attacked by the amine substrate, and, via intermediate states of the substrate- and the product-Schiff bases, is reduced to an aminoresorcinol form, which eventually reaches an equilibrium state with a semiquinone radical. Simultaneously the amine substrate is oxidized to the product aldehyde. In some amine substrates of CAOs from Arthrobacter globiformis (AGAO) and bovine plasma (BSAO),7,8 strong deuterium kinetic isotope effects were observed for the product Schiff base formation. A conserved aspartate residue (Asp298 in AGAO) near the TPQ was suggested to act as a catalytic base. The Asp is also expected to be important for anchoring the substrate and reducing the flexibility of the TPQ ring plane.9–13
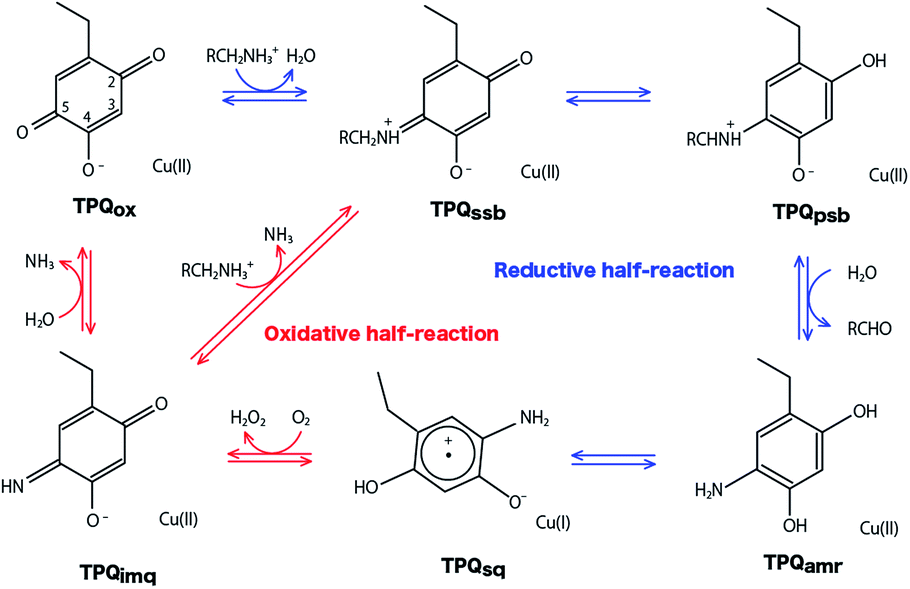 | ||
| Fig. 1 Catalytic cycle of AGAO. Proposed states are resting oxidative form (TPQox), substrate Schiff base (TPQssb), product Schiff base (TPQpsb), aminoresorcinol (TPQamr), semiquinone radical (TPQsq), and iminoquinone (TPQimq).6 | ||
In the oxidative half-reaction, the semiquinone radical form is oxidized to an iminoquinone form (TPQimq) with the formation of H2O2 (Fig. 1). The TPQimq is then hydrolyzed to the oxidative form of TPQ in the resting state (TPQox) with the release of ammonia. In the oxidative half-reaction, the rate-determining step is a double electron transfer to O2.14,15 In this stage, the two electrons are transferred along with two-proton transfers to generate H2O2 from O2. The protons are expected to be transferred between the TPQ and O2 via hydrogen bonds (H-bonds). Direct H-bond between O2 and TPQ and H-bonds via a conserved Tyr residue (Tyr284 in AGAO) are the primary candidates. However, the actual proton transfer pathways are not yet fully determined.16 Asp298 is the only base which can assist the deprotonation occurring during the hydrolysis of TPQimq. Therefore, the protonation states and the H-bond network in the CAO active site are the crucial factors to trigger this multistep catalytic reaction.
We recently provided an accurate determination of the positions of H atoms in AGAO by neutron diffraction (ND) with a resolution of 1.72 Å.17 On the basis of the TPQ protonation state, the neutron scattering length density map revealed the presence of a keto form TPQ and the enolate form, in which the enolate form has been believed to be the sole chemical form in TPQox state. In addition, one H atom was identified at a central position among three protonatable atoms, namely the OD1 and OD2 atoms in Asp298 and O(5) of TPQ. Although a similar triply shared proton was observed in the 5′-methylthioadenosine nucleosidase, these protonation states are uncommon. In order to unravel the nature of these unusual protonation states, precise quantum-mechanics-based simulations are required.
In the present study, the possible protonation states of the TPQ, Asp298, and other titratable residues including the water molecules coordinated to the Cu (Watax, Wateq) in the active site of AGAO are investigated through the direct comparison to the ND data in the TPQox state. For the triply shared proton, we examine the possibility of the presence of an alkali metal cation as an alternative to an actual proton. To this aim, we provide an accurate analysis of the related, potential energy surfaces to energetically locate all the stable conformers. We also made a thorough search for the possible interconversion pathways between the stable conformers. These results provide novel insights into the unusual protonation states and H-bond network in AGAO. In addition, we investigate the structural similarities between the oxidized state and the precursor state, TPQimq state, since the protonation state of TPQimq is also crucial because of a direct interaction with Asp298.
Computational details
The initial structure of AGAO was taken from the recently determined ND structure (PDBID: 6L9C).17 Atomic positions with higher occupation factor in the crystal structure were selected for the construction of the disordered residues, and the protonation states of titratable residues were followed to the ND structure. A one-subunit model containing one TPQ and one Cu atom was prepared. We have checked that the results by using a dimer model are qualitatively the same as those by using the monomer model (detailed results are shown in the ESI†). The modelled AGAO was immersed into a water droplet with a 60 Å radius and the neutrality of the total charge was ensured by placing water molecules with 21 Na+ counter ions. The whole system was relaxed at a classical level within the Amberff99 force field framework. Equilibration was done via molecular dynamics (MD) at 298.15 K for 1 ps, followed by an annealing MD from 298.15 K to 0 K for another ps. The scope of these relatively short equilibration and annealing MD is just the stabilization of undetermined light protons to complement the ND structure and, in this respect, times of the order of 1 ps are sufficient. In both MDs, added solvent water molecules and all the H atoms in AGAO are allowed to move, while the atoms kept fixed are the non-hydrogen atoms of AGAO, O atoms belonging to the crystallographic water molecules and all the atoms inside the QM region including TPQ, Cu cation, and the triply shared proton near Asp298.After the MD equilibration, hybrid quantum mechanics/molecular mechanics (QM/MM) simulations were performed. The QM subsystem includes the side chains of Tyr284, Asp298, Tyr384, Asn381, TPQ382, His431, His433, and His592, the Cu2+ ion and three water molecules within the first hydration shell of the Cu2+, as shown in Fig. 2. The combination of quantum and classical descriptions used in the present work is an unrestricted density functional theory approach of the UB3LYP-D3/DZVP type and an Amberff99 force field. The hybrid exchange-correlation functional B3LYP, accounting for an exact-exchange refinement, is complemented by Grimme's D3 dispersion correction.18 The electron orbitals are expanded on a localized basis set of valence double zeta plus polarized (DZVP) level. Specifically, LANL-2DZ for Cu and 6-31G* for all the other atoms.19–21 The selected UB3LYP/DZVP combination of the functional and basis set grants accurate structure and energetics for several enzymatic reactions.22–24 The Amber force field used for the MM subsystem is same parameter sets used in the full classical MD relaxations.
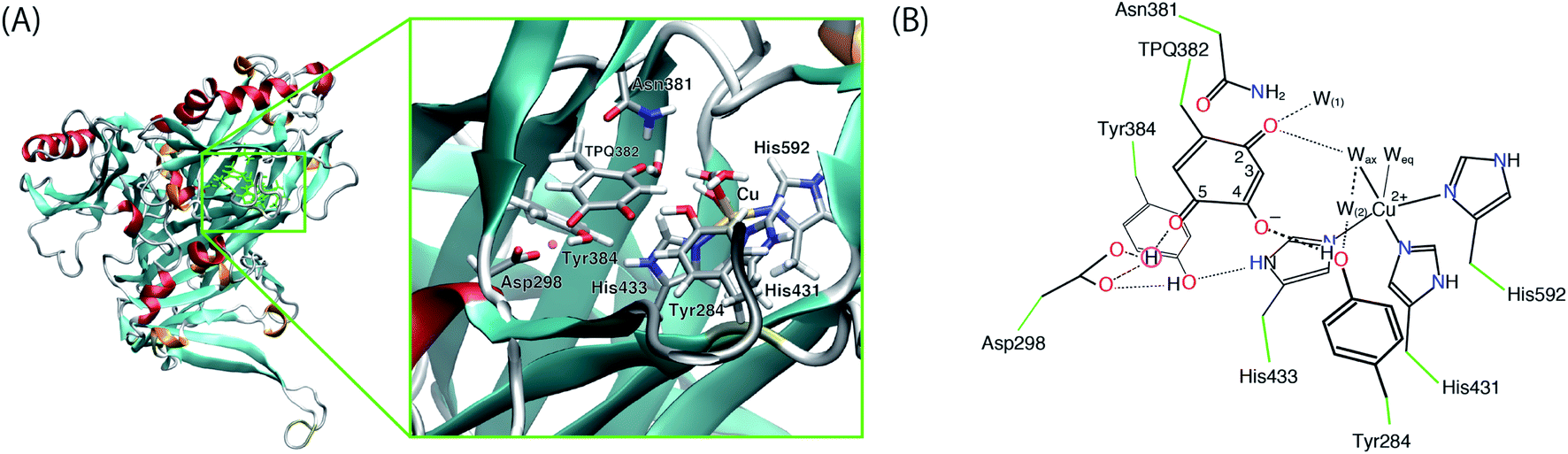 | ||
| Fig. 2 (A) Overview of AGAO and the enlarged view of the active site. The triply shared proton is highlighted in pink. (B) Schematic illustration of the active site of AGAO. The molecular structure in the QM and MM regions used in the present QM/MM study are coloured black and green, respectively. | ||
Geometry optimizations were performed for all atoms within a 15 Å radius from a fixed centre of the QM region. Covalent bonds across the QM and MM regions were saturated by hydrogen link atoms for the QM region. An electronic embedding scheme was adopted, and non-bonded interactions for the QM/MM interactions were explicitly computed avoiding the use of ad-hoc cutoff distances (no-cutoff). Free energies were evaluated upon the inclusion of the enthalpy and entropy corrections estimated from the frequency calculations at room temperature (T = 298.15 K) in the optimized states.
All simulations, MD and QM/MM calculations, were performed with the NWChem 6.5 program package.25 Molecular structures shown in figures were drawn with the VMD program.26
Results and discussion
Protonation states of the crystal structure obtained by neutron diffraction
Using the QM/MM model described in the computational details, four different protonation states were considered: (i) the fully protonated (Full) state, (ii) the singly deprotonated (–H+) one, (iii) the doubly deprotonated (–2H+) one, and (iv) the triply deprotonated (–3H+) one. The scope is to determine the best suited protonation state and H-bond network for the reported crystal structure. In Table 1, the calculated relative free energies and root-mean-square deviations (RMSDs) are summarized for all the stable states above the most stable state by less than 10 kcal mol−1. The results of the Full state were already reported in our previous study.17 In the present paper, instead, other protonation states, along with their H-bond formations, are mainly discussed. The states are labelled according to their relative stability and the total charge of the QM region. For instance, 0 and 1 refer to the most and second-most stable protonation states in the Full state, respectively, whereas 0n− and 1n− refer to those of –nH+. Their protonation states are schematically illustrated in Fig. 3. All the states including the higher energy ones are listed in Table S1 of the ESI.† We found that the closest to the ND structure among all the calculated state is the most stable state of –H+, i.e., the TPQ enolate state (0−). The RMSD for the QM heavy atoms is RMSD(0−) = 0.107 Å, one order of magnitude smaller than the ND resolution, indicating that our simulation did not bring the system to a conformation far from the experimental one. With respect to the relative stability, the second stable state of –H+ (1−) is still close to the ND structure (RMSD(1−) = 0.190 Å, G(1−) = 7.8 kcal mol−1). The major difference between these states (1− and 0−) is the rotation of the carboxy group of Asp298 (Fig. 4). The structural changes in the deprotonated His residues, His431, His433, and His592, are nearly negligible (02−, 22−, 32−, 42−, 13−, 33−), and the total number of deprotonated states increases as they combine to each other as in –2H+ and –3H+. Given these similarities, we shall focus the ongoing discussion on the deprotonation of the His residues. Another matter of debate is the stability of the keto form with respect to the normal protonation of the enolate form. On this aspect we try to shed some light in the next paragraphs.| States | G/kcal mol−1 | RMSD/Å | Protonation state |
|---|---|---|---|
| Fully protonated (Full) | |||
| Asp298 protonated | |||
| 0 | 0 | 0.251 | TPQ382 O(2) protonated |
| 1 | 0.69 | 0.167 | TPQ382 keto form |
| Asn381 side chain rotated | |||
| 2 | 1.95 | 0.264 | TPQ382 keto form |
| 3 | 3.24 | 0.306 | TPQ382 O(4) protonated |
| 4 | 9.87 | 0.375 | TPQ382 O(5) protonated |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Deprotonated (–H+) | |||
| TPQ382 enolate form | |||
| Asp298 protonated | |||
| 0− | 0 | 0.107 | — |
| 1− | 7.84 | 0.190 | Asp298 rotated |
|
|||
| Doubly deprotonated (–2H+) | |||
| TPQ382 enolate form | |||
| Asp298 protonated | |||
| 02− | 0 | 0.238 | His433 deprotonated |
| Tyr384 OH rotated | |||
| 12− | 3.00 | 0.302 | Asp298 rotated |
| His433 deprotonated | |||
| Tyr384 OH rotated | |||
| 22− | 4.16 | 0.132 | His431 deprotonated |
|
|||
| Triply deprotonated (–3H+) | |||
| TPQ382 enolate form | |||
| Asp298 protonated | |||
| 03− | 0 | 0.304 | Asp298 rotated |
| His433 deprotonated | |||
| His431 deprotonated | |||
| Tyr384 OH rotated | |||
| 13− | 2.20 | 0.151 | His433 deprotonated |
| His431 deprotonated | |||
| Tyr384 OH rotated | |||
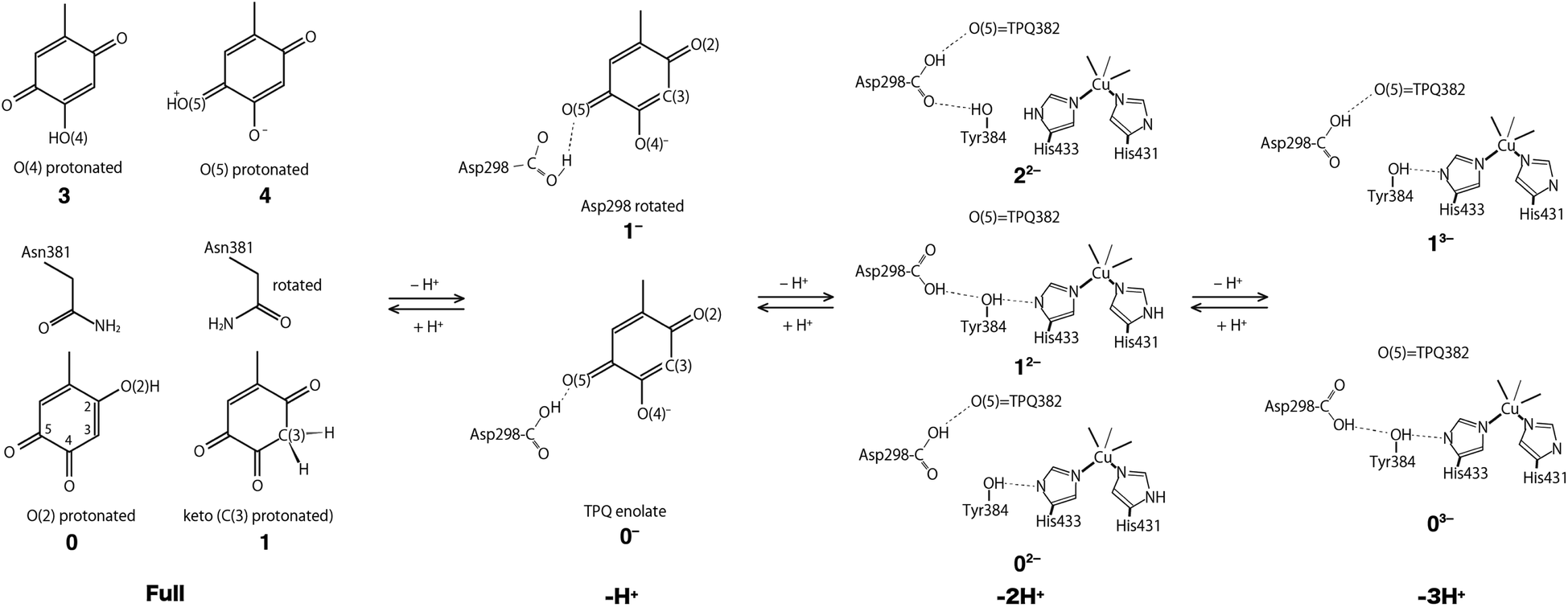 | ||
| Fig. 3 Schematic illustration of the stable states in the fully protonated (Full), singly deprotonated (–H+), doubly deprotonated (–2H+), and triply deprotonated (–3H+) states. All the conformations of Asp298 in the Full states are the same as the one in the 0− state. | ||
 | ||
| Fig. 4 Superimposed view of the QM/MM optimized structures (0− and 1−) in the singly deprotonated (–H+) state compared to the neutron structure (ND). Only the atoms in the QM region are shown. The structures are coloured by element (0−), orange (1−), and green (ND). | ||
Fully protonated (Full) states
In the Full state, TPQ is over-protonated because all the protonatable amino acid residues of the active site are already completely protonated. Among all the possible TPQ protonated states, we found that the O(2) atom is the one that can be most easily protonated. Then the C(3), O(4), and O(5) atoms are, in this order, the other protonatable sites. The relative free energies are G(0) = 0, G(1) = 0.69, G(3) = 3.2, and G(3) = 9.9 kcal mol−1, respectively.17 We can infer that the keto form with a protonated C(3) site can exist in a thermally accessible energetic state in the Full condition. The QM/MM optimized structures of 0 and 1 are superimposed in Fig. S2† to evidence their analogies and differences. The stable keto form is a unique feature found only in the Full state, because the keto forms in the other protonation states become significantly unstable (G(5−) = 34.0 kcal mol−1, G(72−) = 37.0 kcal mol−1, and G(83−) = 48.1 kcal mol−1). In the keto form, the rotated conformation of the Asn381 side chain is more stable by ΔG = 1.3 kcal mol−1 (1 vs. 2). As the H atom of the Asn381 amide group of 2 is close to the H atom of keto C(3), the contiguity between the TPQ keto moiety and Asn381 amide group can be reduced in 1 (RC(3),N(Asn381)(2) = 3.53 Å, RC(3),O(Asn381)(1) = 2.93 Å).Singly deprotonated (–H+) state
In the –H+ state, several plausible states can a priori be identified, such as the TPQ keto form, a Watax deprotonation, and deprotonations of the histidine residues. We remark that for the TPQ keto form, the rotation of Asn381 in the amide group is considered. We found that only the TPQ enolate forms of 0− and 1− are stable, while the other states turn out to be less stable by more than ∼20 kcal mol−1, thus making them unlikely to be realized. From an energetics standpoint, the second stable state 1− presents a rotated Ap298 carboxy group with respect to 0−. In this 1− state, TPQ O(5) is still close to Asp298, and an H-bond between TPQ O(5) and the Asp298 carboxy group is formed (Fig. 4). In the third stable state 2−, the TPQ382 O(2) atom is protonated, and His433 is deprotonated (G(2−) = 19.9 kcal mol−1). The TPQ keto form is energetically higher by G(5−) = 34.0 kcal mol−1, which shows that the TPQ keto form is rather unstable compared to the enolate form. The state with the TPQ keto form and Watax = OH is even more unstable, featuring a free energy of G(7−) = 36.8 kcal mol−1. When other protonation states different from the TPQ enolate form (0− and 1−) are to be formed, a deprotonation is required for one of the amino acid residues followed by the protonation to the TPQ enolate form is required since the amino acid residues near the active site are fully protonated, which translates into a double instability. Hence, it is legitimate to conclude that only the TPQ enolate form (0− and 1−) is stable in this protonation state (–H+). The direct enolate-keto transformation of 0− ↔ 7− is very unlikely to occur within the –H+ state because of the large free energy difference.Doubly deprotonated (–2H+) state
In the –2H+ state, three stable states have been found and all of them are energetically located in a small interval of less than 4 kcal mol−1. We found that the imidazole side chains of His433 and His431 can be easily deprotonated, and some conformers of the Tyr384 and Asp298 side chains co-exist upon the deprotonation of His433. The deprotonation of the His592, Asp298, and Watax require higher energies above 17 kcal mol−1 to be realized, thus the probability of deprotonation of these residues becomes negligible. On the ground of their relative energetics, we get the following order for the deprotonation sites:| His433 ∼ His431 > Asp298 ∼ Watax > His592. |
Interestingly, the deprotonation of Asp298 is as difficult as the deprotonation of Watax. The most stable state in –2H+ (02−) is the His433 deprotonated state, and the direction of H-bonds connecting from His433 to Asp298 via Tyr384 are reversed compared to the most stable state in –H+ (0−). In the stable states of 02− and 12−, the hydroxy group of Tyr384 points to His433. The H-bond of Tyr384 is directed toward Asp298 in 32−, analogously to 0−. The RMSDs of the altered H-bond in 02− and 12− are rather large (RMSD(02−) = 0.238 Å, RMSD(12−) = 0.302 Å) compared to the original conformation in 0− (RMSD(32−) = 0.158 Å).
In the most stable state (02−), Asp298 forms one H-bond with TPQ O(5). Due to the rotation of Asp298 and the H-bond with Tyr384, the H-bond between Asp298 and TPQ is lost (RO,O = 4.03 Å), and TPQ can apart from Asp298 toward Asn381 in 12−. In 12−, one H-bond is formed between the Tyr382 phenol and deprotonated His433 (RO,N = 2.74 Å). The TPQ shift contributes to increase the RMSD value.
The H-bonds of 22− are similar to those of 0− because of the presence of a deprotonated His433 in 22−. These results suggest that the H-bond network in the side chains of Asp298, Tyr384, and His433 is close to the ND structure in both 22− and 0−. The QM/MM optimized structures of 02−, 12−, and 22− are shown in Fig. S3.† As 22− is not clearly stabilized, we were unable to determine the actual deprotonation of His431 in the ND structure within the present QM/MM approach.
Triply deprotonated state (–3H+)
In the –3H+ state, systems carrying deprotonated His431 and His433 are very stable. We found that two conformers for the Asp298 side chain are energetically stable and structurally very close to each other, except for the H-bond of Asp 298. These conformations closely resemble those found in other protonated states (–H+(0− and 1−) and –2H+(02− and 12−)). The state with the rotated Asp298 carboxy group (03−) is slightly lower in energy than the unrotated state (13−) by 2.2 kcal mol−1. The QM/MM optimized structures of 03− and 13− are shown in Fig. S4.† The state in which the Tyr384 form a H-bond with Asp298 (23−) is energetically located at 11.3 kcal mol−1 above the most stable state (03−), in which the hydroxy group of Tyr384 points toward the deprotonated His433. The energetically subsequent state, 33−, is located at 13.2 kcal mol−1 above 03−, and is characterized by deprotonated His431 and His433. The following states (43−, …) are more unstable by more than 20 kcal mol−1, thus being unlikely to exist at ordinary conditions. Specifically, the deprotonations of Asp298 and Watax are not stabilized. Therefore, in –3H+, the stable state is the two-histidine deprotonated state, and Asp298 can take two conformations.Assignment of the triply shared proton: possibility of an alkali metal atom
Based on the ND density map, the experimentally identified proton is located apart from the two O atoms of Asp298 and O(5) of TPQ by distances of 1.56, 2.00, and 1.59 Å, respectively. These distances are clearly beyond any O–H chemical bond, thus it seems that this proton remains unbounded between Asp298 and TPQ.17 During our QM/MM calculations, we always failed to obtain the unusual H-bonded state of the ND structure, even if the starting positions of the H atom are changed. This result is clearly unable to explain the ND observed proton position.17 Rationalizing this uncommon isolated H atom position observed by ND is hard challenge also at a theoretical level. An alternative possibility if that this ND-identified site is not a proton but rather an alkali metal atom cation (M) trapped between the negatively charged Asp298 and TPQ. Even the electron density is not clearly observed, presence of low occupancy of an alkali metal atom is possible.17 In fact, potassium (K+) and sodium (Na+) cations are highly contained in both the crystallization buffer (1.05 M potassium–sodium tartrate in 25 mM HEPES-NaOH, pH = 7.4) and cryoprotection/deuteration buffer (3.0 M deuterated malonate-NaOD, pD = 7.4).17We investigated this possibility by replacing the proton with a Na+ cation in our QM/MM system. With this substitution, we obtained the O–Na distances of 2.12, 2.39, and 2.22 Å for the Asp298 OD1, OD2, and TPQ O(5) atoms, respectively. These longer atomic distances between the O and Na+ atoms approach the values of the ND structure. The distances between Asp298 OD1 and TPQ O(5) were 2.88 and 3.84 Å, respectively for H+ and Na+, to be compared with the value of 2.72 Å in the ND structure. The total RMSDs for the heavy atoms excluding the shared atom (total non-H RMSDs) are 0.107 and 0.283 Å for the H+ and Na+ models, respectively. If a Li+ cation is examined similarly, the O–Li distances becomes shorter (1.84, 1.97, and 1.85 Å). However, the distance between Asp298 OD1 and TPQ O(5) does not shrink (3.41 Å). The total non-H RMSD was 0.220 Å in the case of the presence of Li+. The superimposed structures for the metal substituents are shown in Fig. S5.† In the alkali metal models, the TPQ ring becomes rather far away from Asp298, mainly due to their longer van der Waals atomic radii. These results corroborate the idea that the shared atom detected by ND can be consistently assigned to a H atom, rather than to alkali metal cations.
Two-dimensional-potential energy surface (2D-PES) of the shared proton
In an attempt at rationalizing the unusual position of the shared proton located almost at the central position among the O atoms of the Asp298 carboxy group and TPQ O(5), an extensive PES calculation was done with special attention on the three H+ binding sites. To this aim, we performed constrained optimizations for the QM region, in which the imposed constraints are two distances between the H and two Asp298 carboxy O atoms (OD1, OD2). The contour map of the calculated PES for the shared proton is shown in Fig. 5. The optimized states of 0− and 1− correspond to the local minimum at (x, y) = (1.00 Å, 2.34 Å) and (2.17 Å, 1.00 Å), respectively. On the other hand, the TPQ O(5) protonated state can only be obtained under a constrained optimization upon imposing a constraint for the distance between the H and TPQ O(5) atoms (d(H, O(5)) = 1.0 Å). This protonated state (state X) is located at (x, y) = (1.56 Å, 2.00 Å) on the 2D-PES. The 2D-PES in Fig. 5 shows that the state X is unstable (E = 25.6 kcal mol–1) and there is no local minimum near the state X. Hence, states near X are inevitably relaxed into the state 0−. Another notable result is that the ND structure, labelled as ND in Fig. 5, corresponds to the point at (x, y) = (1.56 Å, 2.00 Å), which is coincidentally very close to X on the 2D-PES, and evidencing the fact that the both states of ND and X are located between the states 0− and 1−.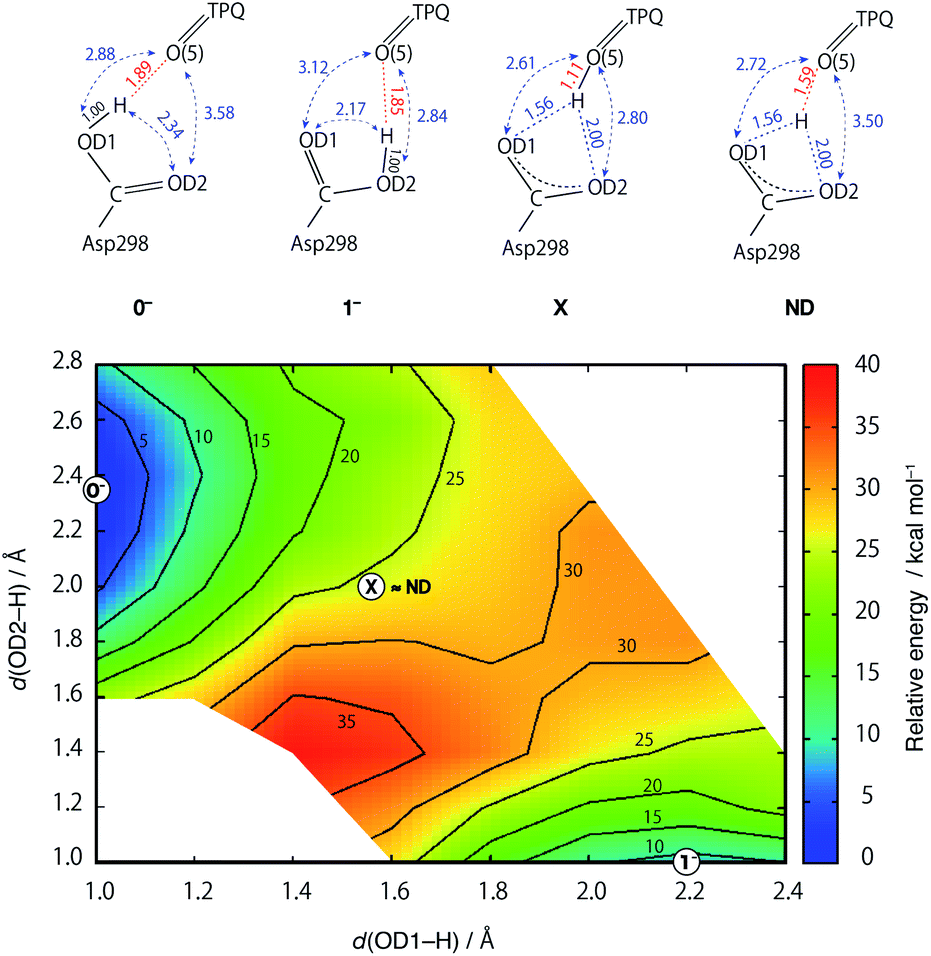 | ||
| Fig. 5 Two-dimensional potential energy surface (2D-PES) of the shared proton as a function of the distances from the O atoms of the Asp298 carboxy groups (x = d(OD1, H), y = d(OD2, H)). The contour lines are drawn at intervals of 5 kcal mol−1 relative to the energy at (x, y) = (1.0, 2.3), corresponding to the 0− state. The 2D-PES was discretized with a mesh of dx = 0.15 and dy = 0.15 Å intervals. | ||
The main structural changes, namely the O(5)–H and O(5)–OD1 distances, are shown in Fig. S6† for the same x and y ranges used in Fig. 5. In the stable minima (0− and 1−), the O(5)–H distances increase up to R(O(5)–H, 0−) = 1.89 Å and R(O(5)–H, 1−) = 1.85 Å, and in the intermediate region between 0− and 1−, the O(5)–H distances shrink (R(O(5)–H, X) = 1.10 Å). Yet, in the ND structure, the distance is longer (R(O(5)–H, ND) = 1.59 Å). The QM/MM results demonstrate that the dissociated H rapidly approaches TPQ O(5), although this is insufficient to achieve an energetically stable condition. As shown in Fig. S7,† the TPQ O(5) and Asp298 OD1 distance becomes slightly shorter in the intermediate region ranging from R(O(5)–OD1, 0−) = 2.88 Å to R(O(5)–OD1, X) = 2.61 Å, and increases up to the values observed in 1− (R(O(5)–OD1, 1−) = 3.12 Å), since, in turn, another distance to OD2 of Asp298 shrinks in 1− (R(O(5)–OD2, 1−) = 2.84 Å). Thus, in the broad area of the 2D region, TPQ O(5) exists close to the carboxy O atom of Asp298 in either of the protonated conformations (0− and 1−). On the other hand, for the ND structure, the two distances R(O(5)–OD1, ND) = 2.72 Å and R(O(5)–OD2, ND) = 3.50 Å, are close to those of 0−. These results indicate that the non-H atoms of ND are closer to the OD1 protonated state (0−) and not to the doubly shared proton of X.
In –2H+, a similar 2D-PES with no local minimum in the intermediate region was also obtained. This turns out to be similar to the 2D-PES for –H+, as shown in Fig. S8–S10.† From the analysis of the 2D-PESs, we could not locate local minimum at the central position among the Asp298 O atoms and TPQ O(5) in both the –H+ and –2H+ states.
Conformational change of the Asp298 carboxy group
Having been able to identify two stable conformations in the singly deprotonated states, two states are separated by high energy barrier of E‡ = ∼30 kcal mol−1 on the 2D-PES. We focused on searching possible interconversion pathways between the states 0− and 1−. Using the nudged elastic band (NEB) method, we inspected the 0− → 1− transition pathway. In particular, we considered two pathways in which a clockwise- (C) or counter clockwise- (CC) rotation of the Asp298 carboxy group around the Cβ–Cγ axis can occur (Fig. 6). The transition barrier results to be lower for the CC rotation by 2.3 kcal mol−1(E‡(CC) = 12.7 kcal mol−1 and E‡(C) = 15.0 kcal mol−1), and the 0− → 1− transition can easily occur upon ordinary thermal fluctuations. We also considered the Asp298 rotation in the other protonation state (–2H+). In –2H+, the two states are very close in energy (G(12−) = 3.0 kcal mol−1), and the transition barrier is calculated to be rather small for both the C and CC rotations (E‡(C) = 5 kcal mol−1, E‡(CC) = 8 kcal mol−1). For the rotated conformation in –2H+ (12−), as the TPQ side chain moves away from the Asp298 carboxy group (R(O(5)-OD1, 12−) = 4.03 Å) forming one H-bond between Tyr384 and Asp298. This makes the RMSD values larger (RMSD(02−) = 0.238 Å and RMSD(12−) = 0.302 Å) with respect to the smaller RMSDs in –H+ (RMSD(0−) = 0.107 Å, RMSD(1−) = 0.190 Å).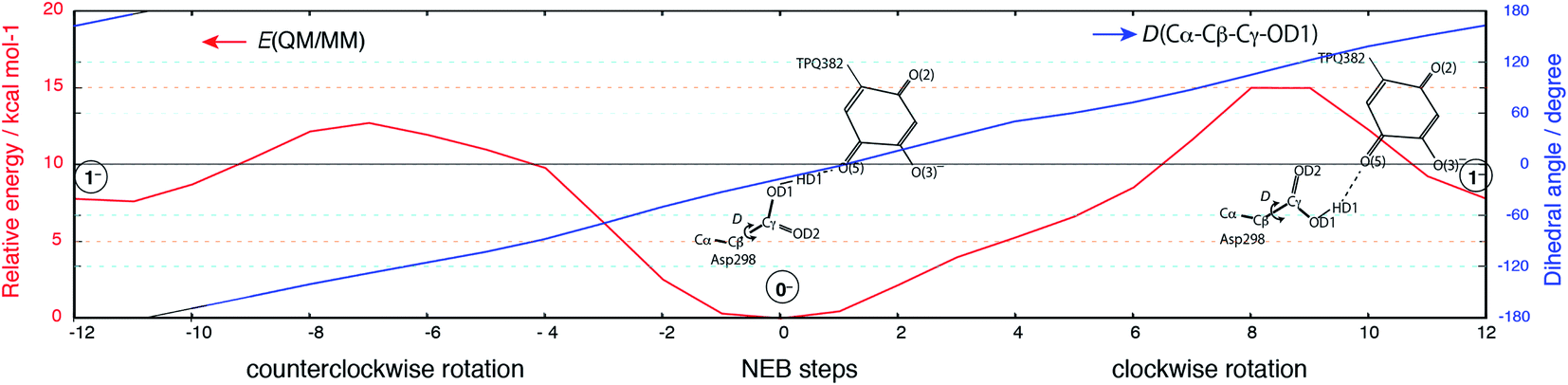 | ||
| Fig. 6 Relative energy and the dihedral angle along the NEB coordinates for the 0− → 1− transition. The steps from 0 to 12 and from 0 to −12 correspond to the clockwise and counterclockwise rotations, respectively. Schematic molecular structures of the Asp298 carboxy group and TPQ moieties are shown in the same panel. | ||
The Asp298 conformations and the low barrier transition pathways suggest that, statistically, some minor populations like the Asp298 rotated states might influence the ND observed shift of the proton to the central position. If the H atom is present in some Asp298 conformations in cooperation with the rotation of the carboxy group, the proton density may is likely to show deviations from the most stable local minima. The H atom in 1− is also close to the triply shared proton of ND (Fig. 4). The distance between the H atoms is 0.45 Å, while the other heavy atoms of Asp298 have approached TPQ O(5). We can infer that the population of the Asp298 rotated states may be related to the central proton observed in ND.
Iminoquinone state (imq)
The imq state is a transient intermediate realized before the last hydrolysis that reverts the system into the resting oxidative state in the oxidative half-reaction (Fig. 1). The crystal structure of the imq state has not yet been determined in AGAO. Here, the imq states were theoretically investigated for comparison with the resting state. By replacing the O(5)2– atom in 0− with a N(5)3– atom, the imq state (imq(02−)) can be obtained. After an optimization of imq(02−) at the QM/MM level, the proton of Asp298 is transferred to the TPQ N(5) atom forming the N(5)H with the deprotonated Asp298. The QM/MM optimized structure of imq(02−) is shown in Fig. 7. We found that the imq(02−) is also very close to the ND structure. In fact, the total non-H RMSD with respect to the ND structure is 0.153 Å. In imq(02−), the tight H-bond between N(5)H and Asp298 induces a curvature of the TPQ plane as observed in 0− and ND structures. In the additionally protonated state of imq, the most stable condition was found to be the protonated Asp298 with TPQ = N(5)H (imq(0−)). The imq keto forms with the deprotonated Asp298 turn out to be very unstable with G(imq(keto−)) = 39.3 kcal mol−1 and G(imq(ketorot−)) = 49.8 kcal mol−1 in the absence of any rotation of the side chain of Asn381 and when the rotation occurs, respectively. Other protonation states for the TPQ O(2) and O(4) are still rather unstable, being characterized by energy difference of G(imq(O(2)H−)) = 42.7 kcal mol−1 and G(imq(O(4)H−)) = 35.3 kcal mol−1. These results indicate that the imq takes only one protonated state in each protonated state (imq(02−) or imq(0−)), and the remaining protonated states, such as the TPQ keto form and TPQ O protonated states, are energetically too high to become accessible, which are very different from the resting oxidative state.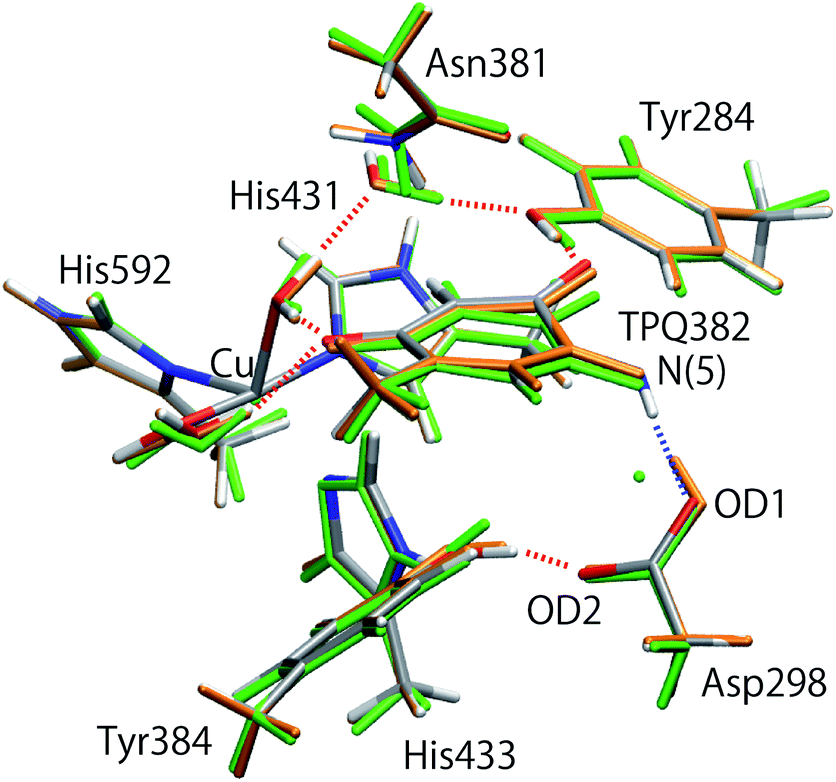 | ||
| Fig. 7 Superimposed view of the QM/MM optimized structure in the iminoquinone state (imq(02−)) compared to the oxidized state (0−) and neutron structure (ND). Only the atoms in the QM region are shown. Structures are coloured by element (imq(02−)), orange (0−), and green (ND). | ||
Conclusions
By resorting to accurate QM/MM methods, we provide a thorough investigation of the protonation states of the active site in AGAO. Our purpose is to analyse the unusual location of protons evidenced by recent ND study.17Among all the models considered here, the one best compatible with the ND data in TPQox is the TPQ enolate form in the singly deprotonated state (–H+). In this state, all the amino acid residues in the active site are fully protonated, and stable conformers are found in the Asp298 carboxy group (0− and 1−). For higher deprotonation (–2H+), the states carrying a deprotonated His433 or His431 are energetically stable. We remark that some conformations are accessible in the His433 deprotonated state upon changes in the H-bond over Tyr384 and Asp298. This is a clear indication that a linking H-bond network exists over TPQ, Asp298, Tyr384, and His433. On the other hand, Watax = OH is unstable and hard to realize even in the deprotonated states. In the fully protonated state (Full), the TPQ keto form can coexist with the most stable state.
The ND detected H-atom position between Asp298 and TPQ (ROH = ∼1.56 Å) has been found to be very unstable and energetically demanding (E > 25 kcal mol−1) at the present QM/MM theoretical level. Indeed, no local minimum could be located at the central position, and the H atom can only be stabilized in one mono-bonded state belonging to either one of the carboxy O atoms of Asp298 (ROH = ∼1.0 Å).
The possibility of an alkali metal (M+) cation instead of the H+ atom has also been ruled out by our analysis. In fact, the RMSD values become very large as the TPQ plane departs away from Asp298, while the distances of H+/M+ to the closest O of Asp298 increase to 1.8 and 2.1 Å for Li+ and Na+, respectively, in comparison to 1.56 Å in ND.
As the Asp298 carboxy group can be rotated by keeping a H-bond with TPQ O(5) in –H+ with a low activation barrier (E‡ = 12.7 kcal mol−1), the conformer 1− is also possible as a minor contribution. Since the position of the H atom in 1− is close to that of the H atom in the ND data, the unusual shared proton can be interpreted as arising from the multiple conformations that Asp 298 can assume.
The iminoquinone state (imq), which is the last transient intermediate state before hydrolysis, has been modelled from a resting state, thus proposing a structural model not yet available in the literature. This imq state is also remarkably close to the ND structure by forming a tight H-bond between Asp298 and TPQ.
As the keto form of TPQ can be formed only in the fully protonated state, we can infer that the state of AGAO in the crystal structure is not a single configuration but rather a mixture of different protonation states and the different conformers. Further investigations related to the catalytic mechanisms in both half-reactions will be pursued to get a deeper insight into the uncommon proton location observed in the ND structure. We underscore that such an anomalous triply shared proton is likely to be characterized by a high reactivity, and, for this reason, can play a key role in the catalytic reactions. The overall scenario we depicted for a shared proton detected at an unusual position in the ND structure is important in at least two respects: (i) on one hand it provides a rational explanation for a functionally important proton, and (ii) on the other hand, provides a paradigm that can be generalized beyond the specific AGAO case, including other enzymes and biomolecular systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JST, PRESTO Grant Number JPMJPR19G6, Japan and JSPS KAKENHI ground numbers 16KT0055, 17H04866, 19H05781, 20H05453, and 20H05448. Numerical calculations were carried out under the support of (1) Multidisciplinary Cooperative Research Program in CCS, University of Tsukuba, and (2) HPCI system research project (project ID: hp190110) using the computational resource of CX400 provided by the Information Technology Center in Nagoya University. M. B. thanks the HPC mesocenter at the University of Strasbourg funded by the Equipex Equip@Meso project (Programme Investissements d'Avenir) and the CPER Alsacalcul/Big Data, and the Grand Équipement National De Calcul Intensif (GENCI) under allocation DARI-A0080906092.References
- M. J. McPherson, M. R. Parsons and C. M. Wilmot, Handbook of metalloproteins, John Wiley & Sons, Ltd, 2001, vol. 2, pp. 1245–1257 Search PubMed.
- D. L. Wertz and J. P. Klinman, Handbook of metalloproteins, John Wiley & Sons, Ltd, 2001, vol. 2, pp. 1258–1271 Search PubMed.
- M. R. Parsons, M. A. Convery, C. M. Wilmot, K. D. S. Yadav, V. Blakeley, A. S. Corner, S. E. V. Phillips, M. J. Pherson and P. F. Knowles, Structure, 1995, 3, 1171–1184 CrossRef CAS.
- M. C. J. Wilce, D. M. Dooley, H. C. Freeman, J. M. Guss, H. Matsunami, W. S. McIntire, C. E. Ruggiero, K. Tanizawa and H. Yamaguchi, Biochemistry, 1997, 36, 16116–16133 CrossRef CAS.
- D. Cai and J. P. Klinman, J. Biol. Chem., 1994, 269, 32039–32042 CAS.
- T. Murakawa, S. Baba, Y. Kawano, H. Hayashi, T. Yano, T. Kumasaka, M. Yamamoto, K. Tanizawa and T. Okajima, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 135–140 CrossRef CAS.
- T. Murakawa, T. Okajima, S. Kuroda, T. Nakamoto, M. Taki, Y. Yamamoto, H. Hayashi and K. Tanizawa, Biochem. Biophys. Res. Commun., 2006, 342, 414–423 CrossRef CAS.
- K. L. Grant and J. P. Klinman, Biochemistry, 1989, 28, 6597–6605 CrossRef CAS.
- M. Mure, S. A. Mills and J. P. Klinman, Biochemistry, 2002, 41, 9269–9278 CrossRef CAS.
- B. Schwartz, E. L. Green, J. Sanders-Loehr and J. P. Klinman, Biochemistry, 1998, 37, 16591–16600 CrossRef CAS.
- J. Plastino, E. L. Green, J. Sanders-Loehr and J. P. Klinman, Biochemistry, 1999, 38, 8204–8216 CrossRef CAS.
- J. M. Murray, C. G. Saysell, C. M. Wilmot, W. S. Tambyrajah, J. Jaeger, P. F. Knowles, S. E. V. Phillips and M. J. McPherson, Biochemistry, 1999, 38, 8217–8227 CrossRef CAS.
- Y.-C. Chiu, T. Okajima, T. Murakawa, M. Uchida, M. Taki, S. Hirota, M. Kim, H. Yamaguchi, Y. Kawano, N. Kamiya, S. Kuroda, H. Hayashi, Y. Yamamoto and K. Tanizawa, Biochemistry, 2006, 45, 4105–4120 CrossRef CAS.
- S. Kishishita, T. Okajima, M. Kim, H. Yamaguchi, S. Hirota, S. Suzuki, S. Kuroda, K. Tanizawa and M. Mure, J. Am. Chem. Soc., 2003, 125, 1041–1055 CrossRef CAS.
- Y. Liu, A. Mukherjee, N. Nahumi, M. Ozbil, D. Brown, A. M. Angeles-Boza, D. M. Dooley, R. Prabhakar and J. P. Roth, J. Phys. Chem. B, 2013, 117, 218–229 CrossRef CAS.
- J. L. DuBois and J. P. Klinman, Biochemistry, 2006, 45, 3178–3188 CrossRef CAS.
- T. Murakawa, K. Kurihara, M. Shoji, C. Shibazaki, T. Sunami, T. Tamada, N. Yano, T. Yamada, K. Kusaka, M. Suzuki, Y. Shigeta, R. Kuroki, H. Hayashi, T. Yano, K. Tanizawa, M. Adachi and T. Okajima, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 10818–10824 CrossRef CAS.
- S. Grimme, S. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- M. Shoji, K. Hanaoka, Y. Ujiie, W. Tanaka, D. Kondo, H. Umeda, Y. Kamoshida, M. Kayanuma, K. Kamiya, K. Shiraishi, Y. Machida, T. Murakawa and H. Hayashi, J. Am. Chem. Soc., 2014, 136, 4525–4533 CrossRef CAS.
- Y. Abe, M. Shoji, Y. Nishiya, H. Aiba, T. Kishimoto and K. Kitaura, Phys. Chem. Chem. Phys., 2017, 19, 9811–9822 RSC.
- T. Tokiwa, M. Shoji, V. Sladek, N. Shibata, Y. Higuchi, K. Kataoka, T. Sakurai, Y. Shigeta and F. Misaizu, Molecules, 2019, 24(1), 76–87 CrossRef.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowaiski, T. P. Straatsma, H. J. J. van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477 CrossRef CAS.
- W. Humphery, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06365g |
| This journal is © The Royal Society of Chemistry 2020 |